

Abstract
Hyperthermic stress is known to protect against myocardial dysfunction after ischemia-reperfusion injury. It is unclear however, what energetic mechanisms are affected by the molecular adaptation to heat stress. We hypothesized that mild hyperthermic stress can increase mitochondrial respiratory enzyme activity, affording protection to mitochondrial energetics during prolonged cardiac preservation for transplantation. Rat hearts were excised after heat-stress or sham treatment and subjected to cold cardioplegic arrest and ischemia followed by reperfusion in an ex vivo perfusion system. Cardiac function, mitochondrial respiratory, and complex activities were assessed before and after ischemia. Heat shock protein (Hsp 32, 60, and 72) expression was increased in heat-stressed hearts. This was associated with increased mitochondrial complex activities in heat-stress versus sham-treated groups for complex I-V. During reperfusion, higher complex activities and respiratory control ratios were observed in heat-stressed versus sham-treated groups. Recovery of ventricular function was improved in heat-stressed hearts. Furthermore, mitochondria in reperfused heat-stressed myocardium exhibited intact membranes with packed, parallel, lamellar cristae, whereas in sham-treated myocardium, mitochondria were severely disrupted. This study provides the first evidence of heat-stress-mediated enhancement of mitochondrial energetic capacity. This is associated with increased tolerance to ischemia-reperfusion injury. Protection by heat stress against myocardial dysfunction may be partially due to enhancement of mitochondrial energetics.
Cardiac transplant viability in the period after reperfusion is dependent primarily on the limitation of ischemia-reperfusion injury occurring as a consequence of organ storage. Myocardial ischemia produces a number of deleterious alterations to the physiological maintenance of intracellular oxygen, calcium, pH, and glucose levels and to osmotic control within cardiomyocytes. 1-3 These changes and the increased free radical generation, shown to occur as a consequence of re-oxygenation of the heart, impose both metabolic and oxidative stress. 4,5 This stress is associated with structural damage and mitochondrial dysfunction characterized by the reduced energy production through the oxidative phosphorylation process and loss of respiratory enzyme activity of complexes I-V located in the inner mitochondrial membrane. 6
Delayed myocardial tolerance to ischemia reperfusion injury can be increased by heat stress that results in de novo synthesis of heat shock proteins (Hsps) 7 and antioxidant enzymes, such as manganese superoxide dismutase. 8 Hsps are a family of highly conserved cytosolic proteins, transiently expressed after exposure of the cell to sublethal environmental stimuli such as hyperthermic and oxidative stress. Several studies have shown that controlled in vivo hyperthermic preconditioning resulting in elevated Hsp levels can improve myocardial functional recovery in isolated ischemic heart models. 9 Each Hsp group has a specificity of function and cellular compartmentation. Cardiac heat-shock protein research, which has focused primarily on the abundant 70-kd group, has identified three main classes: the constitutive 73-kd isoform (Hsc 70), physiologically expressed at high levels in the cytosol and involved in protein-folding functions; an inducible nuclear orientating 72-kd isoform; and a mitochondrial-specific Hsp 75 isoform that is up-regulated after hypoxia. 10-12 Hsps 60 and 10 have similarly been shown in combination to form a mitochondrial-located chaperonin complex conferring protection to ischemia-challenged myocytes. 13 Equally pertinent to myocardial protection, is the 32-kd protein (Hsp 32) an inducible isoform of the free heme-metabolizing enzyme, heme oxygenase (HO-1). HO-1 catalyzes the cleavage of heme rings to release the vasodilator neurotransmitter carbon monoxide, 14 ferrous iron, and biliverdin. This last product, biliverdin is rapidly metabolized by biliverdin reductase to form the cytosolic antioxidant bilirubin. 15 Heme oxygenase-mediated antioxidant defenses are further extended in ischemia compromised tissues by the capacity of HO-1 to export the iron byproduct extracellularly thus reducing the potential for Fe2+-mediated hydroxyl formation. 16
Increased expression of cytosolic heat-shock proteins has been associated with improved myocardial recovery after reperfusion. 17,18 We hypothesized that the heat-stress response in the heart may increase mitochondrial respiratory complex activity, thereby protecting integrated mitochondrial and hence myocardial function. As some heat Hsps are known to be mitochondrial chaperones, we also measured the expression of several Hsps with a view to assessing the relationship to changes in mitochondria. The present study was designed, using a clinically relevant model of cardioplegic arrest, to determine the effect of heat stress applied in vivo on myocardial functional recovery after ischemia and its relation to mitochondrial respiratory chain function. Having verified up-regulation of Hsps 72, 60, and 32 in myocardium of heat-stressed animals, we investigated the capacity of heat shock to improve: 1) myocardial function recovery, 2) mitochondrial function (oxygen consumption), 3) mitochondrial respiratory-chain enzyme activities, and 4) cellular and mitochondrial morphology in the isolated rat heart model subjected to 6 hours of cardioplegic arrest.
Materials and Methods
Animal Care
All animal procedures were performed in accordance with the “Animal (Scientific Procedures) Act (U.K.), 1986” and the “Guide for the Care and Use of Laboratory Animals” (National Institutes of Health publication no. 85-23, revised 1996). The study was approved by the National Heart and Lung Institute’s ethics committee on animal research. Animals were allowed standard laboratory rat diet and water ad libitum.
Induction of Heat Stress
Sprague-Dawley rats (male, 280 to 320 g) were anesthetized using sodium pentobarbitone (50 mg kg−1, i.p.). In heat-treated experimental groups, the rats were then placed in a whole-body temperature-controlled heating blanket (IMS K-Temp control unit; IMS, Cheshire, U.K.) and body temperature was raised to 41.8 to 42.0°C. Body temperature was monitored using a rectal temperature probe and maintained for 25 minutes as described previously. 9 Sham-treated groups were similarly anesthetized but not heat stressed. On recovery, animals were rehydrated with normal saline (10 ml kg−1 i.p.) and allowed to recover for 24 hours.
Heart Perfusion
Sham-treated and hyperthermically treated rats were anesthetized with diethyl ether and sodium heparin (1000 IU/kg) injected via the femoral vein. Hearts were rapidly excised and immersed momentarily in ice-cold filtered (0.45-μm pore size) Krebs-Henseleit (KH) buffer containing (in mmol/L): 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.23 KH2PO4, 24 NaHCO3, 11.1 glucose, and 1.2 CaCl2, pH 7.4. The hearts were cannulated in the Langendorff mode and perfused with KH buffer at 37°C, oxygenated (95%O2/5%CO2) at a constant pressure of 1,000 mm H2O. 19 Hearts were not paced during the entire protocol.
