

Abstract
Parasympathetic paragangliomas (PGLs) represent neuroendocrine tumors arising from chief cells in branchiomeric and intravagal paraganglia, which share several histological features with their sympathetic counterpart sympathoadrenal paragangliomas. In recent years, genetic analyses of the familial form of PGL have attracted considerable interest. However, the majority of paragangliomas occurs sporadically and it remains to be determined whether the pathogenesis of sporadic paraganglioma resembles that of the familial form. Furthermore, data on comparative genetic aberrations are scarce. To provide fundamental cytogenetic data on sporadic and hereditary PGLs, we performed comparative genomic hybridization using directly fluorochrome-conjugated DNA extracted from 12 frozen and 4 paraffin-embedded tumors. The comparative genomic hybridization data were extended by loss of heterozygosity analysis of chromosome 11q. DNA copy number changes were found in 10 (63%) of 16 tumors. The most frequent chromosomal imbalance involved loss of chromosome 11. Six of seven familial tumors and two of nine sporadic tumors showed loss of 11q (86% versus 22%, P = 0.012). Deletions of 11p and 5p were found in two of nine sporadic tumors. We conclude that overall DNA copy number changes are infrequent in PGLs compared to sympathetic paragangliomas and that loss of chromosome 11 may be an important event in their tumorigenesis, particularly in familial paragangliomas.
Parasympathetic paragangliomas (PGLs) are rare, highly vascularized tumors, originating from neural crest-derived chief cells of paraganglia in the head and neck region. They share many histological features with sympathoadrenal PGLs, including pheochromocytomas (PCCs). Metastases of PGLs are uncommon, but may emerge in lymph nodes, lung, and liver. Dependent on the anatomical location the tumor can cause serious symptoms like dysphagia, bradycardia, and hearing loss. Because of this and in view of the risk of progression to malignancy, surgical resection of PGLs is often required.
The carotid body and jugulotympanic paraganglia are the most common sites of origin of parasympathetic PGLs, followed by vagal, laryngeal, and aorticopulmonary paraganglia. 1,2 Although most parasympathetic PGLs occur sporadically, there is a positive family history in a considerable minority (10 to 50%) of cases. 3 Predominance in females, multiple PGLs, and young age of onset are characteristic of familial PGLs, but such features have also been reported in sporadic cases. 4,5 Co-occurrence of parasympathetic PGLs and PCCs, and occurrence in Carney’s syndrome and neurofibromatosis type 1 have been described. 6-9 Flow cytometric analyses revealed DNA aneuploidy in 21 to 50% of parasympathetic PGLs, which did not predict malignant behavior or decreased survival. 10-12 A few immunohistochemical studies have suggested a paracrine/autocrine role for IGF-II, c-myc, bcl-2, and c-jun in PGL pathogenesis. 13-15
In attempts to clarify the genetic mechanisms underlying the development of parasympathetic PGLs, the inherited form of this tumor has so far been the main subject of investigation. Linkage analysis and loss of heterozygosity (LOH) studies in unrelated families provided evidence for the existence of two distinct PGL susceptibility genes. The putative PGL1 gene (11q23) and the PGL2 gene (11q13.1) are both thought to be tumor suppressor genes and maternally imprinted. 16-20 Recently, a third, not maternally imprinted gene was demonstrated to cause PGL in a German family (PGL3). 21
Although substantial progress has thus been made in the identification of genetic changes involved in the development of hereditary PGL, comparative data on genomic changes in sporadic and familial parasympathetic PGLs are not available and it remains to be clarified whether these tumors develop along the same genetic pathways. To characterize cytogenetic alterations, we investigated nine sporadic and seven familial parasympathetic PGLs by comparative genomic hybridization (CGH) analysis. In addition, LOH analysis was performed to confirm CGH results.
Materials and Methods
Patients and Tumor Samples
Sixteen benign tumors from 14 patients with parasympathetic PGLs, diagnosed between 1992 and 1996, were studied. The average age of the patients (nine female) at first presentation was 37 years (range, 29 to 55 years) and the mean size of the tumors was 2.5 ± 1.2 cm. Information on family history and other tumors or relevant conditions was obtained by reviewing medical charts and by interviewing all patients, after an average follow-up period of 7.8 years (94 months). Six of 14 patients had a positive family history for parasympathetic PGL and eight patients had sporadic PGL. A PGL was considered sporadic when there were no first or second degree relatives known with a parasympathetic PGL. Clinical data are summarized in Table 1 ▶ .
