

Abstract
The protective genes that mediate endothelial cell (EC) survival during angiogenesis have not been completely characterized. Here, we show that an antisense oligonucleotide to the apoptosis inhibitor survivin suppressed de novo expression of survivin in ECs by vascular endothelial cell growth factor (VEGF). In contrast, the survivin antisense oligonucleotide did not affect anti-apoptotic bcl-2 levels in endothelium. When assessed in cell death assays, antisense targeting of survivin abolished the anti-apoptotic function of VEGF against tumor necrosis factor-α- or ceramide-induced cell death, enhanced caspase-3 activity, promoted the generation of a ∼17-kd active caspase-3 subunit, and increased cleavage of the caspase substrate, polyADP ribose polymerase. In contrast, the survivin antisense oligonucleotide had no effect on EC viability in the absence of VEGF. Antisense oligonucleotides to platelet-endothelial cell adhesion molecule-1 (PECAM-1, CD31), lymphocyte function-associated molecule-3 (LFA-3, CD58), or intercellular adhesion molecule-1 (ICAM-1, CD54) did not reduce the anti-apoptotic function of VEGF in endothelium. When tested on other angiogenic activities mediated by VEGF, survivin antisense treatment induced rapid regression of three-dimensional vascular capillary networks, but did not affect EC migration/chemotaxis. These data suggest that the anti-apoptotic properties of VEGF during angiogenesis are primarily mediated by the induced expression of survivin in ECs. Manipulation of this pathway may increase EC viability in compensatory angiogenesis or facilitate EC apoptosis and promote vascular regression during tumor angiogenesis.
The preservation of vascular homeostasis during inflammation, immune response, and transplant accommodation depends on the ability of endothelial cells (ECs) to continuously counteract a cellular suicide program, ie, apoptosis. 1 This process involves a sequential cascade activation of intracellular cysteine proteases, ie, caspases, initiated by ligation of cell surface death receptors or by cytoplasmic assembly of cell death initiators, ie, apoptosome, induced after mitochondrial damage. 2 Inhibition of EC apoptosis is also an obligatory prerequisite of angiogenesis, in which multiple receptor-ligand interactions at the EC surface stimulate proliferation, migration, and remodeling of ECs to generate new vascular networks. 3 In this context, antibody or adenoviral targeting of critical angiogenesis regulators, including vascular endothelial cell growth factor (VEGF), 4,5 or the angiopoietin-1 (Ang-1) receptor, Tie-2, 6 resulted in rapid involution of vascular networks in vitro and in vivo, associated with classical morphological and biochemical hallmarks of EC apoptosis. The mechanisms that couple preservation of EC survival to angiogenesis have been the subject of intense investigation. In addition to survival signals mediated by adhesion molecule, ie, integrin, engagement, 7,8 and activation of the phophoinositide 3/Akt pathways, 9,10 angiogenesis has been associated with de novo expression of an heterogeneous set of anti-apoptotic protective genes in the endothelium, 11 which in some cases is mediated via nuclear factor-κB signaling. 12 Specifically, stimulation of ECs by VEGF or Ang-1 resulted in up-regulation of anti-apoptotic bcl-2 and A1 molecules, 13,14 and expression of inhibitor of apoptosis (IAP) proteins, 15 survivin, and XIAP. 16-18
In this study, we used an antisense targeting strategy to dissect the relative contribution of survivin to the anti-apoptotic activities of VEGF in endothelium.
Materials and Methods
EC Culture
Human umbilical vein ECs were maintained in M199 medium containing 20% fetal calf serum (FCS), 50 μg/ml EC growth supplement, 100 μg/ml heparin, 100 μg/ml penicillin, and 100 μg/ml streptomycin (all from Life Technologies, Grand Island, NY) in 5% CO2 at 37°C as described previously. 16 Subconfluent ECs were rendered quiescent by an 18-hour culture in M199 plus 0.1% FCS. Cells were detached with 0.05% trypsin/0.02% ethylenediaminetetraacetic acid (EDTA), seeded in C6-well plates (Costar Corp., New Bedford, MA), grown to 70% confluency, and used between passages 2 and 3.
Gene Targeting by Antisense
Quiescent EC monolayers were incubated with 50 ng/ml of recombinant VEGF (Collaborative Biomedical Products, Bedford, MA) for 24 hours at 37°C in M199 plus 0.1% FCS. At the end of the incubation, ECs were washed, harvested by trypsin/EDTA, and lysed in 0.5% Triton X-100, 0.5% Nonidet P-40, 0.05 mol/L Tris-HCl, 0.15 mol/L NaCl plus protease inhibitors. Protein-normalized aliquots of cell extracts were electrophoresed on sodium dodecyl sulfate-polyacrylamide gradient gels, transferred to nylon membranes (Millipore Corp., Bedford, MA) for 1 hour at 1 A, and immunoblotted with 2 μg/ml of a rabbit antibody to survivin 19 or a mouse monoclonal antibody to bcl-2 (Transduction Laboratories, San Diego, CA), followed by chemiluminescence (Amersham, Arlington Heights, IL) and autoradiography. Samples were sequentially analyzed by Western blotting with a mouse antibody to β-actin to confirm equal protein loading. To determine the contribution of survivin to EC protection mediated by VEGF, 2′-O-methoxyethyl chimeric phosphorothioate oligonucleotides were used. A survivin antisense oligonucleotide with the sequence 5′-TGTGCTATTCTGTGAATT-3′ was characterized previously for its ability to suppress endogenous survivin mRNA expression in T24 bladder and HeLa epithelial carcinoma cells. 20 A scrambled oligonucleotide with the sequence 5′-TAAGCTGTTCTATGTGTT-3′ was used as a control, and also characterized in previous cell culture assays. 20 Oligonucleotides were synthesized with uniform phosphorothioate linkages, and underlined nucleosides correspond to 2′-O-methoxyethyl nucleosides. Antisense oligonucleotides to platelet-endothelial cell adhesion molecule-1 (PECAM-1, CD31), lymphocyte function-associated molecule-3 (LFA-3, CD58), and intercellular adhesion molecule-1 (ICAM-1, CD54) were synthesized as described above and characterized in previous studies. 21 For transfection experiments, increasing concentrations of control or the various antisense oligonucleotides (50 to 500 nmol/L) were mixed with 1 ml of OPTI-MEM and 6 μl of Lipofectin according to the manufacturer’s instructions (Life Technologies), and incubated with serum-starved ECs for 8 hours. The transfection medium was replaced with M199 plus 0.1% FCS for an additional 18 hours followed by VEGF stimulation for 24 hours. Transfection efficiency monitored by fluorescence microscopy using fluorescein isothiocyanate-conjugated oligonucleotides was always >85%. To determine the effect of antisense treatment on survivin mRNA expression, proliferating ECs were transfected with control or the survivin antisense oligonucleotide, harvested after a 24-hour culture at 37°C, and total RNA was extracted using the Qiagen RNeasy reagent, according to the manufacturer’s specifications. Samples were separated on 1% agarose-formaldehyde gels, transferred to Hybond nylon membranes and hybridized with a 32P-random-primed labeled survivin cDNA 22 with visualization of radioactive bands by autoradiography. Northern blots were reprobed with random primed 32P-labeled human G3PDH cDNA to confirm equal loading.