Myocardial Function Assessment
Mechanical function was assessed using an intraventricular balloon inserted via the mitral orifice into the left ventricle to determine systolic pressures, as previously described. 20 The balloon was inflated with water to produce a baseline end diastolic pressure of 10 mmHg and connected to a pressure transducer to record left ventricular pressures and heart rate. Coronary flow was measured using a Skalar electromagnetic MD1401 meter (Skalar, Delft, The Netherlands). Analogue signals were continuously recorded using a Biopaq AcqKnowledge Aquisition MP100 System (Linton Instruments, Norfolk, UK). To characterize cardiac function, left ventricular-developed pressure (LVDP) was assessed from the peak systolic pressure minus end diastolic pressure. Recovery of cardiac function was expressed using the relative recovery of post-ischemic versus pre-ischemic LVDP and pre- and post-ischemic coronary flows at the time points stated.
Mitochondrial Isolation Analysis
After the isolated perfusion period, the rat hearts were immersed in ice-cold isolation medium containing (in mmol/L) 225 mannitol, 75 sucrose, 10 Tris, 2 EGTA, pH 7.2, minced and digested using the protease enzyme, Nagarse (1.5 mg/ml) to enable recovery of both sarcolemmal and interfibrillar mitochondria. Nagarse was removed after an 8-minute period by washing (4×) in isolation medium and the tissue homogenized using four strokes of a Potter-Elvehjem homogenizer. Mitochondria were then isolated using differential centrifugation from the homogenate at 3,500 × g (3 minutes) and pelleted at 11,500 × g (2× 12 minutes) as described previously. 21,22 Mitochondrial protein was measured using a modified Lowry method 23 (Lowry D.C. kit; Biorad, UK).
Mitochondrial oxygen consumption was measured as previously described 6,22 using a water-jacketed Clark-type oxygen electrode (World Precision Instruments, Hertfordshire, UK) and recorded using Biopaq AcqKnowledge software. All experiments were performed at 30°C in 350 μl of respiration medium containing (in mmol/L): 100 KCl, 75 mannitol, 25 sucrose, 10 Tris-HCl, 10 KH2PO4-Tris, and 0.05 ethylenediaminetetraacetic acid (dipotassium salt), pH 7.4. Incubations were performed using 0.5 mg of mitochondrial protein and 0.125 mg of fat-free bovine serum albumin. State 4 respiration was initiated using 5 mmol/L of glutamate plus 5 mmol/L of malate or 5 mmol/L of succinate plus 1 μmol/L of rotenone. State 3 respiration was initiated by the addition of ADP. Respiratory control indices (RCI) were calculated as state 3 rate/state 4 rate. Mitochondrial complex activities were measured in mitochondrial samples lysed by three cycles of freeze thawing. Assays for mitochondrial complex I (NADH-ubiquinone oxidoreductase, EC 1.6.99.3), complex II/III (succinate-ubiquinone/ubiquinol-cytochrome c reductase, EC 1.8.3.1), complex IV (cytochrome c oxidase, EC 1.9.3.1), complex V (ATPase, EC 3.6.1.3), and citrate synthase (EC 4.1.3.7) were performed essentially as described, 24 except that the volumes were scaled down to a final reaction volume of 250 μl. Assays were performed using a SpectraMax-Plus 96-well spectrophotometer (Molecular Devices, Crawley UK) operating in kinetic mode at 30°C. After correction for optical path length in the microplate, results were expressed as nmol/min/mg protein for complexes I, II/III, V and citrate synthase and as the first order rate constant k/min/mg protein for complex IV. 22,25
Stress Protein Expression
Levels of Hsp 32, 60, and 72 were determined by Western immunoblotting, as previously described. 26 Briefly, 50 μg of protein of whole heart homogenates or purified mitochondrial pellets were separated on 10% T (total concentration of acrylamide plus bis-acrylamide in g/100 ml) sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred to Hybond C Super membrane at 500 mA for 1 hour. Blots were probed with antibodies specific to Hsp 32 (OSA-111), Hsp 60 (SPA-806), or inducible Hsp 70 (SPA-820) (Bioquote Ltd., York, UK).
Reactivity of antibodies against the Hsps was visualized using an enhanced chemiluminescence detection system (Amersham, Buckinghamshire, UK). After enhanced chemiluminescence exposure, primary and secondary antibodies were stripped from blots by incubation at 50°C for 30 minutes in a solution of 2% w/v sodium dodecyl sulfate, 62.5 mmol/L of Tris-HCl, pH 6.7, and 100 mmol/L of 2-mercaptoethanol. Membranes were reprobed for the cytosolic marker β-tubulin using a monoclonal antibody. Levels of Hsp 32, 60, and 70 levels were scanned densitometrically on enhanced chemiluminescence films using Quantity One software (P.D.I., NY) and normalized against respective β-tubulin levels.
Transmission Electron Microscopy
The effects of heat stress on ultrastructural morphology were assessed by standard thin-section electron microscopy in pre-ischemic and reperfused myocardium. Heat-stressed and sham-treated hearts were subjected to nonischemic perfusion (35 minutes at 37°C) or to the cardioplegic arrest and reperfusion procedures as previously described. At the end of these procedures, hearts were immediately perfusion-fixed via the aorta with 2.5% glutaraldehyde in KH buffer for 5 minutes. 27,28 Left ventricular tissue was dissected out and sections (1 × 1 × 1 mm) were further fixed for 2 hours in 2.5% glutaraldehyde. Tissue samples were postfixed in 2% osmium tetroxide, en bloc stained with uranyl acetate in 50% ethanol, dehydrated through an ethanol series, and embedded in araldite. Ultra-thin sections were stained with uranyl acetate and lead citrate and examined using a Philips EM301 electron microscope.
Experimental Protocols
To determine the effect of elevated heat-shock protein expression with the protection of cardiac mitochondrial and myocardial mechanical function in isolated perfused hearts, two major experimental groups were used. Hearts derived from heat-stressed and sham-treated groups were subjected to the ex vivo ischemic procedures described and compared to appropriate pre-ischemic controls. Cardiac mitochondrial and myocardial mechanical function changes were studied in sham-treated and heat-stressed hearts before, and after 6 hours of cardioplegic arrest at 4°C. After an initial 30-minute normoxic perfusion at 37°C, pre-ischemic cardiac function was evaluated using heart rate, LVDP, and coronary flow. Hearts from each pre-ischemic group were also collected for mitochondrial function.
Subsequent to the initial perfusion, hearts in the postischemic groups were arrested by infusion of 4°C St. Thomas cardioplegic solution at a constant pressure of 60 mmHg for 2 minutes. Hearts were maintained for 6 hours immersed in St. Thomas cardioplegic solution at 4°C. After the 6-hour ischemic period, hearts were reperfused with oxygenated KH buffer at 37°C for 60 minutes. Post-ischemic cardiac function and mitochondrial parameters were evaluated at the end of the reperfusion period.
Statistical Analyses
Values are presented as group means ± SEM of six animals. Statistical analysis was performed using StatStat V2.03 (SPSS Inc, Erkrath, Germany). Comparison between groups was performed using a one-way analysis of variance for repeated measures followed by Bonferroni test to indicate individual significant differences. Normalized differences between sham- and heat-treated group means were assessed using the nonparametric Mann-Whitney test. P values less than 0.05 were considered significantly different.