Table 1.
Clinical Characteristics and Genetic Findings in Parasympathetic Paragangliomas
| Patient | Age of onset/sex | Location | Size (cm) | Horm. activity | Type | Follow-up data (months) | LOH results | CGH results | |
|---|---|---|---|---|---|---|---|---|---|
| D11S134 | D11S1986 | ||||||||
| 1 | 33 /F | VAG | 4.0 | − | spor. | NED (85) | |B* | |B* | 5p−, 11p− |
| 2 | 32 /M | CAR | 2.5 | − | fam. | MUL (85) | ▪ | ▪ | 11pq− |
| 3 | 30 /F | CAR | nk | − | fam. | MUL (80) | ni | ▪ | n |
| 4 | 45 /F | CAR | 3.0 | − | spor. | MUL (68) | ni | |B* | n |
| 5 | 45 /M | TYMP | 0.5 | − | spor. | NED (66) | — | — | 11pq− |
| 6 | 42 /F | IUG | 1.5 | − | spor. | REC (127) | — | — | n |
| 7 | 31 /F | CAR | 1.5 | − | spor. | NED (63) | — | — | 9p−, 11pq− |
| 8 | 29 /F | CAR | 4.0 | − | fam. | MUL (63) | ni | ▪ | 1p−, 11pq− |
| CAR | 1.8 | − | ni | ▪ | 11pq−, 13q−, 17pq+ | ||||
| 9 | 33 /M | CAR | 1.5 | − | spor. | MUL (61) | |B* | |B* | n |
| CAR | 2.0 | − | |B* | |B* | n | ||||
| 10 | 39 /M | CAR | 4.0 | − | fam. | MUL (56) | ni | |B* | n |
| 11 | 30 /F | CAR | 3.5 | − | fam. | MUL (248) | ▪ | ▪ | 3q−, 11q− |
| 12 | 33 /F | TRACH | 2.0 | − | spor. | NED (96) | |B* | |B* | 3pq− |
| 13 | 55 /F | AO-PULM | 1.2 | + | spor. | NED (96) | |B* | |B* | 5p−, 11p− |
| 14 | 38 /M | AO-PULM | 4.0 | + | fam. | MUL* (120) | ▪ | ▪ | 2pq−, 11pq−, 18q− |
Abbreviations: F, female; M, male; VAG, vagal; CAR, carotid body; TYMP, tympanic; IUG, iugular; TRACH, tracheal; AO-PULM, aorto-pulmonal; nk, not known; fam., familial; spor., sporadic; NED, no evidence of disease; REC, local recurrence; MUL, one or more parasympathetic PGL in other locations including the contralateral carotid body; ni, noninformative; n, no detectable changes; —, no data.
*This patient also presented with carotid body PGL and abdominal masses suggestive of paraganglioma, as detected by computed tomography imaging.
DNA Extraction
Genomic DNA from 12 frozen tumors was isolated using the D-5000 Puregene DNA isolation kit (Gentra Systems, Minneapolis, MN). DNA extraction from four formalin-fixed, paraffin-embedded samples was performed by standard detergent-proteinase K lysis, followed by phenol/chloroform extraction and ethanol precipitation, as described elsewhere. 22 Only tumors with >80% tumor cell content were included in this study.
CGH and Digital Image Analysis
CGH was performed as described. 22 In brief, 1 μg of tumor DNA was labeled with Spectrum Green-dUTPs (Vysis, Downers Grove, IL) by nick translation (BioNick kit; Life Technologies, Basel, Switzerland). The hybridization mixture consisted of 200 ng of Spectrum Green-labeled tumor DNA, 200 ng of Spectrum Red-labeled sex-matched normal reference DNA (Vysis), and 10 to 20 μg human Cot-1 DNA (Life Technologies) dissolved in 10 μl of hybridization buffer (50% formamide, 10% dextran sulfate, 2× standard saline citrate, pH 7.0). Hybridization to normal metaphase spreads (Vysis) took place for 3 days at 37°C. Slides were washed at 45°C three times for 10 minutes in 50% formamide/2× standard saline citrate and two times in 2× standard saline citrate. The chromosomes were counterstained with 4,6-diamidino-2-phenylindole in anti-fade solution for identification.
