

Abstract
Analyses of cancer incidence data in the United States and Western Europe revealed steadily rising rates over the past decades of adenocarcinomas of the esophagus and gastric cardia. Genetic information on gastric cardia adenocarcinoma and its preneoplasias is sparse. We have used comparative genomic hybridization to obtain a genome-wide overview of 20 archival gastric cardia adenocarcinomas and 10 adjacent preneoplastic lesions (4 metaplasias, 1 low-grade dysplasia , 5 high-grade dysplasias). Multiple genetic alterations were discriminated in all adenocarcinomas. Frequent loss (≥25% of all tumors) was detected, in decreasing order of frequency, on 5q, 18q, 4q, 3p, 9p, 2q, 11q, 14q, 21q, 4p, 9q, 16q, 1p, and 8p. Frequent gain (≥25% of all tumors) was disclosed, in decreasing order of frequency, on 20q, 7p, 8q, 1q, 7q, 20p, 17q, 13q, Xp, 6q, 8p, 19q, 5p, 6p, and Xq. Loss of the Y chromosome was found in 60% of male cases. High level amplification was frequently (>10% of all tumors) detected on 7q21, 8p22, 12p11.2, 17q12-q21, and 19q13.1-q13.2. The precursor lesions showed multiple aberrations in all high-grade dysplasias, whereas few genetic changes were discerned in LGD and metaplasias. High level amplifications were also found in high-grade dysplasias, ie, on 7q21, 8p22, and 17q12-q21. Moreover, the percentage of aberrations was not significantly different for invasive carcinomas or high-grade dysplasias. Approximately 70% of the precursor aberrations were also present in the adjacent carcinoma. Minimal overlapping regions in the preneoplasias included loss on 18q12-q21 and gains on 8q23 and 17q12-q21, suggesting involvement of genes residing in these regions. In conclusion, we have (i) created a map of genetic alterations in gastric cardia adenocarcinomas and (ii) provided evidence for the presence of a metaplasia-dysplasia-carcinoma sequence in this poorly understood type of cancer.
The incidence of adenocarcinomas arising around the gastro-esophageal junction has shown steadily rising rates over the past decades in the Western world. 1,2 A five- to sixfold increase was reported between 1970 and 1990. 3,4 Overall, cancer of the esophagus is increasing, and stomach cancer is decreasing. However, when analyzed by histological type and subsite, the picture is very different. In esophagus, squamous cell carcinoma rates have remained stable, whereas a rapid increase of adenocarcinoma is observed. In stomach, cardia cancer shows a very similar pattern to adenocarcinoma of the esophagus, but pyloric-antrum cancer is decreasing. Adenocarcinomas of the gastro-esophageal junction region disproportionately affect white men and occur less frequently among women. 1,5 The simultaneously increased incidence at the different locations suggest that adenocarcinomas of the gastro-esophageal junction are related. Reflux disease has been suggested as an etiological factor not only in esophageal adenocarcinoma, but also in cancer of the gastric cardia. 6 Esophageal and gastric cardia adenocarcinomas have in common that they arise around the gastro-esophageal junction. Esophageal adenocarcinoma is strongly correlated with Barrett’s esophagus. In Barrett’s esophagus, the squamous cell epithelium has undergone metaplastic change to columnar epithelium as a result of long-standing gastro-esophageal reflux. 7,8 Metaplastic change has also been observed at the gastro-esophageal junction, which also might explain the rising frequency at this location. 9 Intestinal metaplasia of the gastric cardia has been reported; however, its relation with malignant transformation is presently not clear. 10,11 Adenocarcinomas of the esophagus and gastric cardia share a poor prognosis, due to aggressive tumor behavior, as well as late detection. 12,13
Cytogenetic studies of series of both gastric and esophageal adenocarcinomas have shown frequent chromosomal rearrangement of 11p13–15 14 and deletion of 3q. 15 In a study of 37 adenocarcinomas in Barrett’s esophagus and gastric cardia, loss of the Y chromosome appeared to be a prominent feature. 16 Further, rearrangements were most frequently seen of chromosome arms 1p, 3q, 11p, and 22q. Genetic abnormalities have been extensively documented in the formerly common pyloric-antrum type of gastric cancer. Patterns of gene amplification have been investigated. 17 Ranzani et al 18 detected loss of heterozygosity (LOH) at 5q, 11p, 17p, and 18q, and with a low frequency also at 7q and 13q. In these gastric cancers deletions often occur at the APC and MCC loci on 5q21. 19 Little is known of LOH in gastric cardia tumors. In a study of adenocarcinoma of the gastric cardia frequent allelic loss was seen on 3p, 4q, 5q, 8p, 9pq, 12q, 13q, 17p, and 18q. 20 A comparison of esophageal and gastric cardia cancers revealed similar patterns of TP53 alterations. 21 Comparative genomic hybridization of gastro-esophageal junction cancers was reported including limited numbers of gastric cardia carcinomas. 22-24 Gain of chromosome 20 was most frequently found. Further, prominent gain was seen on 1q, 7pq, 8q, 13q, 15q, and 17q, and loss was observed on 4pq, 5q, 9p, 14q, and 18q. Recurrent high-level amplifications were disclosed at 8q23–24.1, 17q12–21, and 19q13.1.