Results
Stress Protein Expression Up-Regulation
Figure 1, A and B ▶ , illustrates alterations in Hsp expression in the myocardium of sham- and heat-treated rats. Densitometric analysis of stress proteins confirmed that exposure (25 minutes) to a hyperthermic episode resulted in a significant elevation after 24 hours in the expression of myocardial Hsps 72 and 32 as compared to untreated myocardium. Constitutive Hsp 60 levels were also enhanced (P < 0.05) in the whole heart samples and in purified cardiac mitochondrial pellets (Figure 1C) ▶ derived from heat-treated rats. Anti-β-tubulin antibody did not react with mitochondrial proteins demonstrating that no cytosolic contamination was present in the purified cardiac mitochondrial pellets.
Figure 1.
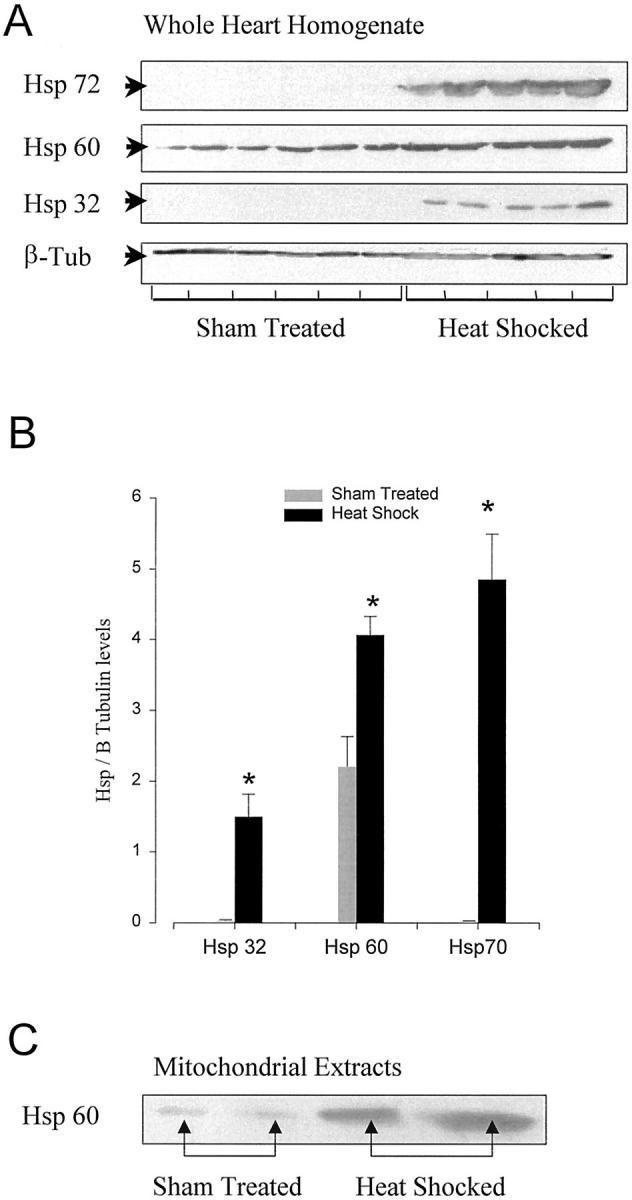
A: Western blot analysis showing overexpression of Hsp 32, 60, and 70 in rat myocardium 24 hours after heat-stress versus sham treatment. Each lane is representative of an individual heart extract. B: Graph showing mean ± SEM of Hsp 32, 60, and 70 levels normalized to β tubulin in ventricular myocardium from sham-treated and heat-stressed rats (*, P < 0.05; n = 5 to 6). C: Western blot analysis of Hsp 60 in representative cardiac mitochondrial samples, showing stronger expression in mitochondria isolated from heat-stressed than from sham-treated myocardium.
Myocardial Functional Recovery
The maximally attained percent recovery of functional values in post-ischemic reperfused myocardium are presented in Table 1 ▶ . No significant differences were seen under baseline conditions in pre-ischemic LVDP, heart rate, or coronary flow between the two groups (sham-treated versus heat-stressed groups).
Table 1.
Recovered Functional Values in Langendorff-Perfused Hearts of Heat-Stressed and Sham-Treated Rat Hearts at 45 Minutes after Reperfusion
| Cardiovascular Parameters | |||
|---|---|---|---|
| LVDP | Heart rate | Coronary flow | |
| Sham (n = 6) | |||
| Post-ischemic | 29.6 ± 2.9% | 81.4 ± 2.5% | 33.6 ± 2.3% |
| Pre-ischemic baseline | 103.7 ± 3.4 mmHg | 279 ± 8.3 bpm | 14.8 ± 0.5 ml min−1 |
| Heat shock (n = 6) | |||
| Post-ischemic | 49.5 ± 3.7%* | 93.7 ± 2.9%* | 61.9 ± 3.8%* |
| Pre-ischemic baseline | 109.2 ± 4.1 mmHg | 284 ± 10.7 bpm | 15.2 ± 0.7 ml min−1 |
Data represent the mean ± SEM. Left ventricular-developed pressure (LVDP), heart rate, and coronary flow recorded at 45 minutes of post-ischemic reperfusion are presented as a percentage of corresponding pre-ischemic baseline values. *Represents P < 0.05 from the values in the sham-treated post-ischemic group.
The data summarized in Table 1 ▶ indicate that heat-stress treatment provided significantly (P < 0.05) improved functional recovery in comparison to the sham-treated group. During reperfusion, the post-ischemic heat-stressed group attained recovery coronary flows of 61.9 ± 3.8%, heart rates of 93.7 ± 2.9%, and LVDP levels of 49.5 ± 3.7% of pre-ischemic baseline values. In the post-ischemic sham-treated group, coronary flow, heart rate, and LVDP recovered to only 33.6 ± 2.3%, 81.4 ± 2.5% and 29.6 ± 2.9%, respectively, of the pre-ischemic baseline values.
Recovery of Cardiac Mitochondrial Respiratory Function
Pre-ischemic state 4 and 3 mitochondrial respiratory rates and RCI values calculated in heat-stressed myocardium show no significant change from values obtained in sham-treated rats. These results confirm that the hyperthermic protocol used in this study did not affect any of the cardiac mitochondrial parameters considered in this study.