Digital images were collected from six to seven metaphases using a Photometrics cooled charge-coupled device camera (Microimager 1400; Xillix Technologies, Vancouver, Canada). The QUIPS software program (Vysis) was used to calculate average green-to-red ratio profiles of at least four observations per autosome and two observations per sex chromosome in each analysis. Gains and losses of DNA sequences were defined as chromosomal regions where the mean green-to-red fluorescence ratio was >1.20 and <0.80, respectively. Overrepresentations were considered amplifications when the fluorescence ratio values in a subregion of a chromosomal arm exceeded 1.5. Because of some false-positive results at chromosomes 1p32-pter, 16p, 19, and 22 found in normal tissues, gains of these known G-C-rich regions were excluded from all analyses.
LOH Analysis
To validate CGH data independently, 12 PGLs of 11 patients of whom normal DNA was available, were analyzed for allelic imbalances of the 11q23 locus using two microsatellite markers D11S1347 and D11S1986 (Research Genetics, Huntsville, AL). 19 Polymerase chain reaction amplification of tumor and germline DNA was performed in reaction mixtures of 50 μl. Each reaction contained 50 to 100 ng of template DNA, 0.2 mmol/L dATP, dTTP, dGTP, dCTP, 20 to 50 pmol of each primer, 1.5 mmol/L Mg2+, 10 mmol/L Tris-HCl, 50 mmol/L KCl, and 1 U Taq DNA polymerase (Amplitaq Gold; Perkin Elmer, Norwalk, CT). An initial denaturation step at 94°C for 5 minutes was followed by 35 cycles of denaturation at 94°C for 45 seconds, annealing at 55°C (for marker D11S1986 at 52°C) for 60 seconds, and extension at 72°C for 60 seconds. A final extension step was performed at 72°C for 10 minutes. Polymerase chain reaction products of tumor and normal DNA from each patient were diluted 1:1 in 10 μl of loading buffer (95% formamide, 20 mmol/L ethylenediaminetetraacetic acid, 0.05% xylene cyanol, 0.05% bromophenol blue) and loaded onto a nondenaturing 6% polyacrylamide gel. Electrophoresis was performed at 40 W for 2.5 hours. The DNA was visualized by silver staining and evaluated as described previously. 23 Allelic loss was considered to be present when the intensity of the signal from one allele was significantly reduced in the tumor DNA when compared with normal DNA by direct visualization. Because PGLs are known to contain admixed normal tissue (sustentacular cells and supportive fibrovascular stroma), a weak band of the lost allele was accepted in the determination of LOH.
Statistical Analysis
The chi-square test for nominal variance was used to calculate the statistical significance of differences in genomic changes between familial and sporadic tumors.
Results
Overview of CGH Findings
Figure 1A ▶ summarizes all DNA copy number changes identified in 16 parasympathetic PGLs. Genetic alterations were observed in 10 (63%) of 16 PGLs, and the average number of chromosome arm aberrations per tumor was 1.75 ± 1.69 (range, 0 to 5). With the exception of one gain of chromosome 17, all aberrations implied chromosomal losses and no amplifications were identified (Table 1) ▶ . The most common chromosome arm copy number change was loss of chromosome 11. Six tumors showed loss of the entire chromosome, one tumor exhibited loss of 11q, and two tumors had a deletion of 11p. Losses of chromosome 11q were strongly associated with familial PGL (86% versus 22% in sporadic PGL) and seemed to be statistically significant (P = 0.012). Other genomic imbalances were rare; one gain was found on chromosome 17 (1 of 16), and losses involved 3q and 5p (both 2 of 16), 1p, 3p, 9p, 13q, and 18q (1 of 16 each). Both tumors with loss of 11p also exhibited loss of 5p and seemed to be sporadic tumors. Representative examples of CGH results are shown in Figure 1B ▶ .