In this study we wanted to document the spectrum of genetic changes in a series of gastric cardia adenocarcinomas and surrounding preneoplastic lesions. We were especially interested in a metaplasia-dysplasia-carcinoma sequence in this type of cancer. Therefore, we collected metaplastic and dysplastic tissues from archival resection specimens of gastric cardia adenocarcinomas. To our knowledge such an investigation has not been reported. Comparative genomic hybridization (CGH) was used to obtain a genome-wide view of chromosomal gains and losses.
Materials and Methods
Patient Material
We collected 20 surgical specimens with adenocarcinomas of the gastric cardia, ie, the center of the tumor was clearly located in the proximal stomach. Barrett’s epithelium was absent in all cases. The presence of Helicobacter pylori infection was evaluated on routine H&E sections, but in difficult cases special stains were used. Special attention was given to the selection of preneoplastic lesions (metaplasia, dysplasia) in the vicinity of the tumor. Intestinal metaplasia was defined as the presence of goblet cells in gastric epithelium. Precursors were selected from 10 resection specimens. In the other 10 cases no preneoplasias could be found, possibly due to overgrowth of these generally large cancers. Fourteen of the tumor specimens were derived from paraffin-embedded, formalin-fixed, materials; six were fresh-frozen. All preneoplastic lesions were paraffin-embedded. Staging of the tumors was performed according to the Union Internationale Contre le Cancer (UICC). 25
Comparative Genomic Hybridization
Isolation of DNA from the formalin-fixed, paraffin-embedded tumor material was performed using standard procedures. Microdissection of the tumor areas was performed using a hollow bore coupled to the microscope. Metaplastic and dysplastic areas were scraped from 10-μm paraffin sections using a stereo microscope and a 0.4 × 12 mm hollow needle. DNA from both fresh-frozen and archival paraffin samples was isolated according to standard protocols, and concentration, purity, and molecular weight of the DNA were estimated. Tumor DNA with a fragment size of <1 kb was labeled with a platinum/biotin complex (bio-ULS), using the ULS biotin labeling kit (Kreatech Diagnostics, Amsterdam, The Netherlands.) 26 Tumor DNA with larger DNA fragment sizes was labeled with biotin by nick translation (Nick Translation System, Gibco BRL, Gaithersburg, MD). Likewise, male reference DNA (Promega, Madison, WI) was labeled by nick translation with digoxigenin (Boehringer Mannheim, Indianapolis, IN). The reaction time and the amount of DNase were adjusted to obtain a matching probe size for reference and tumor DNAs. Molecular weight of both tumor and reference DNA was checked by gel electrophoresis after nick translation, and ranged between 500 and 1500.
CGH was performed as described before. 24,26 In brief, 400 ng of labeled archival tumor DNA, 200 ng of reference DNA, and 15 μg of unlabeled Cot-1 DNA were ethanol-precipitated and dissolved in 10 μl of hybridization mixture (50% formamide, 0.1% Tween-20, and 10% dextran sulfate in 2× standard saline citrate at pH 7.0). The probe mixture was denatured and hybridized to normal male metaphase chromosomes (Vysis, Downers Grove, IL) for 3 days at 37°C. After washing of the slides, fluorescent detection of the biotin- and digoxigenin-labeled DNA probes was accomplished with avidin-fluorescein isothiocyanate and anti-digoxigenin rhodamine, respectively. Samples were counterstained with 4′,6′-diamidino-2-phenyl indole (DAPI) in anti-fade solution.
Images were acquired with an epifluorescent microscope (Leica DM, Rijswijk, The Netherlands) equipped with a CCD camera (Photometrics, Tucson, AZ), three single excitation filters, a multiband pass dichroic mirror, and emission filters. For comparative genomic hybridization analysis Quips XL software (version 3.1.1; Vysis) was used. Loss of DNA sequences was defined as chromosomal regions where the mean green to red ratio was <0.85, whereas gain was defined as chromosomal regions where the ratio was >1.15. These threshold values were based on series of normal controls. A high-level amplification, probably representing an amplicon, was seen as a distinct peak (ratio >1.5). At least 8 to 10 metaphases per sample were used for the analysis of gains and losses.