Figure 2 ▶ summarizes the respiratory data in mitochondria isolated from the two groups of sham-treated and heat-stressed animals. Mitochondria isolated from reperfused control hearts subjected to 6 hours of cardioplegic ischemia showed, as expected, a significantly (P < 0.05) decreased state 3 respiratory rate with increased (P < 0.05) state 4 rate, resulting in a decline in RCI. As can be seen however, reperfused cardiac mitochondrial respiratory parameters were substantially salvaged by the hyperthermic treatment protocols used in this study. Both glutamate plus malate (NAD+)- and succinate (FAD)-driven state 4 rates were significantly (P < 0.05) reduced in the reperfused heat-stressed hearts from the values obtained in the sham-treated group (Figure 2, A and B) ▶ . At the same time, heat stress significantly (P < 0.05) ameliorated the reperfusion-mediated fall in malate NAD+-linked state 3 rate seen in the sham-treated hearts (Figure 2, C and D) ▶ . RCI values were observed (Figure 2, E and F) ▶ to recover significantly (P < 0.05) in heat-stressed myocardium as a consequence of preserving state 3 and 4 rates.
Figure 2.
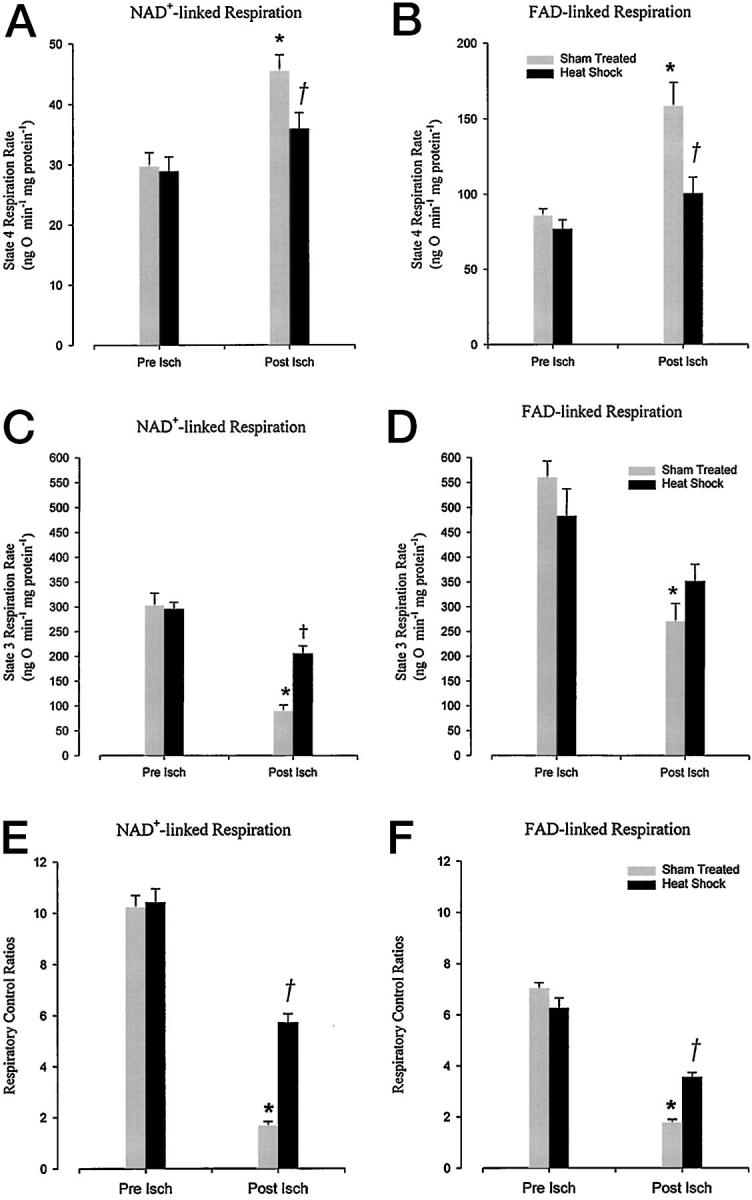
Effects of 6 hours of cold storage and crystalloid reperfusion on state 4 respiration rate (A and B), state 3 respiration rate (C and D), and respiratory control indices (E and F) in cardiac mitochondria isolated from sham-treated and heat-stressed rats. Respiratory activity is assessed in the presence of glutamate and malate (NAD+-linked.) or succinate (FAD-linked.). Each column represents the mean ± SEM of cardiac mitochondrial parameters after 30 minutes of perfusion (Pre Isch) and 60 minutes reperfusion (Post Isch). *, Statistical significance at P < 0.05 against sham-treated pre-ischemic controls; †, P < 0.05 versus sham-treated post-ischemic tissue (n = 6).
Increased Pre-Ischemic and Post-Ischemic Mitochondrial Enzyme Activities in Stressed Rat Hearts
Hyperthermic treatment raised individual mitochondrial respiratory chain enzyme activities (Figure 3) ▶ . Although, only complex I, IV, and V activity were shown to be significantly (P < 0.05) increased from the sham-treated values, complex II-III respiratory enzymes showed a slight increase of activity, which was not statistically significant (P < 0.06). Reperfusion of cardioplegically arrested myocardium after 6 hours of cold cardioplegic storage significantly (P < 0.05) reduced mitochondrial enzyme activities (Figure 3) ▶ . The results obtained in this study showed that, whereas the complex activities were all reduced at 1 hour after reperfusion, complex I (NADH-CoQ reductase) and complex V (oligomycin-sensitive ATPase) decreased markedly. Significantly, all of the complexes in the hyperthermic group studied showed higher (P < 0.05) enzyme activity levels after 6 hours of storage and reperfusion myocardium than shown in the sham-treated group under identical conditions. It is possible that complex IV activities in vivo may have been decreased as a consequence of heme oxygenase-derived CO binding. 29 Any effects of CO binding on complex IV activity measured in our spectrophotometric assay may be difficult to ascertain however, because of CO being driven off the complex IV-binding site by light from room light sources.
Figure 3.

Effect of heat-stress versus sham treatment on the activity of mitochondrial complex I (A), complex II-III (B), complex IV (C), and on complex V (oligomycin-sensitive) (D). Each column represents the mean ± SEM of activity of six experiments assessed at either 30 minutes of Langendorff perfusion (Pre Isch) or after 60 minutes reperfusion (Post Isch) after cardioplegic arrest and 6 hours of storage. *, Statistical significance at P < 0.05 against sham-treated pre-ischemic controls; †, P < 0.05 versus sham-treated post-ischemic tissue.
Loss of mitochondrial matrix integrity, as defined by the decrease of citrate synthase marker from the reperfused sham-treated mitochondrial isolate, was also significantly (P < 0.05) protected from ischemia-reperfusion injury in heat-stressed animals (Figure 4) ▶ . Unlike the membrane-bound complexes, no significant increase in citrate synthase activity was observed in heat-treated pre-ischemic myocardium.
Figure 4.

Effect of heat-stress versus sham treatment on mitochondrial citrate synthase activity. Columns represents the mean ± SEM of activity assessed at either 30 minutes of Langendorff perfusion (Pre Isch) or after 60 minutes reperfusion (Post Isch) after cardioplegic arrest and 6 hours of storage. *, Statistical significance at P < 0.05 against sham-treated pre-ischemic controls; †, P < 0.05 versus sham-treated post-ischemic tissue (n = 6).