Determination of EC Apoptosis
ECs were transfected with increasing concentrations of control or the various antisense oligonucleotides, stimulated with 50 ng/ml VEGF for 16 hours at 37°C, and incubated in the presence of 25 μmol/L of C-6 ceramide or the combination of tumor necrosis factor (TNF)-α (10 ng/ml; Endogen, Woburn, MA) plus cycloheximide (10 μg/ml, Sigma Chemical Co., St. Louis, MO) for an additional 12 hours at 37°C. At the end of the incubation, ECs (floaters plus attached cells) were harvested, fixed in 70% ethanol, stained with 10 μg/ml propidium iodide plus 100 μg/ml RNase A and 0.05% Triton X-100 in phosphate-buffered saline (PBS), pH 7.4, and analyzed for DNA content by flow cytometry as described previously. 16 In other experiments, transfected ECs stimulated with VEGF and incubated with C-6 ceramide for 12 hours at 37°C were harvested, washed in PBS, pH 7.4, and fixed in 4% paraformaldehyde containing 0.25% Triton X-100 for 10 minutes at 22°C. Cell nuclei were stained with 6.5 μg/ml 4,6-diamidino-2-phenylindole (DAPI, Sigma), 16% polyvinyl alcohol (Air Products and Chemicals, Allentown, PA), and 40% glycerol. The percentage of apoptotic cells was calculated by counting the average cells with nuclear morphology of apoptosis (chromatin condensation, DNA fragmentation) in three independent high-power fields (×400), each containing ∼150 cells using a Zeiss fluorescent microscope.
Caspase Activation
ECs transfected with the various oligonucleotides were stimulated with 50 ng/ml VEGF, incubated with C-6 ceramide as described above, and detergent-solubilized cell extracts were assayed for caspase-3-dependent hydrolysis of the fluorogenic substrate Ac-DEVD-AMC (N-acetyl-Asp-Glu-Val-Asp-aldehyde; Pharmingen, San Diego, CA). Fluorescence emissions were quantitated on a spectrofluorometer with excitation wavelength of 360 nm and emission of 460 nm. For biochemical markers of caspase activation, transfected ECs treated with VEGF plus ceramide were lysed in 0.25% Triton X-100, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L dithiothreitol, 20 mmol/L HEPES, plus protease inhibitors. Protein-normalized aliquots of the various cell extracts were separated by sodium dodecyl sulfate-gel electrophoresis, transferred to nylon membranes (Millipore Corp.), and immunoblotted with a 1:5000 dilution of a rabbit antibody to caspase 3 (Transduction Laboratories), or a 1:1000 dilution of a mouse antibody to PolyADP ribose polymerase (Pharmingen, San Diego, CA), followed by chemiluminescence and autoradiography.
EC Migration
Migration assays were performed using a Boyden chamber (Neuroprobe) as described. 23 Briefly, quiescent ECs were transfected with control or the survivin antisense oligonucleotide, stimulated with VEGF, and detached using 0.05% trypsin and 0.53 mmol/L EDTA. Twenty thousand cells were suspended in M199 medium containing 0.1% bovine serum albumin and added to the lower chamber. Polycarbonate filters (8-μm diameter) were coated with 100 μg/ml type I collagen. The top half of the chamber was attached and the chamber was incubated in an inverted position for 2 hours at 37°C. Increasing concentrations (1 to 500 ng/ml) of VEGF or d-erythro-sphyngosine-1-phosphate (SPP-1; Calbiochem, La Jolla, CA) were separately added to the upper chamber and incubated for an additional 5 hours at 37°C. At the end of the incubation, cells were fixed in 70% ethanol and nonmigrating ECs on the upper surface of the filter were removed. Migrated cells were stained with Giemsa and counted at ×400 magnification in three random fields per well. 23 Each experiment was performed in triplicate and migration was expressed as the total number of cells counted per well.
Three-Dimensional Capillary Formation
Monolayers of quiescent ECs (80% confluency) in C6-well plates were transfected with 500 nmol/L of control or the survivin antisense oligonucleotide. After an 8-hour incubation, the transfection medium was replaced with M199 medium containing 0.1% FCS for an additional 18 hours at 37°C. Rat-tail type I collagen (3 mg/ml; Becton Dickinson, Bedford, MA) in 1/10 volume of 10× Dulbecco’s modified Eagle’s medium was neutralized with sterile 1 mol/L NaOH and kept on ice. Suspended ECs were added to the collagen suspension to a final concentration of 1 × 10 6 cells/ml of collagen. Ten drops (0.1 ml each) of the EC-collagen mixture were added to a 35-mm plate. Plates were placed in a humidified incubator at 37°C, and the EC-collagen mixtures were allowed to gel for 10 minutes, after which 3 ml of M199 medium containing 20% FCS, 50 μg/ml EC growth supplement, 100 μg/ml heparin, 100 μg/ml penicillin, and 100 μg/ml streptomycin were added to each plate. ECs were allowed to form capillary-like vascular tubes throughout a 24-hour incubation in the presence of 16 nmol/L phorbol myristate acetate (PMA, Sigma). Addition of PMA in this experimental protocol results in a potent morphogenic effect promoting the formation of endothelial tube-like structures, which closely mimics capillary formation in vivo via a PKC-, MAPK-, and PI3-kinase-dependent pathway. 24 As determined in previous studies, PMA withdrawal under these conditions is associated with rapid regression of capillary structures and EC apoptosis, in vitro. 24 After an additional 24-hour incubation, ECs were washed three times in PBS, pH 7.4, and supplemented with fresh M199 growth medium in the presence or in the absence of 50 ng/ml VEGF. The cultures were examined by phase-contrast microscopy for the presence of capillary-like vascular tubes during an additional 48-hour incubation at 37°C as described. 25
Results
Antisense Inhibition of VEGF-Induced Expression of Survivin in EC
Previous studies demonstrated that a survivin 2′-O-methoxyethyl chimeric phosphorothioate antisense oligonucleotide (5′-TGTGCTATTCTGTGAATT-3′) inhibited survivin mRNA and protein expression in HeLa and T24 cancer cell lines. 20 Consistent with these observations, increasing concentrations of the survivin antisense oligonucleotide suppressed survivin mRNA expression in proliferating ECs in a dose-dependent manner, by Northern blotting (Figure 1A) ▶ . In contrast, comparable concentrations of a control scrambled oligonucleotide (5′-TAAGCTGTTCTATGTGTT-3′), did not decrease survivin mRNA levels in ECs (Figure 1A) ▶ . In parallel experiments, VEGF stimulation resulted in an approximate fourfold increased survivin expression in quiescent endothelium (Figure 1B) ▶ , and in agreement with previous observations. 16-18 Pretreatment of ECs with the survivin antisense, but not control oligonucleotide, suppressed VEGF induction of survivin in a concentration-dependent manner by Western blotting (Figure 1B) ▶ . In contrast, transfection with control or the survivin antisense oligonucleotide did not reduce antiapoptotic bcl-2 expression in endothelium by Western blotting 13,14 (Figure 1C) ▶ .
