

Abstract
Lewis rats, on recovery from monophasic clinical experimental allergic encephalomyelitis (EAE), can be induced to develop repeated paralytic relapses with a graded reduction in clinical severity following intraperitoneal administration of IL-12. By the time of the third relapse, the number and size of inflammatory cuffs in the spinal cord were reduced with the makeup of the cellular infiltrate shifting to a significantly increased number of B cells. Serum levels of myelin basic protein (MBP)-specific IgG1 and IgG2b were found to rise over time while MBP and MBP peptide-positive macrophages and microglia became evident in perivascular cuffs and in spinal cord parenchyma, indicative of myelin phagocytosis. Axonal death was observed in semithin and EM sections of spinal cord in third relapse animals in association with iNOS and tPA immunostaining throughout gray and white matter. These neurotoxic or excitotoxic agents may contribute to axonal damage directly or indirectly by activated microglia and macrophages, leading to limited damage of the axonal-myelin unit.
The inducer and effector cells in experimental allergic encephalomyelitis (EAE), an experimental model of the demyelinating disorder multiple sclerosis (MS), are comprised of CD4+ T cells and macrophages in the inflammatory perivascular cuffs predominant in the spinal cord. 1-3 T cells that transfer EAE are characteristic of T-helper (Th)-1 type 4-6 while recovery from the disease correlates with a surge in the expression of Th2 type cytokines. 7 Rats that recover from the acute form of EAE are usually resistant to reinduction, which may result from loss of effector T cells via apoptosis 8 or the appearance of suppressor T cells. 9
Interleukin (IL)-12 is a potent cytokine capable of inducing the release of interferon (IFN)-γ from natural killer cells and T cells, augmenting cell-mediated immune responses in vitro and in vivo. 10-14 In patients with acute and progressive MS, IL-12 production by peripheral blood mononuclear cells has been shown to increase significantly 15,16 via an activated CD4+ T cell receptor-mediated pathway, involving the CD40 ligand. 17 IL-12 mRNA is increased in macrophages in active MS plaques and in peripheral blood mononuclear cells from patients with relapsing-remitting MS. 18 Additionally, transfer of lymph node cells from proteolipid protein primed animals in the presence of IL-12 has been shown to exacerbate clinical signs of EAE 19 and we have recently reported that IL-12 administration reinduces paralytic EAE in Lewis rats. 20
Proteolysis of myelin is a key step in demyelination, the prominent cellular route being via macrophages, which are observed with ingested myelin in MS plaques. 21 In acute rat EAE, little or no demyelination has been observed, 22 whereas in chronic relapsing models in both rat and mouse, extensive myelin loss is prominent, particularly in the spinal cord. 23-26 Plasminogen activators can degrade myelin by the action of plasmin, which is found to be increased in MS lesions. 21 Of the serine proteases, tissue-type plasminogen activator (tPA) has also been implicated in neuronal plasticity and degeneration. 27,28 tPA null mice have been shown to be resistant to neuronal damage following injections of neurotoxins into the hippocampus while normal mice suffered neurodegeneration. 29,30 In MS lesions, tPA expression together with plasminogen activator inhibitor−1 is prominent in foamy macrophages, 31 and preliminary experiments have shown that tPA is also localized on demyelinated axons (D. Gveric, unpublished observations).
The synthetic enzyme of NO, inducible nitric oxide synthase (iNOS), which has been described in active demyelinating MS lesions 32 and in macrophages of the inflammatory cuff in EAE, 20 is found to be associated with an increased CNS lesion load of macrophage-enriched perivascular cuffs in an IL-12-induced relapse of EAE. 20 Proinflammatory cytokines are thought to mediate axonal damage by the formation of reactive oxygen intermediates such as NO, which also facilitates damage of the highly susceptible myelin membrane. 32,33 Exposure to high concentrations of NO has also been shown to cause axonal degeneration and neuronal death. 34-37
The aim of this study was to investigate if repeat relapses could be obtained with IL-12, and whether serial relapses lead to axonal and/or myelin damage. This may then serve as a useful model for understanding the interactions involved in relapsing/remitting MS.
Materials and Methods
Animals and Induction of Active EAE
Female Lewis rats (180 to 200 g; Charles River, Kent, UK) were housed in pairs in a standard animal facility, allowed free access to food and water. Animals, which showed excess weight loss, were fed moist rodent chow. Rats were immunized in each hind foot with a mixture of purified guinea pig myelin basic protein (MBP) (final concentration 1 mg/ml), emulsified in Freund’s complete adjuvant (MBP-CFA) containing Mycobacterium tuberculosis H37Ra (final concentration 5 mg/ml; Difco Laboratories, Detroit, Michigan) in a final volume of 50 μl.
Assessment of Clinical EAE
Animals were weighed and monitored daily for clinical signs of EAE using the following criteria: 0, no clinical signs; 0.5, loss of tonicity in distal half of tail; 1, flaccid tail and weight loss; 2, hind limb hypotonia; 2.5, paralysis of one hind limb; 3, complete hind limb paralysis; 4, moribund; and 5, death.