Preservation of Cardiac Morphology
Sham-treated, pre-ischemic heart contained abundant mitochondria in rows alongside the myofibrils (Figure 5A) ▶ . Myofibril appearance was regular and transverse tubules distinct. Where atypical structure was observed in occasional mitochondria of control tissues, this was attributed to suboptimal fixation. Reperfusion of the sham-treated myocardium after cold ischemia (6 hours) resulted in severe disruption of the myofibrils and disorganization of mitochondrial distribution (Figure 5B) ▶ . This appearance was representative of a large proportion (>80%) of the fields examined. Heat-stress pretreatment produced no overall change in the normal ultrastructural appearance of pre-ischemic myocardium (Figure 5C) ▶ , and post-ischemic pretreated hearts gave a very similar ultrastructural appearance (Figure 5D) ▶ to pre-ischemic myocardium.
Figure 5.
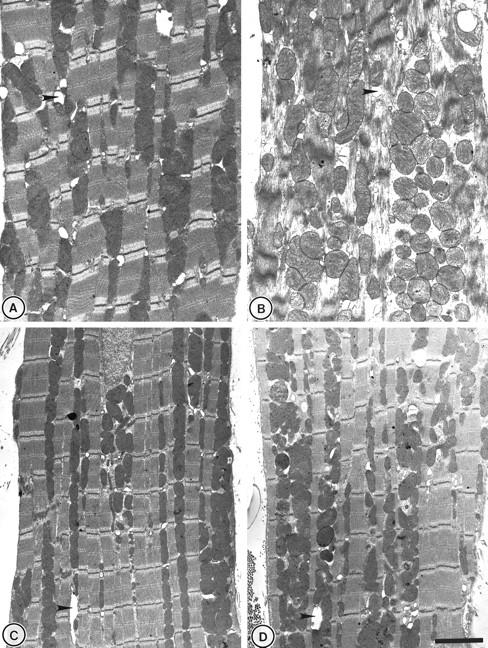
Ultrastructural examination of perfused rat heart. A: Typical myocardial structure in sham-treated myocardium after 35 minutes of perfusion with intact mitochondria interspersed around regular myofibril structures. B: Mitochondrial swelling and derangement accompanied by structural disruption to myotubule formation in the reperfused sham-treated myocardium. C and D: Heat-stressed pre-ischemic and reperfused myocardium. The overall appearance resembles that seen in pre-ischemic sham-treated controls. Arrows indicate transverse tubule formation. Scale bar, 2 μm.
Inspection at a higher magnification revealed further detail of mitochondrial structure (Figure 6) ▶ . In pre-ischemic controls, mitochondria exhibited intact double membranes with tightly packed, parallel lamellar cristae, characteristic of normal ultrastructural morphology (Figure 6A) ▶ . Mitochondria of hearts subjected to cold ischemia and reperfusion, were severely disrupted, swollen, and empty in appearance (Figure 6B) ▶ . In contrast, the appearance of the reperfused heat-stressed myocardium closely resembled that of the pre-ischemic group in that mitochondrial structures (Figure 6C) ▶ were preserved in the majority of fields examined.
Figure 6.
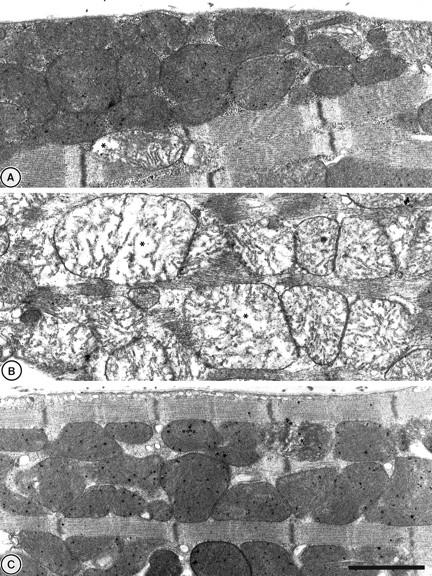
At higher magnifications mitochondria structure in sham-treated tissues (A) generally exhibited intact double membranes with tightly packed lamellar cristae. After the same sham treatment, reperfusion (B) resulted in a large proportion (>80%) of the sections studied, displaying swollen mitochondria with cristae distortion and loss of surrounding membrane. Mitochondria in reperfused myocardium from heat-stressed animals (C) were generally intact with little appearance of the damage seen in the reperfused sham-treated group. *, Examples of swollen mitochondria. Scale bar, 1 μm.
Discussion
The data obtained in this study demonstrate, for the first time that, the activities of mitochondrial respiratory complexes are increased, and integrated mitochondrial function is protected against reperfusion-induced damage in hyperthermically treated hearts. Hearts obtained from heat-stressed rats in the present study have demonstrated a significant improvement in reperfused myocardial performance over sham-treated hearts after cardioplegic arrest and 6 hours of cold storage. This result concurs with previous observations in hearts subjected to hyperthermia that noted a decreased rate of high-energy phosphate depletion during ischemia followed by an increased recovery rate with maintained endothelial function and vasodilatation during reperfusion. 9,14,17,30-32 Our study shows that the improved mechanical recovery seen in reperfused hearts expressing elevated levels of Hsps 72, 60, and 32, occurs in association with protected, integrated mitochondrial respiratory activity. Further evidence of this association is shown in the increased level of Hsp 60 reactivity in mitochondria from heat-stressed myocardium. In examining the hypothesis that this protection is derived as a consequence of a heat-stress-mediated preservation of the respiratory complexes, the data obtained show that the individual enzyme activities of complexes I-V are also augmented in both pre- and post-ischemic myocardium.
Mitochondrial dysfunction has been documented in various models of ischemia-reperfusion in relation to the content and activity of proteins involved in ATP production by oxidative phosphorylation. 6,33 Reperfusion of sham-treated myocardium after exposure to cold cardioplegic ischemia for 6 hours consistently decreased state 3 rate and uncoupled mitochondrial respiration using both NAD+- and FAD-linked substrates. State 4 respiration rate was simultaneously raised, suggestive of an increased membrane proton leak. 34 These data together with the loss of citrate synthase activity from the mitochondrial fraction confirm an increase in membrane permeability with diminished phosphorylation capacity in the reperfused myocardium. 35 This result is supported by the post-ischemic ultrastructural appearance of the mitochondrial organelles in situ, in heat-stressed and sham-treated myocardium. Our study shows that recovery of RCI (consistent with ATP restoration after reperfusion) is much greater in heat-stressed than in the sham-treated hearts. The increase in pre-ischemic level however, of mitochondrial protein activity was not reflected in state 3 and 4 rates. Oxygen consumption and phosphorylation rates during physiological respiration state 3, have previously been shown to be controlled by a combination of respiratory chain enzyme and ATP synthase activity. 24 Studies using specific inhibitors of the individual mitochondrial complexes demonstrate that the activities of several of the complexes can be in excess of that needed for maximal rates of fully integrated mitochondrial oxidative phosphorylation. 36 This excess enzyme capacity may account for the lack of effect of whole-body hyperthermia on respiration states 3 and 4 in pre-ischemic myocardium.