Results
Gastric Cardia Adenocarcinomas
We have used CGH to obtain a genome-wide overview of 20 archival gastric cardia adenocarcinomas (17 males, 3 females) and 10 adjacent preneoplastic lesions: 4 metaplasias, 1 low-grade dysplasia (LGD), and 5 high-grade dysplasias (HGD; see Table 1 ▶ ). Intestinal metaplasia was found in the vicinity of 6 tumors (30%), whereas H. pylori-associated gastritis was present in 3 cases (15%). Multiple genetic alterations were discriminated in all adenocarcinomas (Figure 1A) ▶ . Frequent loss (≥25% of all tumors) was detected, in decreasing order of frequency, on 5q (55%), 18q (55%), 4q (50%), 3p (45%), 9p (45%), 2q (40%), 11q (35%), 14q (35%), 21q (35%), 4p (30%), 9q (30%), 16q (30%), 1p (25%), and 8p (25%). Frequent gain (≥25% of all tumors) was disclosed, in decreasing order of frequency, on 20q (80%), 7p (70%), 8q (60%), 1q (55%), 7q (55%), 20p (45%), 17q (40%), 13q (35%), Xp (35%), 6q (30%), 8p (30%), 19q (30%), 5p (25%), 6p (25%), and Xq (25%). Loss of the Y chromosome was found in 60% of male cases. Minimal overlapping regions for loss were assigned to 3p14, 5q21, 5q31-q33, 8p21-p22, 9p21, 11q24-q25, 14q23-q24, 18q12-q21, and 21q21, whereas minimal overlapping regions for gain were assigned to 1q41-q42, 7p12-p14, 7q21, 8p22, 8q21.3-q23, 12p11.2, 13q12-q14, 17q12-q21, 19q13.1-q13.2, and 20q11.2-q13.1. Recurrent (>10% of all tumors) high-level amplification (HLA) was detected on 7q21, 8p22, 12p11.2, 17q12-q21, and 19q13.1-q13.2, illustrating the genomic instability in these carcinomas.
Table 1.
Patient and Tumor Data
| Sex/age | Grade* | Stage† | Precursor | HP-gastritis | |
|---|---|---|---|---|---|
| 1 | M /75 | G2 | T1N0M0 | HGD | − |
| 2 | M /75 | G3 | T2N2M0 | LGD | − |
| 3 | M /63 | G3 | T2N0M0 | MET | + |
| 4 | M /50 | G1 | T1N0M0 | HGD | − |
| 5 | F /75 | G2 | T2N2M0 | HGD | − |
| 6 | M /71 | G3 | T2N1M0 | HGD | − |
| 7 | M /67 | G2 | T2N0M0 | HGD | − |
| 8 | F /71 | G3 | T3N1M0 | MET | + |
| 9 | M /54 | G3 | T3N1M0 | MET | − |
| 10 | M /32 | G2 | T3N2M0 | MET | − |
| 11 | M /63 | G1 | T2N1M0 | n.a. | − |
| 12 | M /57 | G2 | T2N1M0 | n.a. | − |
| 13 | M /54 | G3 | T3N1M0 | n.a. | − |
| 14 | M /75 | G2 | T3N1M0 | n.a. | − |
| 15 | M /58 | G3 | T3N2M0 | n.a. | − |
| 16 | M /69 | G2 | T2N0M0 | n.a. | − |
| 17 | M /45 | G3 | T2N0M0 | n.a. | − |
| 18 | M /56 | G1 | T3N1M0 | n.a. | − |
| 19 | M /70 | G3 | T3N1M0 | n.a. | − |
| 20 | F /72 | G3 | T3N1M0 | n.a. | + |
*G1, well differentiated; G2, moderately differentiated; G3, poorly differentiated.
†Tumor-node-metastasis classification according to the UICC, 25 in brief: T1, tumor invades mucosa or submucosa; T2, muscularis propria or gastric subserosa; T3, gastric serosa or adjacent esophageal adventitia. N0, no lymph node metastasis; N1, 1–6 positive nodes; N2, 7–15 positive nodes. M0, no distant metastases.
MET, metaplasia; LGD, low-grade dysplasia; HGD, high-grade dysplasia; n.a., not available (possibly due to overgrowth of the generally large cancer); HP-gastritis, Helicobacter pylori-associated gastritis.
Figure 1.

A: Chromosomal ideograms showing the summary of DNA copy number changes, detected by CGH in 20 gastric cardia adenocarcinomas. Losses are displayed (as bars) on the left of the ideogram, gains are shown on the right. Frequent loss is seen on 1p, 2q, 3p, 4pq, 5q, 8p, 9pq, 11q, 14q, 16q, 18q, 21q, and Y. Frequent gain is detected at 1q, 5p, 6pq, 7pq, 8pq, 13q, 17q, 19q, 20pq, and Xpq. High level amplification (HLA; marked by an open bar) was frequently detected on 7q21, 8p22, 12p11.2, 17q12-q21, and 19q13.1-q13.2. B: Chromosomal ideograms showing the summary of DNA copy number changes, detected by CGH in 10 preneoplastic lesions (4 metaplasias, 1 low-grade dysplasia, 5 high-grade dysplasias). Frequent loss can be discriminated on 2q, 4p, 5q, 9p, 18q, 21q, and Y; frequent gain is seen on 6p, 7q, 8q, 13q, 17q, and 20q. HLAs were found only in high-grade dysplasia on 7q21, 8p22, and 17q12-q21.