Figure 1.
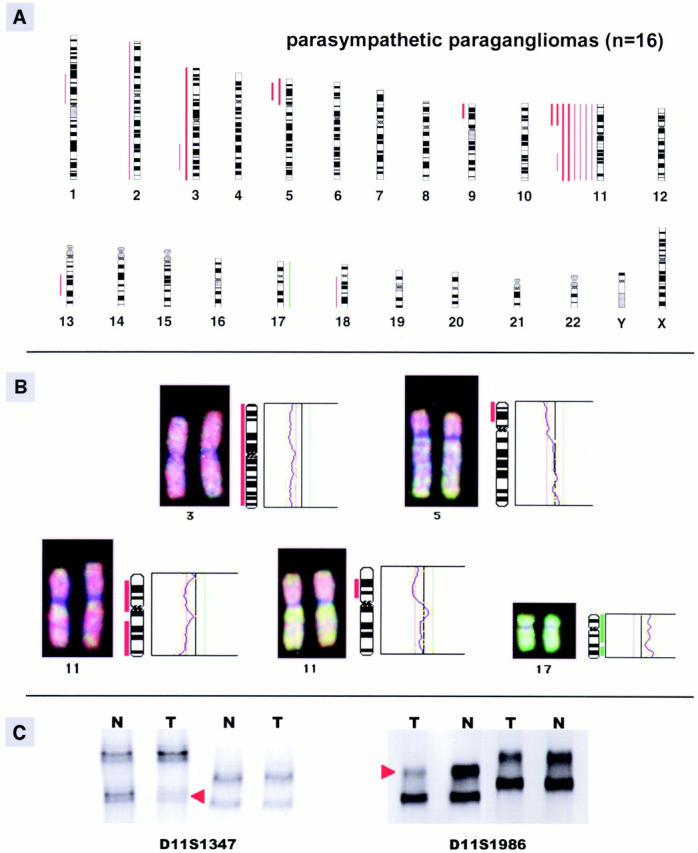
A: Summary of all DNA copy number changes detected by CGH in 16 (seven familial and nine sporadic) parasympathetic PGLs. The vertical green lines on the right side of the chromosome ideograms indicate gains, the red lines on the left side indicate losses of the corresponding chromosomal region. Findings in sporadic PGLs are indicated as solid bars. B: Individual examples of CGH digital images (left) and fluorescent ratio profiles (right) illustrating genomic alterations of chromosome 3 (loss), 5 (loss of 5p), 11 (loss of the entire chromosome and of 11p), and 17 (gain). C: Example of LOH analysis of a familial PGL, showing a band markedly diminished in tumoral (T) DNA in comparison with normal (N) DNA (allelic loss) in two microsatellite markers at 11q23, D11S1986, and D11S1347 (red arrowheads).
Comparison of CGH and LOH Results
Twelve tumors of 11 patients of whom normal DNA was available were examined by microsatellite analysis. We could confirm the CGH results of chromosome 11q in all tumors that were analyzed and detected LOH of this region in one additional tumor without apparent loss of 11q in CGH analysis (Table 1) ▶ . Altogether, most deletions of chromosome 11q were found in familial PGLs (6 of 7 familial PGLs versus 2 of 9 sporadic PGLs). Representative examples of LOH results are shown in Figure 1C ▶ .
Discussion
This study represents the first comprehensive, genome-wide analysis of chromosomal aberrations in sporadic and familial parasympathetic PGLs. Our results indicate that DNA copy number changes are infrequent in these tumors and that tumor suppressor genes on chromosome 11 may play a critical role in the tumorigenesis of familial PGLs. Sporadic PGLs may develop along different genetic pathways.
Cancer, as a genetic disease, is believed to arise from an accumulation of genetic aberrations that promote clonal selection of cells with increasingly aggressive behavior. It is proposed that most cancer cell genotypes demonstrate essential alterations in cell physiology. This is reflected by the finding of multiple chromosomal imbalances in the majority of human cancers. However, our results indicate that DNA copy number changes are infrequent in parasympathetic PGLs. This is consistent with their characteristically slow growth rate and benign behavior and indicates that some cell-signaling mechanisms, generally disrupted in cancer cells, may still be intact in PGL tumor cells.