Figure 1.
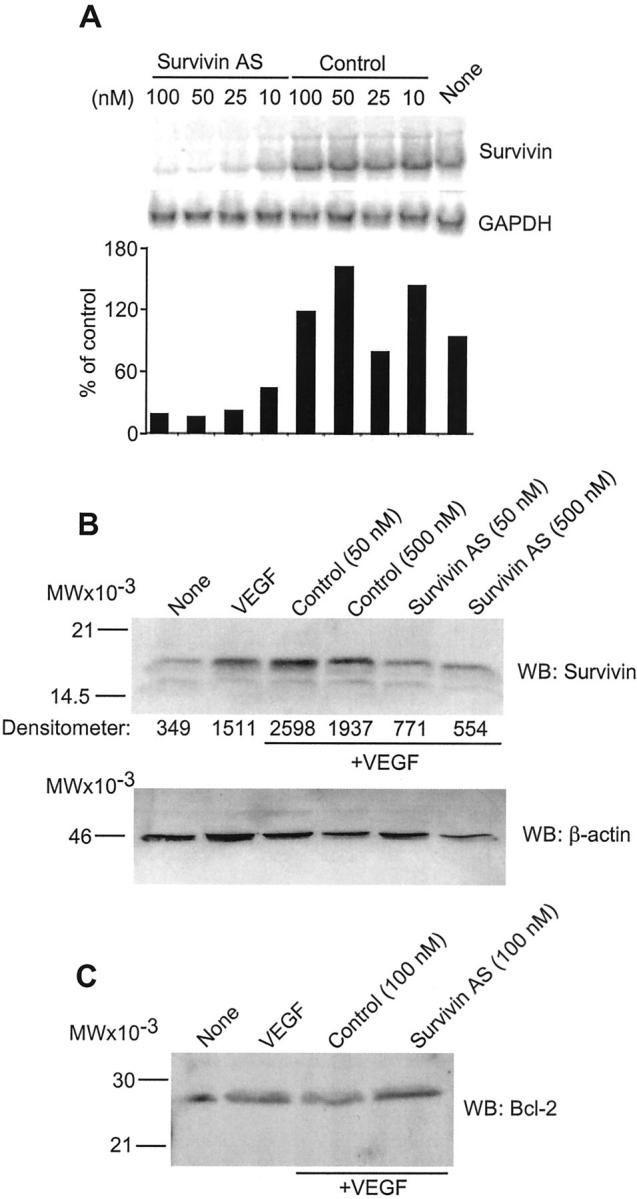
Antisense inhibition of survivin expression in ECs. A: ECs were transfected with the indicated increasing concentrations of control-scrambled (control) or the survivin antisense oligonucleotide (survivin AS) followed by hybridization with cDNAs for survivin or GAPDH. Densitometric quantitation of hybridizing bands under the various conditions tested is shown at the bottom. B: Quiescent ECs were stimulated with VEGF (50 ng/ml) in the presence of the indicated oligonucleotide concentrations and protein-normalized cell extracts were immunoblotted with an antibody to survivin or β-actin followed by chemiluminescence. C: The experimental conditions are the same as in B, except that EC extracts treated with control or the survivin antisense oligonucleotide were analyzed with an antibody to bcl-2 by Western blotting. For panels B and C, molecular weight markers in kd are indicated on the left. WB, Western blot.
Antisense Targeting of Survivin Suppresses the Anti-Apoptotic Activities of VEGF in ECs
Exposure of quiescent ECs to C-6 ceramide resulted in induction of apoptosis as determined by chromatin condensation and DNA fragmentation, by DAPI nuclear staining (Figure 2, A and B) ▶ . Addition of VEGF attenuated ceramide-induced EC apoptosis and restored normal nuclear morphology (Figure 2, A and B) ▶ . Under these experimental conditions, transfection of ECs with the survivin antisense oligonucleotide completely reversed the anti-apoptotic function of VEGF against ceramide-induced apoptosis, whereas the control oligonucleotide was without effect (Figure 2, A and B) ▶ . Similarly, treatment with ceramide or the combination of TNF-α plus cycloheximide resulted in an approximate sevenfold increase in EC apoptosis, as determined by the appearance of a cell fraction with hypodiploid, ie, sub-G1, DNA content, by propidium iodide staining and flow cytometry (Figure 2, C and D) ▶ . Addition of VEGF counteracted both ceramide- and TNF-α-induced apoptosis in ECs by ∼45% (Figure 2, C and D) ▶ . However, and consistent with the data presented above, EC transfection with the survivin antisense, but not control oligonucleotide, suppressed VEGF cytoprotection against both cell death-inducing stimuli, and restored EC apoptosis to levels observed in the absence of VEGF (Figure 2, C and D) ▶ .
Figure 2.