Metaplasia-Dysplasia-Carcinoma Data
The precursor lesions showed multiple aberrations in all HGDs, whereas few genetic (mostly non-frequent) changes were discerned in LGD and metaplasias (Figure 1B) ▶ . Frequent gains included 8q (40%), 6p (30%), 7q (30%), 13q (30%), 17q (30%), and 20q (30%), whereas frequent losses were seen on 18q (50%), Y (40%), 2q (30%), 4p (30%), 5q (30%), 9p (30%), and 21q (30%). HLAs were found only in HGD: two on 17q12-q21, one on 7q21, and one on 8p22. The mean percentage of aberrations in the 20 adenocarcinomas appeared only slightly higher than in the 6 dysplasias (11.9 vs. 10.6, respectively). This was caused by the 5 HGDs in this set of dysplasias (Table 2) ▶ . The 4 metaplasias showed an average of 0.7 alterations per lesion. Approximately 70% of the precursor aberrations were also present in the adjacent carcinoma, illustrating the presence of a metaplasia-dysplasia-adenocarcinoma sequence in gastric cardia cancer (Table 2 ▶ and Figure 2 ▶ ). A high percentage of concurrent alterations were seen in HGD and invasive cancer, despite the evident differences in histomorphology (Figure 2, C–F) ▶ . Overlapping alterations were not only seen in HGD, but also in LGD (case 2), and even in intestinal metaplasia (cases 3 and 10). Minimal overlapping regions in the preneoplasias included loss on 18q12-q21 and gains on 8q23 and 17q12-q21, suggesting involvement of genes in these regions in the development of cancer of the proximal stomach.
Table 2.
Genetic Aberrations in Preneoplastic Lesions of the Gastric Cardia
| Patient | Precursor | Aberrations | |
|---|---|---|---|
| 1 | HGD | loss: | 2q21-q22, 4p14-pter, 5p13-pter, 5q12-q33, 9p13-pter, 11q21-qter, 12q21 |
| gain: | 6p11.2-p21.3, 7q32-q34, 8q23, 19p13.1-pter, 19q13.1-q13.2 | ||
| 2 | LGD | loss: | n.a. |
| gain: | 17q21-qter | ||
| 3 | MET | loss: | 18q12-q21 |
| gain: | X | ||
| 4 | HGD | loss: | 1p21-p31, 1q23-qter, 2p12-pter, 2q13-q31, 3p13-pter, 4, 9p13-pter, 18, Y |
| gain: | 6p11.2-p22, 6q12-q15, 7q11.2-q22, 13q12-q22, 17q11.2-q21 (HLA 17q12-q21), Xq12-qter | ||
| 5 | HGD | loss: | 12, 18q12-qter, 19, 21q11.2-qter, 22 |
| gain: | 8q22-qter, 13, 20 | ||
| 6 | HGD | loss: | 2p23-pter, 5q12-qter, 9p13-pter, 9q21-q32, 15q11.2-q24, 17p12, 18q11.2-qter, 21, 22, Y |
| gain: | 1q32-qter, 5p14-pter, 7p11.2-pter, 7q11.2-q22 (HLA 7q21), 8 (HLA 8p22-q23), 12p11.2-p12, 20 | ||
| 7 | HGD | loss: | 1p13-pter, 1q41-q42, 2q31-qter, 3, 4, 5q12-qter, 8p21-pter, 10q22-qter, 14q13-qter, 16q13-qter, 18q12-qter, 20p11.2-p12, 21, Y |
| gain: | 5p12-pter, 6p11.2-p23, 6q12-q24, 7p11.2-pter, 7q11.2, 8q11.2-qter, 9p23, 9q13-q21, 13, 17q11.2-q24 (HLA 17q12-q21), 20q11.2-qter | ||
| 8 | MET | loss: | n.a. |
| gain: | n.a. | ||
| 9 | MET | loss: | n.a. |
| gain: | n.a. | ||
| 10 | MET | loss: | 19p13.2-pter |
| gain: | n.a. | ||
Alterations shared with the concurrent invasive cancer are printed in bold.
MET, metaplasia; LGD, low-grade dysplasia; HGD, high-grade dysplasia; HLA, high-level amplification; n.a., no aberrations.
Figure 2.
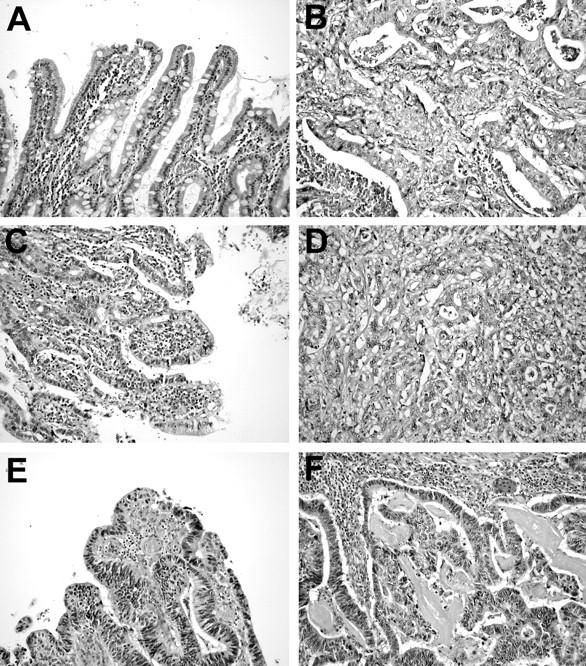
Photomicrographs illustrating pairs of preneoplastic lesions (A, C, and E) and adenocarcinoma (B, D, and F) of the gastric cardia. Case 3 showed intestinal metaplasia (A) adjacent to poorly differentiated adenocarcinoma (B). Precursor and cancer shared gain of the X chromosome, whereas in this metaplastic tissue also loss on 18q was disclosed by CGH. Case 6 displayed high-grade dysplasia (C) and poorly differentiated adenocarcinoma (D). Many alterations (12 of 17) detected by CGH in the dysplasia were also discerned in the invasive cancer. Case 7 demonstrated HGD (E) and moderately differentiated adenocarcinoma (F). CGH revealed a basically similar pattern of chromosomal aberrations, despite the clear differences in histomorphology of the two lesions, ie, a non-invasive histology in HGD versus the invasive growth pattern of the adenocarcinoma.