In a considerable proportion of PGLs, we found a normal DNA copy number profile. To exclude the possibility of false-negative results, we included in our analysis only samples with at least >80% tumor cells. Nonetheless, it should be borne in mind that the lower limit of CGH-based detection is ∼10 Mb. Indeed, we found additional loss of 11q by LOH analysis in one tumor.
Only one PGL in this study showed a DNA copy number gain. This gain could not be related to one of the proteins found to be up-regulated in PGLs. 13-15 Therefore, in parasympathetic PGL, up-regulation of growth stimulating factors is probably caused by other events, such as genetic mutations or rearrangements.
Loss of 11q was the most common chromosomal aberration in our series of PGLs, with a remarkable difference in incidence between familial and sporadic tumors (86% versus 22%, respectively; P = 0.012). This may point toward distinct tumorigenic pathways in these subgroups of PGLs.
Co-occurrence of parasympathetic PGLs and sympathoadrenal PGLs (including PCCs) has been described. 6,8 Because these tumors share many histological characteristics, they may be thought to result from similar genetic changes. However, comparison of CGH data of benign parasympathetic and sympathoadrenal PGLs shows that these types are genetically different. In benign sympathoadrenal PGLs, genetic changes are much more frequent and 11q losses are less frequent compared to the parasympathetic PGLs in this study. 24,25 Yet, it remains to be tested whether the same tumor suppressor gene(s) on 11q are involved in the tumorigenesis of both parasympathetic PGLs and PCCs.
The long arm of chromosome 11 contains two well-defined critical regions that each harbor a putative PGL disease gene, PGL1 at 11q23 and PGL2 at 11q13. 16,18,26 Recently, Baysal and co-workers 27 detected germline mutations of the mitochondrial succinate dehydrogenase complex II subunit d (SDHD) gene (11q23) co-segregating with tumor occurrence in PGL families, indicating that this is the putative PGL1 gene. The mitochondrial complex II is an important enzyme complex in the aerobic respiratory chains of mitochondria. Loss of function of this complex may cause cellular hypoxia and increased superoxide levels. Chronic hypoxia is known to be an important cause of carotid body hyperplasia and conceivably plays a role in tumor initiation and progression of carotid body PGLs. In this respect, it is of interest that two of nine sporadic PGLs showed loss of 5p, the gene locus of another component (the flavoprotein subunit) of the mitochondrial succinate dehydrogenase complex II. 28 These tumors also demonstrated loss of 11p, but not of 11q. This may imply that in the pathogenesis of some sporadic PGLs, other components of the mitochondrial complex II are involved in their tumorigenesis.
In conclusion, our study demonstrates that chromosomal imbalances are infrequent in parasympathetic PGLs compared to their sympathetic counterpart, PCCs. We observed a high frequency of 11q loss in familial but not in sporadic PGLs, pointing toward differences in the genetic evolution of familial and sporadic PGLs. Genetic changes on chromosome 11p and 5p may be involved in sporadic PGLs.
More genetic data are needed to assess the concept and aspects of genetic differences between familial and sporadic parasympathetic PGLs. Further analysis of the candidate tumor suppressor gene SDHD and other genes of the mitochondrial complex II is required to determine their involvement in the development of these tumors.
Acknowledgments
We thank Monica Seijbel, Erasmus University Medical Center Rotterdam, for excellent secretarial assistance; and Claudia Matter and Alex Scheidweiler, University of Zürich, Switzerland, for outstanding technical assistance.
Footnotes
Address reprint requests to Ronald R. de Krijger, M.D., Ph.D., Erasmus University Medical Center Rotterdam, Josephine Nefkens Institute, P. O. Box 1738, 3000 DR, Rotterdam, the Netherlands. E-mail: dekrijger@path.azr.nl.
Supported by the Foundation “De Drie Lichten,” the Jan Dekker en Dr. Ludgardina Bouwman Foundation, and the Dutch Cancer Society (Koningin Wilhelmina Fonds) the Netherlands.