Inhibition of VEGF cytoprotection by survivin targeting. A: Quiescent ECs were transfected with control or the survivin antisense oligonucleotide, treated with VEGF (50 ng/ml), and exposed to C-6 ceramide (25 μmol/L). Cell nuclei were stained with 6.5 μg/ml of DAPI (Sigma), 16% polyvinyl alcohol, and 40% glycerol and scored morphologically for apoptosis (chromatin condensation, DNA fragmentation) using a Zeiss fluorescent microscope. Photographs of phase contrast or DAPI staining of each field are from a representative experiment out of at least three independent determinations. B: The experimental conditions are the same as in A. Data are expressed as percentage of apoptosis as determined by nuclear morphology by DAPI staining, and represent the mean ± SEM of three independent experiments. C and D: ECs transfected with control or the survivin antisense oligonucleotide were incubated with C-6 ceramide (25 μmol/L) (C), or the combination of TNF-α (10 ng/ml) plus cycloheximide (10 μg/ml) (D), for 12 hours at 37°C. Cells were analyzed for DNA content by propidium iodide staining and flow cytometry. The percentage of apoptotic cells with hypodiploid, ie, sub-G1, DNA content is indicated per each condition tested. Data are representative of an experiment of at least two independent determinations. Comparable transfection efficiencies were demonstrated by fluorescence microscopy of ECs transfected with fluorescein isothiocyanate-conjugated oligonucleotides.
Next, we asked if suppression of VEGF cytoprotection by survivin targeting was associated with biochemical hallmarks of apoptosis in ECs. Treatment of quiescent ECs with ceramide resulted in increased caspase-3 catalytic activity, as determined by hydrolysis of the fluorogenic substrate DEVD-AMC, and in a reaction entirely suppressed by the caspase-3 inhibitor, DEVD-CHO (Figure 3A) ▶ . This was associated with proteolytic cleavage of ∼32-kd proform caspase-3 and generation of active subunits of ∼19 and ∼17 kd (Figure 3B) ▶ , 26 and cleavage of the ∼115-kd caspase substrate polyADP ribose polymerase (PARP) to an ∼85-kd apoptotic fragment (Figure 3C) ▶ . Consistent with its survival properties in EC, addition of VEGF reduced ceramide-induced caspase-3 activity, nearly completely inhibited the generation of ∼17-kd active caspase-3 subunit, and of ∼85-kd PARP fragment (Figure 3; A, B, and C ▶ ). Under these experimental conditions, EC transfection with the survivin antisense, but not control oligonucleotide, restored caspase-3 catalytic activity, which was associated with proteolytic generation of a ∼17-kd active caspase-3 subunit and apoptotic fragmentation of PolyADP ribose polymerase (Figure 3, A, B, and C) ▶ .
Figure 3.
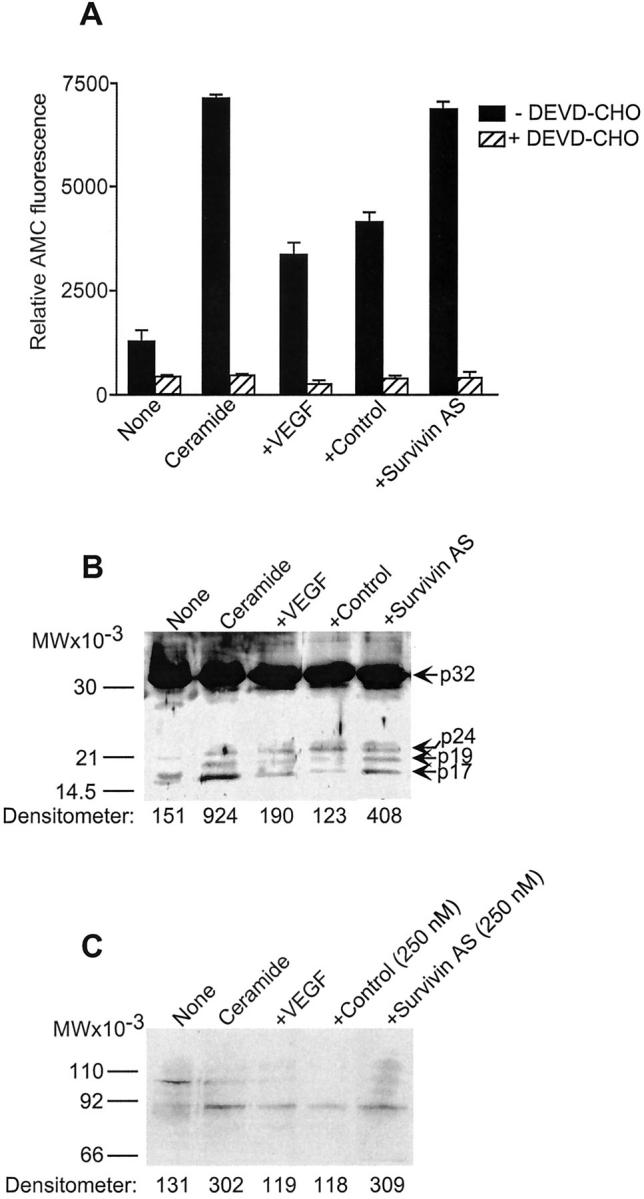
Modulation of caspase activity by survivin targeting. A: The conditions are as described in Figure 2 ▶ . Ceramide-treated ECs stimulated with VEGF and transfected with control or the survivin antisense oligonucleotide were analyzed for caspase-3 activity by hydrolysis of the fluorogenic substrate DEVD-AMC in the presence or absence of the caspase inhibitor DEVD-CHO. Data are the mean ± SD of two independent determinations. B: EC extracts under the various indicated conditions were analyzed for caspase-3 proteolytic cleavage with an antibody to caspase-3 by Western blotting. The positions of ∼32-kd proform caspase-3, of the intermediate product of ∼24 kd, and of active subunits of ∼17 and ∼19 kd are indicated. C: The experimental conditions are as in B, except that EC extracts were analyzed for proteolytic cleavage of the caspase-3 substrate, PolyADP ribose polymerase, by Western blotting. Densitometric quantitation under the various conditions tested was performed on the 17-kd active caspase-3 subunit (B) or the apoptotic ∼85-kd PARP fragment (C).