Mitochondrial oxidative phosphorylation is dependent on the integration of the polypeptide complexes of the respiratory chain with ATP synthase assembled on the inner membrane (Figure 7) ▶ . The data collected in this study shows that, post-ischemic mitochondrial function is protected in myocardium expressing high levels of Hsp 72, 60, and 32. We suggest that this protected mitochondrial function arises as a consequence of elevated pre-ischemic mitochondrial enzyme activities. The relationship between Hsp expression and mitochondrial complex activity presented here supports previous studies, 10,37 showing that up-regulation of molecular chaperone complexes (Hsp72 or Hsp 60/10) in the myocardium contribute to the preservation of mitochondrial integrity. This may be achieved primarily through facilitation of nuclear-encoded protein import and assembly in the mitochondrial matrix. 38 Indeed, translocation of the β-subunit of the F1F0 ATPase (complex V) into mitochondria of Saccharomyces cerevisiae has been shown to be dependent on the presence of cytosolic Hsp72 (Figure 7) ▶ . 39 Consequently, mitochondrial enzyme activity may be increased at the translational level. Therefore, up-regulation of inducible Hsp 70 expression should effectively sustain state 3 respiration rates. Previous reviews have also postulated an involvement of Hsp 72 in preventing electron leak between complexes III and IV by binding and reducing cytochrome c loss from mitochondrial membranes (Figure 7) ▶ , thereby averting an increase in state 4 respiration rates and induction of cytochrome c linked apoptosis. 40,41 Certainly, Hsp 60/10 over-expression and stress-induced chaperonin-like molecules such as Tcm62p have been shown to markedly protect both nuclear and mitochondrial encoded proteins including primary dehydrogenases from ischemia damage. 13,42 However, protection of mitochondrial proteins from oxidative stress need not necessarily stem exclusively from a chaperoning action of Hsps. Mitochondrial enzyme activity may be also be simultaneously protected by antioxidant systems derived from the activation of heat-stress-mediated scavenging mechanisms. Under normal physiological conditions, superoxide anions (O2−·), are generated by the mitochondrial respiratory chain as a consequence of electron leak onto molecular oxygen. 24,43 Ischemic damage to the myocardium simultaneously results in increased nitric oxide production and exacerbation of O2−· radical production with consequent increases in peroxynitrite and hydroxyl radical formation. As mitochondrial enzyme complexes consist of polypeptides encompassing iron-sulfur clusters, they can become subject to oxidative deactivation by reactive oxygen and nitrogen species, thereby reducing enzyme activity. 44,45 By up-regulating HO-1 (Hsp 32) expression and activity, levels of the antioxidant bilirubin, known to be a peroxynitrite scavenger, 46 and shown to be cytoprotective at nanomolar concentrations, are increased. 15,47 Additionally, oxidative stress-induced changes in cellular glutathione redox status have recently been demonstrated to be responsible for the induction of Hsp 70. 48 Consequently, we propose that levels and activity of respiratory enzymes may be augmented in this model of normoxic and ischemic-reperfused myocardium by an Hsp-mediated up-regulation of protein production and simultaneous dissipation of free radicals. Recent studies by our group pursuing this theme, have confirmed that selective viral-mediated Hsp 72 gene transfection of transplanted hearts, 49 results in elevated myocardial complex I activities (Jayakumar J, Suzuki S, Sammut I, et al, submitted for publication). The two phenomena of Hsp up-regulation and elevated mitochondrial enzyme activities are therefore closely associated.
Figure 7.

Diagrammatic representation of the electron transport chain. Specific complexes are denoted by the numerals I to V. Electrons are transferred from NADH and FADH 2 to the electron-transport chain carriers with H+ release. H+ is exported across the inner mitochondrial membrane before diffusing back into the matrix via complex V thereby coupling oxidation to phosphorylation. The diagram shows the proposed mechanisms of mitochondrial respiratory complex protection from ischemia-reperfusion injury in hyperthermically treated myocardium. Up-regulated heme oxygenase-derived antioxidant systems offer protection of respiratory proteins against oxidative stress damage to the respiratory complexes whereas Hsp chaperonins are shown to assist in folding and translocation of both nuclear and mitochondrial encoded respiratory protein subunits.
The contingency of a direct hyperthermic effect on the pretranslational expression of genes encoding for mitochondrial enzymes is debatable. No evidence has been presented to date of an increased gene transcription of entire supramolecular complexes other than subunits 50 after heat stress. Mammalian mitochondrial DNA encodes for only part of the subunits of complexes I (7 subunits), III (cytochrome b), IV (3 subunits), and V (2 subunits), whereas cytochrome c1, F1F0 ATPase (β subunit) and all of the subunits of complex II and citrate synthase are encoded for by the nuclear DNA. 51,52 A regulatory Sp1 site does appear to be a common activating element in a number of the nuclear genes involved in mitochondrial biogenesis. 53 However, a biogenesis regulatory mechanism whereby both mitochondrial and nuclear genomes need to be tightly coordinated in their response to changes in metabolic conditions remains unknown.
In summary, this study provides the first evidence, confirming that in vivo heat stress enhances mitochondrial energetics, while protecting myocardial function against ischemia-reperfusion injury. Heat-stress mediated enhancement of mitochondrial enzyme activity and prevention of the loss of mitochondrial potential to drive oxidative-phosphorylation may confer resistance to mitochondrial-linked apoptosis. This energetic enhancement would also support a protection of cardiac high-energy phosphate supply. The present findings offer the exciting possibility that pharmacological modulation 54,55 of heat-stress proteins in organ grafts may play a central role in the future of clinical transplantation.
Acknowledgments
We thank Miss Nicola Ruth for her technical assistance.
Footnotes
Address reprint requests to Ivan A. Sammut Ph.D., NHLI at Imperial College School of Medicine, Heart Science Centre, Harefield Hospital, Harefield, Middlesex UB9 6JH, UK. E-mail: i.sammut@ic.ac.uk.
Supported by British Heart Foundation Grant PG99173. R.T.S. is a Visiting Professor at the Medical University of Gdansk, Poland..