Discussion
In this study we have created a genome-wide map of genetic alterations in gastric cardia adenocarcinomas. Furthermore, we have provided evidence of the presence of a metaplasia-dysplasia-carcinoma sequence in this type of cancer. Intestinal metaplasia of the gastro-esophageal junction is found in approximately 10 to 40% of patients without long segments of Barrett’s mucosa. Spechler et al 9 reported that 15 to 20% of adults undergoing elective upper endoscopy had segments of intestinal metaplasia at the gastro-esophageal junction that were not recognized by endoscopy. Trudgill et al 27 also concluded that intestinal metaplasia at the junction is a common finding. In our study a high percentage of overlapping alterations was found between adenocarcinomas and preneoplasias. Interestingly, in two of the metaplasias aberrations were already discerned: In case 10 a non-frequent alteration that was also seen in the adenocarcinoma, in case 3 two changes, one of which was present in the cancer. Comparable numbers of chromosomal aberrations, as well as a very high percentage of overlap, were found in HGDs and invasive carcinomas despite clearly non-invasive versus invasive growth patterns. Thus, a discrepancy seems present between phenotypic and genotypic characteristics in HGD, leading us to speculate that the apparently non-invasive histomorphology of HGD might actually represent well-differentiated adenocarcinoma.
We have disclosed frequent alterations in gastric cardia cancers and precursors, and minimal overlapping regions could be assigned at multiple locations. Below we will discuss the most prevalent and relevant candidate genes for these frequent chromosomal regions of gain or loss in the adenocarcinomas 28,29 (also listed on the genecards database of the Weizmann Institute at http://bioinfo.weizmann.ac.il/cards).
Chromosomal Loss
In the literature only one detailed study has been reported describing LOH solely in gastric cardia adenocarcinomas. 20 Recurrent allelic losses (>50%) were seen on 3p, 4q, 5q, 8p, 9p, 9q, 12q, 13q, 17p, and 18q. In our series the most frequent losses were noted on chromosome arms 5q and 18q (both 55% of cases), whereas less frequent deletions were found on 1p, 2q, 3p, 4p, 4q, 8p, 9p, 9q, 11q, 14q, 16q, and 21q. Thus, a good concordance is present between these two studies using different methodologies. A discrepancy between the two series is our low percentage of deletions on distal 17p within the TP53 (tumor protein p53) region. It might be attributed to the lower sensitivity of CGH, as compared to LOH analysis. 30 Furthermore, the frequency of TP53 alterations might be somewhat lower in gastric cardia cancers than in esophageal (Barrett’s) adenocarcinomas. 31 The determination of two regions of loss on 5q might point to two sites for putative tumor suppressors. The 5q21 region contains the mutated in colorectal cancer (MCC) and adenomatous polyposis coli (APC) genes, which have been shown to be involved in Barrett-related cancers. The loss at 5q31-q33 might involve α-catenin (CTNNA1, locus at 5q31), which is part of the E-cadherin-catenin complex. The loss at 18q12-q21 might be related to DCC (deleted in colorectal carcinoma) and/or DPC4 (deleted in pancreatic carcinoma), genes known to be altered in gastrointestinal cancers. A high frequency of loss is observed on chromosome 4, but it was not possible to assign a common region on each of the two chromosome arms. This could be due to a high density of tumor suppressor genes on this chromosome, which is in concordance with a LOH study of chromosome 4q in Barrett’s adenocarcinoma. 32
Chromosomal Gain
The most prominent gain was found on chromosome 20, especially 20q (80%), with a minimal region at 20q11.2-q13.1. HLA was detected on 20q13.1. It is noteworthy that in a recent CGH array investigation of breast carcinomas, CYP24, encoding vitamin D24 hydroxylase, and located at 20q13.2, was discriminated as a putative oncogene. 