Specificity of Antisense Targeting of Survivin
Next, we wished to determine whether antisense treatment affected a constitutive versus VEGF-inducible population of survivin molecules in ECs. Incubation of ECs in 0% serum for 24 hours resulted in increased caspase-3 activity by DEVD hydrolysis as compared with continuously growing cultures (Figure 4A) ▶ , and appearance of an apoptotic cell fraction with hypodiploid DNA content by propidium iodide staining and flow cytometry (Figure 4B) ▶ . Addition of ceramide to these cells further increased caspase-3 activity and generation of ECs with hypodiploid DNA content (Figure 4, A and B) ▶ . However, in the absence of VEGF, and at variance with the data presented above, transfection of ECs with control or the survivin antisense oligonucleotide did not further enhance caspase-3 activity, or the generation of apoptotic cells in the presence or absence of ceramide (Figure 4, A and B) ▶ . These data suggest that antisense targeting of survivin affects EC viability only in the presence of VEGF stimulation.
Figure 4.
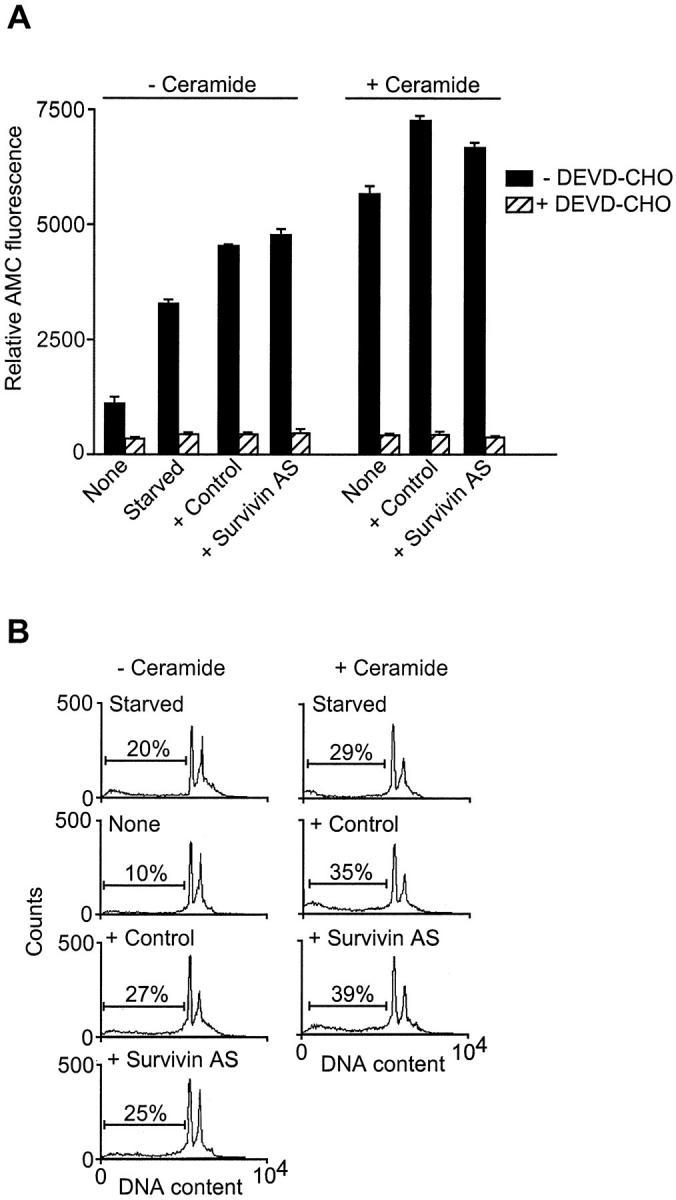
Effect of survivin targeting on quiescent ECs. ECs grown in 0% FCS for 24 hours were transfected with the indicated oligonucleotides and incubated in the absence (− ceramide) or presence (+ ceramide) of C-6 ceramide. Cell extracts were analyzed for caspase-3 activity by hydrolysis of the fluorogenic substrate DEVD-AMC, in the presence or absence of DEVD-CHO (A), or by DNA content by propidium iodide staining and flow cytometry (B). Data are expressed as the mean ± SD of three independent experiments. In B, the percentage of apoptotic cells with hypodiploid, subG1, DNA content is indicated.
In other experiments, EC transfection with antisense oligonucleotides to PECAM-1 (CD31), LFA-3 (CD58), or ICAM-1 (CD54) characterized in previous studies 21 did not significantly reduce the anti-apoptotic effect of VEGF against ceramide-induced EC apoptosis (Figure 5) ▶ , thus confirming the specificity of the survivin antisense approach. In contrast, EC transfection with the survivin antisense, but not control oligonucleotide, completely blocked the cytoprotective effect of VEGF in ceramide-treated cultures (Figure 5) ▶ , in agreement with the data presented above.
Figure 5.

Effect of antisense oligonucleotides to EC adhesion molecules on VEGF cytoprotection. The experimental conditions are as in Figure 2 ▶ , except that quiescent ECs were transfected with the indicated antisense oligonucleotides, stimulated with VEGF, and exposed to 25 μmol/L of C-6 ceramide. Cells were analyzed for nuclear morphology by DAPI staining after a 12-hour culture at 37°C. Data are the mean ± SEM of at least four independent experiments. **, P < 0.005; ***, P < 0.0009. ns, not significant.
Role of Survivin in VEGF-Induced EC Migration and Remodeling
The potential role of survivin targeting on other angiogenic responses induced by VEGF 3 was next investigated. First, stimulation with VEGF or SPP-1 resulted in EC chemotaxis and migration in a specific and concentration-dependent manner (Figure 6) ▶ , in agreement with previous observations. 23 However, transfection of VEGF-stimulated ECs with inhibitory concentrations of control or the survivin antisense oligonucleotide failed to decrease EC migration in response to VEGF or SPP-1 (Figure 6) ▶ .
Figure 6.