References
- 1.Jennings RB, Ganote CE: Mitochondrial structure and function in acute myocardial ischemic injury. Circ Res 1976, 38:I80-I91 [PubMed] [Google Scholar]
- 2.Jennings RB, Schaper J, Hill ML, Steenbergen C, Jr, Reimer KA: Effect of reperfusion late in the phase of reversible ischemic injury. Changes in cell volume, electrolytes, metabolites, and ultrastructure. Circ Res 1985, 56:262-278 [DOI] [PubMed] [Google Scholar]
- 3.Khandoudi N, James F, Feuvray D: Influence of intracellular pH on mitochondrial calcium during ischaemia of the isolated rat heart. Histochem J 1989, 21:99-106 [DOI] [PubMed] [Google Scholar]
- 4.Ferrari R: Metabolic disturbances during myocardial ischemia and reperfusion. Am J Cardiol 1995, 76:17B-24B [PubMed] [Google Scholar]
- 5.De Vecchi E, Pala MG, Di Credico G, Agape V, Paolini G, Bonini PA, Grossi A, Paroni R: Relation between left ventricular function and oxidative stress in patients undergoing bypass surgery. Heart 1998, 79:242-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sammut IA, Thorniley MS, Simpkin S, Fuller BJ, Bates TE, Green CJ: Impairment of hepatic mitochondrial respiratory function following storage and orthtopic transplantation of rat livers. Cryobiology 1998, 36:49-60 [DOI] [PubMed] [Google Scholar]
- 7.Heads RJ, Latchman DS, Yellon DM: The molecular basis of adaptation to ischemia in the heart: the role of stress proteins and anti-oxidants in the ischemic and reperfused heart. EXS 1996, 76:383-407 [DOI] [PubMed] [Google Scholar]
- 8.Yamashita N, Hoshida S, Nishida M, Igarashi J, Taniguchi N, Tada M, Kuzuya T, Hori M: Heat shock-induced manganese superoxide dismutase enhances the tolerance of cardiac myocytes to hypoxia-reoxygenation injury. J Mol Cell Cardiol 1997, 29:1805-1813 [DOI] [PubMed] [Google Scholar]
- 9.Jayakumar J, Smolenski RT, Gray CC, Goodwin A, Kalsi KK, Amrani M, Yacoub MH: Influence of heat stress on myocardial metabolism and functional recovery after cardioplegic arrest: a 31P N.M.R. study. Eur J Cardiothorac Surg 1998, 12:467-474 [DOI] [PubMed] [Google Scholar]
- 10.Cornelussen RN, Garnier AV, van der Vusse GJ, Reneman RS, Snoeckx LHEH: Biphasic effect of heat stress pretreatment on ischemic tolerance of isolated rat hearts. J Mol Cell Cardiol 1998, 30:365-372 [DOI] [PubMed] [Google Scholar]
- 11.Knowlton AA, Kapadia S, Torre-Amione G, Durand JB, Bies R, Young J, Mann DL: Differential expression of heat shock proteins in normal and failing human hearts. J Mol Cell Cardiol 1998, 30:811-818 [DOI] [PubMed] [Google Scholar]
- 12.Matouschek A, Azem A, Ratliff K, Glick BS, Schmid K, Schatz G: Active unfolding of precursor proteins during mitochondrial protein import. EMBO J 1997, 16:6727-6736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau S, Patnaik N, Sayen MR, Mestril R: Simultaneous overexpression of two stress proteins in rat cardiomyocytes and myogenic cells confers protection against ischemia-induced injury. Circulation 1997, 96:2287-2294 [DOI] [PubMed] [Google Scholar]
- 14.Sammut IA, Foresti R, Clark JE, Exon DJ, Vesely MJ, Sarathchandra P, Green CJ, Motterlini R: Carbon monoxide is a major contributor to the regulation of vascular tone in aortas expressing high levels of haeme oxygenase-1. Br J Pharmacol 1998, 125:1437-1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dore S, Takahashi M, Ferris CD, Hester LD, Guastella D, Snyder SH: Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci USA 1999, 96:2445-2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poss KD, Tonegawa S: Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci USA 1997, 94:10919-10924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yellon DM, Pasini E, Cargnoni A, Marber MS, Latchman DS, Ferrari R: The protective role of heat stress in the ischaemic and reperfused rabbit myocardium. J Mol Cell Cardiol 1992, 24:895-907 [DOI] [PubMed] [Google Scholar]
- 18.Amrani M, Corbett J, Allen NJ, O’Shea J, Boateng SY, May AJ, Dunn MJ, Yacoub MH: Induction of heat-shock proteins enhances myocardial and endothelial functional recovery after prolonged cardioplegic arrest. Ann Thorac Surg 1994, 57:157-160 [DOI] [PubMed] [Google Scholar]
- 19.Smolenski RT, Yacoub MH, Seymour A-ML: Reduced purine catabolite production in the postischemic rat heart: a 31P NMR assessment of cytosolic metabolites. Magma 1994, 2:417-420 [Google Scholar]
- 20.Suzuki K, Sawa Y, Kaneda Y, Ichikawa H, Shirakura R, Matsuda H: In vivo gene transfection with heat shock protein 70 enhances myocardial tolerance to ischemia-reperfusion injury in rat. J Clin Invest 1997, 99:1645-1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassani RA, Fagian MM, Bassani JW, Vercesi AE: Changes in calcium uptake rate by rat cardiac mitochondria during postnatal development. J Mol Cell Cardiol 1998, 30:2013-2023 [DOI] [PubMed] [Google Scholar]
- 22.Sammut IA, Burton K, Balogun E, Sarathchandra P, Brooks KJ, Bates TE, Green CJ: Time-dependent impairment of mitochondrial function after storage and transplantation of rabbit kidneys. Transplantation 2000, 69:1265-1275 [DOI] [PubMed] [Google Scholar]
- 23.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951, 193:265-275 [PubMed] [Google Scholar]
- 24.Bates TE, Heales SJ, Davies SE, Boakye P, Clark JB: Effects of 1-methyl-4-phenylpyridinium on isolated rat brain mitochondria: evidence for a primary involvement of energy depletion. J Neurochem 1994, 63:640-648 [DOI] [PubMed] [Google Scholar]
- 25.Almeida A, Brooks KJ, Sammut I, Keelan J, Davey GP, Clark JB, Bates TE: Postnatal development of the complexes of the electron transport chain in synaptic mitochondria from rat brain. Dev Neurosci 1995, 17:212-218 [DOI] [PubMed] [Google Scholar]
- 26.Latif N, Baker CS, Dunn MJ, Rose ML, Brady P, Yacoub MH: Frequency and specificity of antiheart antibodies in patients with dilated cardiomyopathy detected using SDS-PAGE and Western blotting. J Am Coll Cardiol 1993, 22:1378-1384 [DOI] [PubMed] [Google Scholar]
- 27.Kaprielian RR, Gunning M, Dupont E, Sheppard MN, Rothery SM, Underwood R, Pennell DJ, Fox K, Pepper J, Poole-Wilson PA, Severs NJ: Downregulation of immunodetectable connexin43 and decreased gap junction size in the pathogenesis of chronic hibernation in the human left ventricle. Circulation 1998, 97:651-660 [DOI] [PubMed] [Google Scholar]