33 High-frequency gain was also noted on 8q with a minimally gained region at 8q21.3-q23, which is slightly proximal to the MYC (v-myc myelocytomatosis oncogene) locus. Expression studies have shown that, eg, epidermal growth factor receptor, Kirsten rat sarcoma viral oncogene homologue, or v-erb-b2 oncogene (HER2-neu) are involved in the malignant transformation of Barrett’s esophagus, 34 which may explain the gains and amplifications at 7p12-p14, 12p11.2, and 17q12-q21. The amplification on 7q21 is associated more with hepatocyte growth factor (HGF) than with the more distally located MET oncogene, encoding for the HGF receptor. HGF serum levels were reported to correlate significantly with the aggressiveness of gastric carcinomas. 35 Immunohistochemistry showed that MET was overexpressed in about half of gastric cancers, whereas gene amplification was detected by Southern blot hybridization in about 10% of them. 36 The recurrent HLA at 8p might be associated with cathepsin B, which appears to be amplified and overexpressed in Barrett-related carcinomas. 37 HLA at 19q13.1-q13.2 might involve cyclin E, which has recently been described as the candidate gene of a 19q12 amplicon in adenocarcinomas of the gastro-esophageal junction. 38
The genetic spectrum of imbalances in gastric cardia cancers, disclosed by CGH, is very similar to the distribution of alterations seen in Barrett-related adenocarcinomas. 24,39 It is also in keeping with allelotyping and in situ hybridization studies of esophageal adenocarcinoma. 40-42 Furthermore, the pattern of gains and losses in the preneoplastic lesions shows a high degree of similarity to the genetic changes seen in Barrett’s esophagus 39,43-46 , ie, losses in cardia cancer precursor lesions are frequently seen on 4p, 5q, 9p, and 18q, whereas gains are repeatedly found on 7q, 8q, 17q, and 20q. This illustrates, moreover, that adenocarcinomas at these two locations have much in common, although a minor proportion of proximal stomach cancers might be related to H. pylori infection. However, our data strongly suggest a shared etiology of gastro-esophageal junction adenocarcinomas arising either in the distal esophagus (Barrett’s esophagus) or in the gastric cardia.
Footnotes
Address reprint requests to Dr. H. van Dekken, Department of Pathology, Josephine Nefkens Institute, Erasmus University Rotterdam, P.O. Box 1738, 3000 DR Rotterdam, The Netherlands.
Supported by Dutch Cancer Society grants EUR 97–1478 and EUR 97–1404.
References
- 1.Blot WJ, Devesa SS, Kneller RW, Fraumeni JF, Jr: Rising incidence of adenocarcinoma of the esophagus and gastric cardia. JAMA 1991, 265:1287-1289 [PubMed] [Google Scholar]
- 2.Powell J, McConkey CC: The rising trend in esophageal adenocarcinoma and gastric cardia. Eur J Cancer Prev 1992, 1:265-269 [DOI] [PubMed] [Google Scholar]
- 3.Pera M, Cameron AJ, Trastek VF, Carpenter HA, Zinsmeister HR: Increasing incidence of adenocarcinoma of the esophagus and esophagogastric junction. Gastroenterology 1993, 104:510-513 [DOI] [PubMed] [Google Scholar]
- 4.Locke GR, III, Talley NJ, Carpenter HA, Harmsen WS, Zinsmeister HR, Melton LJ, III: Changes in the site- and histology-specific incidence of gastric cancer during a 50-year period. Gastroenterology 1995, 109:1750-1756 [DOI] [PubMed] [Google Scholar]
- 5.Blot WJ, Devesa SS, Fraumeni JF, Jr: Letter: continuing climb in rates of esophageal adenocarcinoma. JAMA 1993, 270:1320. [PubMed] [Google Scholar]
- 6.Chow WH, Blot WJ, Vaughan TL, Risch HA, Gammon MD, Stanford JL, Dubrow R, Schoenberg JB, Mayne ST, Farrow DC, Ahsan H, West AB, Rotterdam H, Fraumeni JF, Jr: Body mass index and risk of adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst 1998, 21:150-155 [DOI] [PubMed] [Google Scholar]
- 7.Spechler SJ, Goyal RK: Barrett’s esophagus. New Engl J Med 1986, 315:362-371 [DOI] [PubMed] [Google Scholar]
- 8.Haggitt RC: Barrett’s esophagus, dysplasia and adenocarcinoma. Hum Pathol 1994, 25:982-993 [DOI] [PubMed] [Google Scholar]
- 9.Spechler SJ, Zeroogian JM, Antonioli JA, Wang HH, Goyal RK: Prevalence of metaplasia at the gastro-esophageal junction. Lancet 1994, 344:1533-1536 [DOI] [PubMed] [Google Scholar]
- 10.Morales TG, Sampliner RE, Bhattacharyya A: Intestinal metaplasia of the gastric cardia. Am J Gastroenterol 1997, 92:414-418 [PubMed] [Google Scholar]
- 11.Ruol A, Parenti A, Zaninotto G, Merigliano S, Costantini M, Cagol M, Alfieri R, Bonavina L, Peracchia A, Ancona E: Intestinal metaplasia is the common precursor of adenocarcinoma in Barrett esophagus and adenocarcinoma of the gastric cardia. Cancer 2000, 88:2520-2528 [DOI] [PubMed] [Google Scholar]