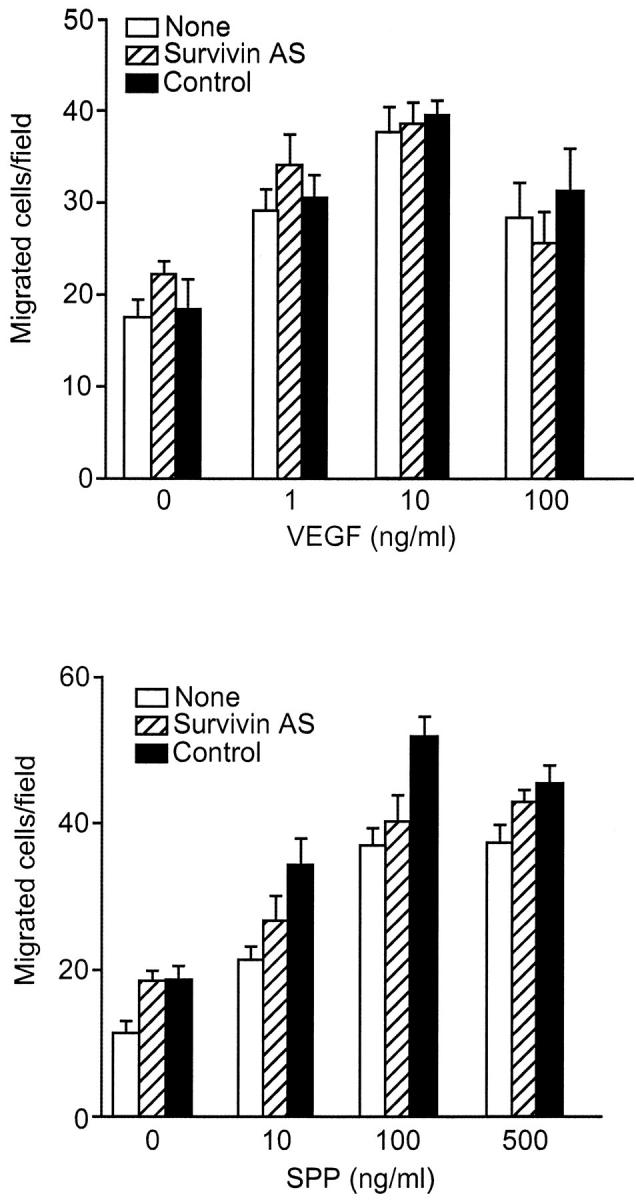
Effect of survivin targeting on VEGF-induced EC migration/chemotaxis. ECs were transfected with control or the survivin antisense oligonucleotide, stimulated with VEGF, and exposed to the indicated increasing concentrations of VEGF or control SPP-1 in a Boyden chamber. After a 5-hour incubation at 37°C, migrated cells were counted microscopically by Giemsa. Data are the mean ± SEM of triplicates of a representative experiment out of two independent determinations.
Secondly, we used a morphogenic model of PMA-induced capillary tube formation in vitro to determine the effect of survivin targeting on the remodeling phase of VEGF-induced angiogenesis. 24 In this system, withdrawal of PMA after capillary formation results in rapid EC apoptosis and involution of vascular tube structures in a reaction prevented by VEGF, which stabilizes three-dimensional capillary networks. 24 Accordingly, addition of VEGF to EC-collagen gels after PMA withdrawal resulted in the persistence of viable three-dimensional capillary-like structures throughout a 3-day culture at 37°C (Figure ▶ 7). 24 In contrast, no viable capillaries were formed in the absence of PMA (not shown), and withdrawal of VEGF resulted in rapid involution of three-dimensional vascular networks (Figure 7) ▶ . 24 Under these experimental conditions, transfection of ECs with control oligonucleotide did not affect the ability of VEGF to maintain EC viability and stabilize vascular capillaries after PMA withdrawal in vitro (Figure 7) ▶ . In contrast, transfection of EC with the survivin antisense oligonucleotide completely suppressed the anti-apoptotic function of VEGF in preserving EC viability, and resulted in complete involution of three-dimensional vascular networks throughout a 3-day culture (Figure 7) ▶ .
Figure 7.
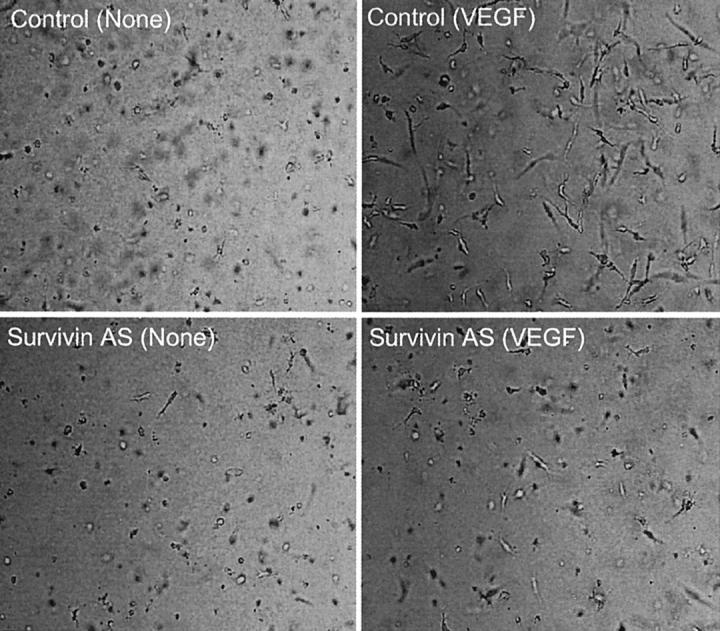
Effect of survivin targeting on three-dimensional capillary formation. Three-dimensional capillary networks were formed by culturing ECs in collagen gels in the presence of PMA, and stabilized in the absence (none) or presence of VEGF. ECs were transfected with control or the survivin antisense oligonucleotide and stability/regression of vascular capillary networks under the various conditions was analyzed by phase contrast microscopy throughout a 3-day culture at 37°C. Pictures are representative of one experiment out of at least three independent determinations. Original magnification, ×100.
Discussion
In this study, we have shown that antisense targeting of IAP family protein, 15 survivin, 22 selectively abrogated the anti-apoptotic properties of VEGF in endothelium. This resulted in ablation of VEGF cytoprotection against ceramide- or TNF-α-induced apoptosis, enhanced caspase-3 activity, generation of ∼17-kd active caspase-3 subunit, and involution of three-dimensional capillary-like networks in vitro. In contrast, other angiogenic functions of VEGF, including EC migration and chemotaxis were not affected by the survivin antisense, and, survivin targeting had no effect on cell viability of quiescent endothelium in the absence of VEGF.