References
- 1.Lack EE, Cubilla AL, Woodruff JM: Paragangliomas of the head and neck region. A pathologic study of tumors from 71 patients Hum Pathol 1979, 10:191-218 [DOI] [PubMed] [Google Scholar]
- 2.Tischler AS: The adrenal medulla and extra-adrenal paraganglia. Functional Endocrine Pathology. Edited by K Kovacs and SL Asa. Boston: Blackwell Scientific Publications, 1990, pp 509–545
- 3.van der Mey AG, Maaswinkel-Mooy PD, Cornelisse CJ, Schmidt PH, van de Kamp JJ: Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet 1989, 2:1291-1294 [DOI] [PubMed] [Google Scholar]
- 4.Gardner P, Dalsing M, Weisberger E, Sawchuk A, Miyamoto R: Carotid body tumors, inheritance, and a high incidence of associated cervical paragangliomas. Am J Surg 1996, 172:196-199 [DOI] [PubMed] [Google Scholar]
- 5.McCaffrey TV, Meyer FB, Michels VV, Piepgras DG, Marion MS: Familial paragangliomas of the head and neck. Arch Otolaryngol Head Neck Surg 1994, 120:1211-1216 [DOI] [PubMed] [Google Scholar]
- 6.Jensen JC, Choyke PL, Rosenfeld M, Pass HI, Keiser H, White B, Travis W, Linehan WM: A report of familial carotid body tumors and multiple extra-adrenal pheochromocytomas. J Urol 1991, 145:1040-1042 [DOI] [PubMed] [Google Scholar]
- 7.Pritchett JW: Familial concurrence of carotid body tumor and pheochromocytoma. Cancer 1982, 49:2578-2579 [DOI] [PubMed] [Google Scholar]
- 8.DeAngelis LM, Kelleher MB, Post KD, Fetell MR: Multiple paragangliomas in neurofibromatosis: a new neuroendocrine neoplasia. Neurology 1987, 37:129-133 [DOI] [PubMed] [Google Scholar]
- 9.Carney JA: Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 1999, 74:543-552 [DOI] [PubMed] [Google Scholar]
- 10.Barnes L, Taylor SR: Vagal paragangliomas: a clinical, pathological, and DNA assessment. Clin Otolaryngol 1991, 16:376-382 [DOI] [PubMed] [Google Scholar]
- 11.Sauter ER, Hollier LH, Bolton JS, Ochsner JL, Sardi A: Prognostic value of DNA flow cytometry in paragangliomas of the carotid body. J Surg Oncol 1991, 46:151-153 [DOI] [PubMed] [Google Scholar]
- 12.van der Mey AG, Cornelisse CJ, Hermans J, Terpstra JL, Schmidt PH, Fleuren GJ: DNA flow cytometry of hereditary and sporadic paragangliomas (glomus tumours). Br J Cancer 1991, 63:298-302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li SL, Goko H, Xu ZD, Kimura G, Sun Y, Kawachi MH, Wilson TG, Wilczynski S, Fujita-Yamaguchi Y: Expression of insulin-like growth factor (IGF)-II in human prostate, breast, bladder, and paraganglioma tumors. Cell Tissue Res 1998, 291:469-479 [DOI] [PubMed] [Google Scholar]
- 14.Jyung RW, LeClair EE, Bernat RA, Kang TS, Ung F, McKenna MJ, Tuan RS: Expression of angiogenic growth factors in paragangliomas. Laryngoscope 2000, 110:161-167 [DOI] [PubMed] [Google Scholar]
- 15.Wang DG, Johnston CF, Barros D’Sa AA, Buchanan KD: Expression of apoptosis suppressing gene bcl-2 in human carotid body tumours. J Pathol 1997, 183:218-221 [DOI] [PubMed] [Google Scholar]
- 16.Baysal BE, van Schothorst EM, Farr JE, Grashof P, Myssiorek D, Rubinstein WS, Taschner P, Cornelisse CJ, Devlin B, Devilee P, Richard CW: Repositioning the hereditary paraganglioma critical region on chromosome band 11q23. Hum Genet 1999, 104:219-225 [DOI] [PubMed] [Google Scholar]