- 28.Ko YS, Yeh HI, Haw M, Dupont E, Kaba R, Plenz G, Robenek H, Severs NJ: Differential expression of connexin43 and desmin defines two subpopulations of medial smooth muscle cells in the human internal mammary artery. Arterioscler Thromb Vasc Biol 1999, 19:1669-1680 [DOI] [PubMed] [Google Scholar]
- 29.Greenwood C, Wilson MT, Brunori M: Studies on partially reduced mammalian cytochrome oxidase. Reactions with carbon monoxide and oxygen. Biochem J 1974, 137:205-215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornelussen R, Spiering W, Webers JH, de Bruin LG, Reneman RS, van der Vusse GJ, Snoeckx LH: Heat shock improves ischemic tolerance of hypertrophied rat hearts. Am J Physiol 1994, 267:H1941-H1947 [DOI] [PubMed] [Google Scholar]
- 31.van der Vusse GJ, Cornelussen RN, Roemen TH, Snoeckx LH: Heat stress pretreatment mitigates postischemic arachidonic acid accumulation in rat heart. Mol Cell Biochem 1998, 185:205-211 [DOI] [PubMed] [Google Scholar]
- 32.Amrani M, Corbett J, Boateng SY, Dunn MJ, Yacoub MH: Kinetics of induction and protective effect of heat-shock proteins after cardioplegic arrest. Ann Thorac Surg 1996, 61:1407-1411 [DOI] [PubMed] [Google Scholar]
- 33.Henke W, Jung K: Ischemia decreases the content of the adenine nucleotide translocator in mitochondria of rat kidney. Biochim Biophys Acta 1991, 1056:71-75 [DOI] [PubMed] [Google Scholar]
- 34.Borutaite V, Mildaziene V, Brown GC, Brand MD: Control and kinetic analysis of ischemia-damaged heart mitochondria: which parts of the oxidative phosphorylation system are affected by ischemia? Biochim Biophys Acta 1995, 1272:154-158 [DOI] [PubMed] [Google Scholar]
- 35.Veitch K, Hombroeckx A, Caucheteux D, Pouleur H, Hue L: Global ischaemia induces a biphasic response of the mitochondrial respiratory chain. Anoxic pre-perfusion protects against ischaemic damage. Biochem J 1992, 281:709-715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davey GP, Clark JB: Threshold effects and control of oxidative phosphorylation in nonsynaptic rat brain mitochondria. J Neurochem 1996, 66:1617-1624 [DOI] [PubMed] [Google Scholar]
- 37.Yamashita N, Hoshida S, Taniguchi N, Kuzuya T, Hori M: Whole-body hyperthermia provides biphasic cardioprotection against ischemia/reperfusion injury in the rat. Circ 1998, 98:1414-1421 [DOI] [PubMed] [Google Scholar]
- 38.Herrmann JM, Stuart RA, Craig EA, Neupert W: Mitochondrial heat shock protein 70, a molecular chaperone for proteins encoded by mitochondrial DNA. J Cell Biol 1994, 127:893-902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becker J, Walter W, Yan W, Craig EA: Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol 1996, 16:4378-4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samali A, Orrenius S: Heat shock proteins: regulators of stress response and apoptosis. Cell Stress Chaperones 1998, 3:228-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simpkins CO, Fogarty KW, Nhamburo P: Reduction of cytochrome C by fragments of heat shock protein 70. Life Sci 1993, 52:1487-1492 [DOI] [PubMed] [Google Scholar]
- 42.Klanner C, Neupert W, Langer T: The chaperonin-related protein Tcm62p ensures mitochondrial gene expression under heat stress. FEBS Lett 2000, 470:365-369 [DOI] [PubMed] [Google Scholar]
- 43.Hansford RG, Hogue BA, Mildaziene V: Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr 1997, 29:89-95 [DOI] [PubMed] [Google Scholar]
- 44.Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC: Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA 1999, 96:846-851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szabo C, O’Connor M, Salzman AL: Endogenously produced peroxynitrite induces the oxidation of mitochondrial and nuclear proteins in immunostimulated macrophages. FEBS Lett 1997, 409:147-150 [DOI] [PubMed] [Google Scholar]
- 46.Minetti M, Mallozzi C, Di Stasi AM, Pietraforte D: Bilirubin is an effective antioxidant of peroxynitrite-mediated protein oxidation in human blood plasma. Arch Biochem Biophys 1998, 352:165-174 [DOI] [PubMed] [Google Scholar]
- 47.Abraham NG, Drummond GS, Lutton JD, Kappas A: The biological significance and physiological role of heme oxygenase. Cell Physiol Biochem 1998, 6:129-168 [Google Scholar]
- 48.Calabrese V, Testa G, Ravagna A, Bates TE, Stella AM: HSP70 induction in the brain following ethanol administration in the rat: regulation by glutathione redox state. Biochem Biophys Res Commun 2000, 269:397-400 [DOI] [PubMed] [Google Scholar]
- 49.Jayakumar J, Suzuki K, Khan M, Smolenski RT, Farrell A, Latif N, Raisky O, Abunasra H, Sammut IA, Murtuza B, Amrani M, Yacoub MH: Gene therapy for myocardial protection: transfection of donor hearts with heat shock protein 70 gene protects cardiac function against ischemia-reperfusion injury. Circulation 2000, 102:III302-III306 [DOI] [PubMed] [Google Scholar]
- 50.Luis AM, Alconada A, Cuezva JM: The alpha regulatory subunit of the mitochondrial F1-ATPase complex is a heat-shock protein. Identification of two highly conserved amino acid sequences among the alpha-subunits and molecular chaperones. J Biol Chem 1990, 265:7713-7716 [PubMed] [Google Scholar]
- 51.Harding AE, Holt IJ, Cooper JM, Schapira AH, Sweeney M, Clark JB, Morgan-Hughes JA: Mitochondrial myopathies: genetic defects. Biochem Soc Trans 1990, 18:519-522 [DOI] [PubMed] [Google Scholar]
- 52.Johns DR: The other human genome: mitochondrial DNA and disease. Nat Med 1996, 2:1065-1068 [DOI] [PubMed] [Google Scholar]
- 53.Zaid A, Li R, Luciakova K, Barath P, Nery S, Nelson BD: On the role of the general transcription factor Sp1 in the activation and repression of diverse mammalian oxidative phosphorylation genes. J Bioenerg Biomembr 1999, 31:129-135 [DOI] [PubMed] [Google Scholar]
- 54.Vigh L, Literati PN, Horvath I, Torok Z, Balogh G, Glatz A, Kovacs E, Boros I, Ferdinandy P, Farkas B, Jaszlits L, Jednakovits A, Koranyi L, Maresca B: Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med 1997, 3:1150-1154 [DOI] [PubMed] [Google Scholar]
- 55.Hartsfield CL, Alam J, Choi AM: Transcriptional regulation of the heme oxygenase 1 gene by pyrrolidine dithiocarbamate. FASEB J 1998, 12:1675-1682 [DOI] [PubMed] [Google Scholar]