One of the cornerstone requirements for new blood vessel formation, ie, angiogenesis, is the continuous preservation of endothelium viability, a multifaceted process that involves activation of pro-survival signaling pathways, 9,10 optimal cell adhesion mechanisms, 7,8 and de novo expression of specialized anti-apoptotic genes. 11 In this context, previous studies identified survivin as one of the target genes induced by VEGF in endothelium, which was associated with prominent up-regulation of survivin in newly formed blood vessels during angiogenesis in vivo. 16 Using selective antisense targeting, the data presented here suggest that most of the anti-apoptotic properties of VEGF are mediated via the de novo induction of survivin in endothelium. Several lines of evidence corroborate the specificity of the antisense approach used here. First, transfection with the survivin antisense had no effect on EC viability in the absence of VEGF, in agreement with the undetectable levels of survivin in quiescent endothelium in vitro and in vivo. 16 Secondly, a panel of antisense oligonucleotides targeting critical adhesion molecules expressed on ECs and including ICAM-1 (CD54), PECAM-1 (CD31), or LFA-3 (CD58) did not decrease the anti-apoptotic function of VEGF in endothelium. Thirdly, survivin targeting did not modify the levels of anti-apoptotic bcl-2, which has also been potentially implicated in VEGF cytoprotection. 13,14 Moreover, a similar antisense targeting strategy has been recently used to further dissect cell death pathways in endothelium and identify differential roles of bcl-XL or A1 in mediating EC cytoprotection. 27
Differently from other members of the IAP gene family, 15 survivin has recently attracted attention for its cell cycle-regulated expression in G2/M and localization to the mitotic apparatus, which suggested a potential role at the interface between apoptosis and cell division control. 28 Although an evolutionary conserved role of survivin in the regulation of mitosis and cytokinesis has been recognized, 20,29,30 the mechanisms by which survivin may control apoptosis are far from elucidated. However, physical proximity/association between survivin and initiator/effector caspases has been reported by several groups in vitro and in vivo. 31-33 Moreover, interference with expression/function of survivin by antisense (see below) or dominant-negative mutant was sufficient to cause increased caspase-3 activity and apoptosis at the G2/M transition. 20,28 This suggests that survivin, alone or in combination with as yet unidentified regulatory molecules, may influence the assembly/function of the cell death machinery and affect downstream caspase processing/topography 2 in a cell cycle-dependent manner. Consistent with this view, interference with mitotic phosphorylation of survivin on Thr 34 by p34cdc2-cyclin B1 resulted in mislocalization of initiator caspase-9 and apoptosis of cells traversing mitosis. 33 Although this pathway is clearly operative in proliferating cells and thus consistent with the mitogenic effect of VEGF on endothelium, the data presented here suggest that survivin may also mediate EC protection independently of cell cycle progression. In this context, antisense suppression of survivin abrogated the anti-apoptotic function of VEGF in a morphogenic, nonproliferative model of three-dimensional capillary formation in vitro. 24 Although the molecular requirements of survivin-dependent cytoprotection in nonproliferating endothelium remain to be elucidated, previous studies demonstrated that EC stimulation with Ang-1, which provides a nonproliferative, stabilizing signal in angiogenesis also resulted in increased survivin expression and apoptosis inhibition via an Akt-dependent pathway. 18
Differently from other apoptosis regulators of the bcl-2 34 or IAP 15 gene families, survivin is undetectable in normal adult tissues but becomes abundantly expressed in most human cancers in vivo. 22 Identified in genome-wide searches as the top fourth “transcriptome” found in common neoplasms, 35 expression of survivin has been associated with abbreviated survival rates, unfavorable prognosis, and accelerated recurrences. 36 This is consistent with the general view that dysregulation of apoptosis constitutes a critical pathogenic factor in cancer, aberrantly extending cell viability and facilitating the insurgence of transforming mutations and resistance to therapy. 37 Using an antisense approach similar to that described here, targeting survivin in cancer cell lines was sufficient, alone, to induce a profound dysregulation of mitosis, 20 associated with spontaneous apoptosis and sensitization of cancer cells to chemotherapy-induced cell death. 38,39 The data presented here extend these observations and suggest that manipulation of the survivin pathway may be also beneficial to promote EC apoptosis during tumor angiogenesis, thus potentially accelerating regression of newly formed blood vessels and reducing the incidence of metastatic disease. 40 The selected expression of survivin in tumor cells and angiogenically stimulated ECs may provide a high degree of specificity for potential survivin antagonists to enhance both anti-angiogenic and anti-neoplastic therapeutic strategies.
Footnotes
Address reprint requests to Dario C. Altieri, M.D., Yale University School of Medicine, BCMM426B, 295 Congress Ave., New Haven, CT 06536. E-mail: dario.altieri@yale.edu.
Supported by National Institutes of Health grants HL54131 and CA78810 (to D. C. A.), HL61371 and HL64793 (to W. C. S.), HL51014 (to J. S. P.), and HL10112 (to M. M.).