- 12.Blomjous JG, Hop WCJ, Langenhorst BL, ten Kate FL, Eykenboom WM, Tilanus HW: Adenocarcinoma of the gastric cardia: recurrence and survival after resection. Cancer 1992, 70:569-574 [DOI] [PubMed] [Google Scholar]
- 13.Wijnhoven BPL, Siersema PD, Hop WCJ, van Dekken H, Tilanus HW: Adenocarcinomas of the distal oesophagus and gastric cardia are of one clinical entity. Br J Surg 1999, 86:529-535 [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez E, Rao PH, Ladanyi M, Altorki N, Albino AP, Kelsen DP, Jhanwar SC, Chaganti RSK: 11p13–15 is a specific region of chromosomal rearrangement in gastric and esophageal adenocarcinomas. Cancer Res 1990, 50:6410-6416 [PubMed] [Google Scholar]
- 15.Rao PH, Mathew S, Kelsen DP, Chaganti RSK: Cytogenetics of gastric and esophageal adenocarcinomas. 3q Deletion as a possible primary chromosomal change. Cancer Genet Cytogenet 1995, 81:139-143 [DOI] [PubMed] [Google Scholar]
- 16.Menke-Pluymers MBE, van Drunen E, Vissers KJ, Mulder AH, Tilanus HW, Hagemeier-Hausman AMMJ: Cytogenetic analysis of Barrett’s mucosa and adenocarcinoma of the distal esophagus and cardia. Cancer Genet Cytogenet 1996, 90:109-117 [DOI] [PubMed] [Google Scholar]
- 17.Houldsworth J, Cordon-Cardo C, Ladanyi M, Kelsen DP, Chaganti RSK: Gene amplification in gastric and esophageal adenocarcinomas. Cancer Res 1990, 50:6417-6422 [PubMed] [Google Scholar]
- 18.Ranzani GN, Renault B, Pellegata NS, Fattorini P, Magni E, Bacci F, Amadori D: Loss of heterozygosity and K-ras gene mutations in gastric cancer. Hum Genet 1993, 92:244-249 [DOI] [PubMed] [Google Scholar]
- 19.Tamura G, Maesawa C, Suzuki Y, Ogasawara S, Terashima M, Saito K, Satodate R: Primary gastric carcinoma cells frequently lose heterozygosity at the APC and MCC genetic loci. Jpn J Cancer Res 1993, 84:1015-1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gleeson CM, Sloan JM, McGuigan JA, Ritchie AJ, Weber JL, Russell SE: Allelotype analysis of adenocarcinoma of the gastric cardia. Br J Cancer 1997, 76:1455-1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gleeson CM, Sloan JM, McManus DT, Maxwell P, Arthur K, McGuigan JA, Ritchie AJ, Russell SE: Comparison of p53 and DNA content abnormalities in adenocarcinoma of the oesophagus and gastric cardia. Br J Cancer 1998, 77:277-286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Rifai W, Harper JC, Cummings OW, Hyytinen ER, Frierson HF, Jr, Knuutila S, Powell SM: Consistent genetic alterations in xenografts of proximal stomach and gastro-esophageal junction adenocarcinomas. Cancer Res 1998, 58:34-37 [PubMed] [Google Scholar]
- 23.Moskaluk CA, Hu J, Perlman PJ: Comparative genomic hybridization of esophageal and gastroesophageal adenocarcinomas shows consensus areas of DNA gain and loss. Genes Chromosomes Cancer 1998, 22:305-311 [PubMed] [Google Scholar]
- 24.van Dekken H, Geelen E, Dinjens WNM, Wijnhoven BPL, Tilanus HW, Tanke HJ, Rosenberg C: Comparative genomic hybridization of cancer of the gastro-esophageal junction: deletion of 14q31–32.1 discriminates between esophageal (Barrett’s) and gastric cardia adenocarcinomas. Cancer Res 1999, 59:748-752 [PubMed] [Google Scholar]
- 25.Hermanek P, Hutter RVP, Sobin LH, Wagner G, Wittekind Ch: TNM Atlas International Union Against Cancer/Union internationale Contre le Cancer (UICC), 4th ed. 1997:pp 81-92 Springer-Verlag, Berlin
- 26.Alers JC, Rochat J, Krijtenburg PJ, van Dekken H, Raap AK, Rosenberg C: Universal linkage system (ULS): an improved method for labeling archival DNA for comparative genomic hybridization. Genes Chromosomes Cancer 1999, 25:301-305 [DOI] [PubMed] [Google Scholar]
- 27.Trudgill NJ, Suvarna SK, Kapur KC, Riley SA: Intestinal metaplasia at the squamocolumnar junction in patients attending for diagnostic gastroscopy. Gut 1997, 41:585-589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knuutila S, Björkqvist A-M, Autio K, Tarkkanen M, Wolf M, Monni O, Szymanska J, Larramendy ML, Tapper J, Pere H, El-Rifai W, Hemmer S, Wasenius V-M, Vidgreb V, Zhu Y: DNA copy number amplifications in human neoplasms: review of comparative genomic hybridization studies. Am J Pathol 1998, 152:1107-1123 [PMC free article] [PubMed] [Google Scholar]