- 17.Heutink P, van Schothorst EM, van der Mey AG, Bardoel A, Breedveld G, Pertijs J, Sandkuijl LA, van Ommen GJ, Cornelisse CJ, Oostra BA, Weber JL, Devilee P: Further localization of the gene for hereditary paragangliomas and evidence for linkage in unrelated families. Eur J Hum Genet 1994, 2:148-158 [DOI] [PubMed] [Google Scholar]
- 18.Mariman EC, van Beersum SE, Cremers CW, Struycken PM, Ropers HH: Fine mapping of a putatively imprinted gene for familial non-chromaffin paragangliomas to chromosome 11q13.1: evidence for genetic heterogeneity. Hum Genet 1995, 95:56-62 [DOI] [PubMed] [Google Scholar]
- 19.van Schothorst EM, Jansen JC, Bardoel AF, van der Mey AG, James MJ, Sobol H, Weissenbach J, van Ommen GJ, Cornelisse CJ, Devilee P: Confinement of PGL, an imprinted gene causing hereditary paragangliomas, to a 2-cM interval on 11q22–q23 and exclusion of DRD2 and NCAM as candidate genes. Eur J Hum Genet 1996, 4:267-273 [DOI] [PubMed] [Google Scholar]
- 20.Devilee P, van Schothorst EM, Bardoel AF, Bonsing B, Kuipers-Dijkshoorn N, James MR, Fleuren G, van der Mey AG, Cornelisse CJ: Allelotype of head and neck paragangliomas: allelic imbalance is confined to the long arm of chromosome 11, the site of the predisposing locus PGL. Genes Chromosom Cancer 1994, 11:71-78 [DOI] [PubMed] [Google Scholar]
- 21.Niemann S, Muller U: Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 2000, 26:268-270 [DOI] [PubMed] [Google Scholar]
- 22.Richter J, Jiang F, Gorog JP, Sartorius G, Egenter C, Gasser TC, Moch H, Mihatsch MJ, Sauter G: Marked genetic differences between stage pTa and stage pT1 papillary bladder cancer detected by comparative genomic hybridization. Cancer Res 1997, 57:2860-2864 [PubMed] [Google Scholar]
- 23.Komminoth P, Kunz E, Hiort O, Schroder S, Matias-Guiu X, Christiansen G, Roth J, Heitz PU: Detection of RET proto-oncogene point mutations in paraffin-embedded pheochromocytoma specimens by nonradioactive single-strand conformation polymorphism analysis and direct sequencing. Am J Pathol 1994, 145:922-929 [PMC free article] [PubMed] [Google Scholar]
- 24.Edstrom E, Mahlamaki E, Nord B, Kjellman M, Karhu R, Hoog A, Goncharov N, Teh BT, Backdahl M, Larsson C: Comparative genomic hybridization reveals frequent losses of chromosomes 1p and 3q in pheochromocytomas and abdominal paragangliomas, suggesting a common genetic etiology. Am J Pathol 2000, 156:651-659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dannenberg H, Speel EJ, Zhao J, Saremaslani P, van Der Harst E, Roth J, Heitz PU, Bonjer HJ, Dinjens WN, Mooi WJ, Komminoth P, de Krijger RR: Losses of chromosomes 1p and 3q are early genetic events in the development of sporadic pheochromocytomas. Am J Pathol 2000, 157:353-359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milunsky J, DeStefano AL, Huang XL, Baldwin CT, Michels VV, Jako G, Milunsky A: Familial paragangliomas: linkage to chromosome 11q23 and clinical implications. Am J Med Genet 1997, 72:66-70 [DOI] [PubMed] [Google Scholar]
- 27.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, Cornelisse CW, Devilee P, Devlin B: Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287:848-851 [DOI] [PubMed] [Google Scholar]
- 28.Morris AA, Farnsworth L, Ackrell BA, Turnbull DM, Birch-Machin MA: The cDNA sequence of the flavoprotein subunit of human heart succinate dehydrogenase. Biochim Biophys Acta 1994, 1185:125-128 [DOI] [PubMed] [Google Scholar]