References
- 1.Karsan A, Harlan JM: Modulation of endothelial cell apoptosis: mechanisms and pathophysiological roles. J Atheroscler Thromb 1996, 3:75-80 [DOI] [PubMed] [Google Scholar]
- 2.Hengartner MO: The biochemistry of apoptosis. Nature 2000, 407:770-776 [DOI] [PubMed] [Google Scholar]
- 3.Risau W: Mechanisms of angiogenesis. Nature 1997, 386:671-674 [DOI] [PubMed] [Google Scholar]
- 4.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E: Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995, 1:1024-1028 [DOI] [PubMed] [Google Scholar]
- 5.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK: Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci USA 1996, 93:14765-14770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin P, Buxton JA, Acheson A, Radziejewski C, Maisonpierre PC, Yancopoulos GD, Channon KM, Hale LP, Dewhirst MW, George SE, Peters KG: Antiangiogenic gene therapy targeting the endothelium-specific receptor tyrosine kinase Tie2. Proc Natl Acad Sci USA 1998, 95:8829-8834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruegg C, Yilmaz A, Bieler G, Bamat J, Chaubert P, Lejeune FJ: Evidence for the involvement of endothelial cell integrin αVβ3 in the disruption of the tumor vasculature induced by TNF and IFN-γ. Nat Med 1998, 4:408-414 [DOI] [PubMed] [Google Scholar]
- 8.Isik FF, Gibran NS, Jang YC, Sandell L, Schwartz SM: Vitronectin decreases microvascular endothelial cell apoptosis. J Cell Physiol 1998, 175:149-155 [DOI] [PubMed] [Google Scholar]
- 9.Fujio Y, Walsh K: Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J Biol Chem 1999, 274:16349-16354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N: Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 1998, 273:30336-30343 [DOI] [PubMed] [Google Scholar]
- 11.Bach FH, Hancock WW, Ferran C: Protective genes expressed in endothelial cells: a regulatory response to injury. Immunol Today 1997, 18:483-486 [DOI] [PubMed] [Google Scholar]
- 12.Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J: Nuclear factor (NF)-κB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor α-induced apoptosis. J Exp Med 1998, 188:211-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerber HP, Dixit V, Ferrara N: Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem 1998, 273:13313-13316 [DOI] [PubMed] [Google Scholar]
- 14.Nor JE, Christensen J, Mooney DJ, Polverini PJ: Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol 1999, 154:375-384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deveraux QL, Reed JC: IAP family proteins—suppressors of apoptosis. Genes Dev 1999, 13:239-252 [DOI] [PubMed] [Google Scholar]
- 16.O’Connor DS, Schechner JS, Adida C, Mesri M, Rothermel AL, Li F, Nath AK, Pober JS, Altieri DC: Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol 2000, 156:393-398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran J, Rak J, Sheehan C, Saibil SD, LaCasse E, Korneluk RG, Kerbel RS: Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem Biophys Res Commun 1999, 264:781-788 [DOI] [PubMed] [Google Scholar]
- 18.Papapetropoulos A, Fulton D, Mahboubi K, Kalb RG, O’Connor DS, Li F, Altieri DC, Sessa WC: Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J Biol Chem 2000, 275:9102-9105 [DOI] [PubMed] [Google Scholar]
- 19.Grossman D, McNiff JM, Li F, Altieri DC: Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J Invest Dermatol 1999, 113:1076-1081 [DOI] [PubMed] [Google Scholar]
- 20.Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S, Villa A, Marchisio PC, Altieri DC: Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat Cell Biologic 1999, 1:461-466 [DOI] [PubMed] [Google Scholar]
- 21.Baker BF, Lot SS, Condon TP, Cheng-Flournoy S, Lesnik EA, Sasmor HM, Bennett CF: 2′-O-(2-methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J Biol Chem 1997, 272:11994-12000 [DOI] [PubMed] [Google Scholar]
- 22.Ambrosini G, Adida C, Altieri DC: A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997, 3:917-921 [DOI] [PubMed] [Google Scholar]
- 23.Morales-Ruiz M, Fulton D, Sowa G, Languino LR, Fujio Y, Walsh K, Sessa WC: Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ Res 2000, 86:892-896 [DOI] [PubMed] [Google Scholar]
- 24.Ilan N, Mahooti S, Madri JA: Distinct signal transduction pathways are utilized during the tube formation and survival phases of in vitro angiogenesis. J Cell Sci 1998, 111:3621-3631 [DOI] [PubMed] [Google Scholar]
- 25.Papapetropoulos A, Garcia-Cardena G, Dengler TJ, Maisonpierre PC, Yancopoulos GD, Sessa WC: Direct actions of angiopoietin-1 on human endothelium: evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Lab Invest 1999, 79:213-223 [PubMed] [Google Scholar]
- 26.Salvesen GS, Dixit VM: Caspases: intracellular signaling by proteolysis. Cell 1997, 91:443-446 [DOI] [PubMed] [Google Scholar]
- 27.Ackermann EJ, Taylor JK, Narayana R, Bennett CF: The role of antiapoptotic Bcl-2 family members in endothelial apoptosis elucidated with antisense oligonucleotides. J Biol Chem 1999, 274:11245-11252 [DOI] [PubMed] [Google Scholar]
- 28.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC: Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396:580-584 [DOI] [PubMed] [Google Scholar]
- 29.Fraser AG, James C, Evan GI, Hengartner MO: Caenorhabditis elegans inhibitor of apoptosis protein (IAP) homologue BIR-1 plays a conserved role in cytokinesis. Curr Biol 1999, 9:292-301 [DOI] [PubMed] [Google Scholar]
- 30.Uren AG, Beilharz T, O’Connell MJ, Bugg SJ, van Driel R, Vaux DL, Lithgow T: Role for yeast inhibitor of apoptosis (IAP)-like proteins in cell division. Proc Natl Acad Sci USA 1999, 96:10170-10175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC: IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res 1998, 58:5315-5320 [PubMed] [Google Scholar]
- 32.Kobayashi K, Hatano M, Otaki M, Ogasawara T, Tokuhisa T: Expression of a murine homologue of the inhibitor of apoptosis protein is related to cell proliferation. Proc Natl Acad Sci USA 1999, 96:1457-1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Connor DS, Grossman G, Plescia J, Li F, Zhang H, Tognin S, Villa A, Marchisio PC, Altieri DC: Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci USA 2000, 97:13103-13107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams JM, Cory S: The Bcl-2 protein family: arbiters of cell survival. Science 1998, 281:1322-1326 [DOI] [PubMed] [Google Scholar]
- 35.Velculescu VE, Madden SL, Zhang L, Lash AE, Yu J, Rago C, Lal A, Wang CJ, Beaudry GA, Ciriello KM, Cook BP, Dufault MR, Ferguson AT, Gao Y, He TC, Hermeking H, Hiraldo SK, Hwang PM, Lopez MA, Luderer HF, Mathews B, Petroziello JM, Polyak K, Zawel L, Zhang W, Zhang X, Zhou W, Haluska FG, Jen J, Sukumar S, Landes GM, Riggins GJ, Vogelstein B, Kinzler KW: Analysis of human transcriptomes. Nat Genet 1999, 23:387-388 [DOI] [PubMed] [Google Scholar]
- 36.Altieri DC, Marchisio PC: Survivin apoptosis: an interloper between cell death and cell proliferation in cancer. Lab Invest 1999, 79:1327-1333 [PubMed] [Google Scholar]
- 37.Reed JC: Dysregulation of apoptosis in cancer. J Clin Oncol 1999, 17:2941-2953 [DOI] [PubMed] [Google Scholar]
- 38.Chen J, Wu W, Tahir SK, Kroeger PE, Rosenberg SH, Cowsert LM, Bennett F, Krajewski S, Krajewska M, Welsh K, Reed JC, Ng S-C: Down-regulation of survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia 2000, 2:236-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olie RA, Simoes-Wust AP, Baumann B, Leech SH, Fabbro D, Stahel RA, Zangemeister-Wittke U: A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res 2000, 60:2805-2809 [PubMed] [Google Scholar]
- 40.Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86:353-364 [DOI] [PubMed] [Google Scholar]