- 29.Knuutila S, Aalto Y, Autio K, Bjorkqvist A-M, El-Rifai W, Hemmer S, Huhta T, Kettunen E, Kiuru-Kuhlefelt S, Larramendy ML, Lushnikova T, Monni O, Pere H, Tapper J, Tarkkanen M, Varis A, Wassenius V-M, Wolf M, Zhu Y: DNA copy number losses in human neoplasms. Am J Pathol 1999, 155:683-694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirchhoff M, Gerdes T, Rose H, Maahr J, Ottesen AM, Lundsteen C: Detection of chromosomal gains and losses in comparative genomic hybridization analysis bases on standard reference intervals. Cytometry 1998, 31:163-173 [PubMed] [Google Scholar]
- 31.Taniere P, Martel-Planche G, Maurici D, Lombard-Bohas C, Scoazec J-Y, Montesano R, Berger F, Hainaut P: Molecular and clinical differences between adenocarcinomas of the esophagus and of the gastric cardia. Am J Pathol 2001, 158:33-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rumpel CA, Powell SM, Moskaluk CA: Mapping of genetic deletions on the long arm of chromosome 4 in human esophageal adenocarcinomas. Am J Pathol 1999, 154:1329-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Albertson DG, Ylstra B, Segraves R, Collins C, Dairkee SH, Kowbel D, Kuo W-L, Gray JW, Pinkel D: Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate gene. Nat Genet 2000, 25:144-146 [DOI] [PubMed] [Google Scholar]
- 34.Jankowski JA, Wright NA, Meltzer SJ, Triadafilopoulos G, Geboes K, Casson AG, Kerr D, Young LS: Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Pathol 1999, 54:965-973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han SU, Lee JH, Kim WH, Cho YK, Kim MW: Significant correlation between serum level of hepatocyte growth factor and progression of gastric carcinoma. World J Surg 1999, 23:1176-1180 [DOI] [PubMed] [Google Scholar]
- 36.Nakajima M, Sawada H, Yamada Y, Watanabe A, Tatsumi M, Yamashita J, Matsuda M, Sakaguchi T, Hirao T, Nakano H: The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer 1999, 85:1894-1902 [DOI] [PubMed] [Google Scholar]
- 37.Hughes SJ, Glover TW, Zhu XX, Kuick R, Thoraval D, Orringer MB, Beer DG, Hanash S: A novel amplicon at 8p22–23 results in overexpression of cathepsin B in esophageal adenocarcinoma. Proc Natl Acad Sci USA 1998, 95:12410-12415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin L, Prescott MS, Zhu Z, Singh P, Chun SY, Kuick RD, Hanash RM, Orringer MB, Glover TW, Beer DG: Identification and characterization of a 19q12 amplicon in esophageal adenocarcinomas reveals cyclin E as the best candidate gene for this amplification. Cancer Res 2000, 60:7021-7027 [PubMed] [Google Scholar]
- 39.Walch AK, Zitzelsberger HF, Bruch J, Keller G, Angermeier D, Aubele MM, Mueller J, Stein H, Braselmann H, Siewert JR, Höfler H, Werner M: Chromosomal imbalances in Barrett’s adenocarcinoma and the metaplasia-dysplasia-carcinoma sequence. Am J Pathol 2000, 156:555-566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnadath KK, Tilanus HW, Alers JC, Mulder AH, van Dekken H: Detection of genetic changes in Barrett’s adenocarcinoma and Barrett’s esophagus by DNA in situ hybridization and immunohistochemistry. Cytometry 1994, 15:176-184 [DOI] [PubMed] [Google Scholar]
- 41.Barrett MT, Galipeau PC, Sanchez CA, Emond MJ, Reid BJ: Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene 1996, 12:1873-1878 [PubMed] [Google Scholar]
- 42.Dolan K, Garde J, Gosney J, Sissons M, Wright T, Kingsnorth AN, Walker SJ, Sutton R, Meltzer SJ, Field JK: Allelotype analysis of oesophageal adenocarcinoma: loss of heterozygosity occurs at multiple sites. Br J Cancer 1998, 78:950-957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blount PL, Meltzer SJ, Yin J, Huang Y, Krasna MJ, Reid BJ: Clonal ordering of 17p and 5q allelic losses in Barrett’s dysplasia and adenocarcinoma. Proc Natl Acad Sci USA 1993, 90:3221-3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krishnadath KK, Tilanus HW, van Blankenstein M, Hop WCJ, Teijgeman R, Mulder AH, Bosman FT, van Dekken H: Accumulation of genetic abnormalities during neoplastic progression in Barrett’s esophagus. Cancer Res 1995, 55:1971-1976 [PubMed] [Google Scholar]
- 45.Wu TT, Watanabe T, Heitmiller R, Zahurak M, Forastiere AA, Hamilton SR: Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction. Am J Pathol 1998, 153:287-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riegman PHJ, Vissers KJ, Alers JC, Geelen E, Hop WCJ, Tilanus HW, van Dekken H: Genomic alterations in malignant transformation of Barrett’s esophagus. Cancer Res 2001 (in press) [PubMed]