IL-12 Administration
Recombinant murine IL-12 (Genetics Institute, Cambridge, MA; batch number MRB630717292A; specific activity of 4.6 × 10 6 U/mg; endotoxin contamination <0.641 EU/mg, measured in the Limulus amoebocyte assay) was used in all experiments. Rats were allowed to recover from the primary bout of disease, and IL-12 (3 μg/rat, 0.2 ml) was administered i.p. on days 17, 19, and 21 post inoculation (p.i.)). 20 The animals were then allowed to recover fully before being administered a second and third dose of IL-12 (3 μg/rat, 0.2 ml i.p.).
Immunocytochemistry
Rats were culled at different stages after immunization and the brains and spinal cords were removed and rapidly frozen on solid CO2. Ten-micrometer-thick longitudinal frozen sections of a 1 cm portion of the cervical and lumbar regions of spinal cords were cut, adhered to Vectabond coated slides (Vector Laboratories, Peterborough, UK), and processed for immunocytochemistry, as described before. 20 Monoclonal antibodies were used to examine the immunopathology of the relapse and demyelination using the following antibodies: anti-rat OX42 (CD11b, 1:1000, microglia); anti-rat CD2 (OX34, 1:500) and anti-rat ED1 (1:500) (all from Serotec, Oxford, UK); clone 6 (1:200, Affiniti Research Products, Exeter, UK) specific for iNOS; monoclonal anti-human MBP (119–131) (Serotec); monoclonal QD9 and rabbit EP, both markers of degenerating myelin; 38 monoclonal anti rat CD4 and CD8 (Serotec); and monoclonal horseradish peroxidase-labeled anti rat IgG1 and IgG2a (Zymed Labs, San Francisco, CA); anti-human tPA (1:100 Biogenesis, Poole, UK).
Tissues were stained by both a fluorescent and an avidin-biotin diaminobenzidine enhancement method. For the fluorescent method, tissues were fixed in ethanol for 1 minute at room temperature followed by incubation with the primary antibodies overnight at 4°C in a humidified chamber before washing in phosphate-buffered saline (PBS) and incubating with fluorescein isothiocyanate-labeled goat anti-mouse IgG (Fab specific; Sigma, Poole, Dorset, UK), diluted 1:100 in PBS, for 1 hour at room temperature. Sections were washed in PBS and counterstained with 1% propidium iodide for 10 minutes at room temperature. Finally, sections were washed in PBS and mounted in Citifluor (Citifluor Ltd., London, UK) and viewed using a fluorescent microscope. For the diaminobenzidine enhancement method, primary antibody was incubated as described above followed by washes in PBS and incubation with either biotinylated anti-mouse or anti-rabbit IgG (both from Vector Labs, Peterborough, UK) at a dilution of 1:200 for 30 minutes at room temperature. Sections were then washed in PBS and peroxidase-labeled avidin-biotin complex solution was added to the sections for 45 minutes, washed in PBS, and peroxidase activity detected by placing the slides in a solution of 3,3′-diaminobenzidine (Sigma) containing 0.01% hydrogen peroxide for 5 minutes. Rinsed sections were counterstained in Mayer’s hematoxylin for 30 seconds, washed in running tap water, dehydrated through a graded series of alcohols, cleared in xylene, and mounted in DPX (BDH, Poole, UK).
Histopathological Evaluation
Initially, the number of inflammatory cuffs in the cervical, thoracic, lumbar, and sacral regions of the spinal cord was counted from sections stained with hematoxylin and eosin to determine whether there were regional differences in cuff numbers.
The number of cuffs in the 1-cm portion of cervical and representative lumbar spinal cords were then counted. Individual cuffs were then scored as: 1, perivascular inflammation three or fewer cells deep; 2, more than three cells deep; 3, parenchymal infiltrate. The histopathological score was calculated for each animal by adding the scores for all cuffs in a section, and taking a mean of three individual sections. The number of T cells (CD2+) was determined in three randomly selected cuffs from cervical spinal cord of three individual animals in each group, and expressed as a percentage of the total number of hematoxylin-stained nuclei. This was repeated on serial sections for macrophages (ED1+), CD4+, CD8+, and B-cells. The number of iNOS and MBP+ cuffs was expressed as a percentage of the total cuff number on serial sections.
Ultrastructural Analysis
Animals were deeply anesthetized with a sublethal dose of Saggital (Rhone, Meriux, France) and prepared for electron microscopy by intracardiac perfusion of 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer containing 0.05 mol/L CaCl2. The spinal cords were removed and fixed further in perfusion buffer. Samples were then rinsed in 0.1 mol/L cacodylate buffer and postfixed in 1% osmium tetroxide for 1 hour at room temperature, followed by washes in cacodylate buffer, dehydrated through a graded series of ethanol, and embedded in epoxy resin. Semithin sections were cut and stained with 1% toluidine blue and analyzed by light microscopy. Ultra-thin sections were cut and placed on copper grids before being stained with lead citrate and uranyl acetate. Sections were then viewed under an electron microscope set at 10 kV.
Adjacent toluidine blue stained sections (n = 2) from 2 individual third relapse animals were used to count the number of dead axons in both the cervical and lumbar regions of the spinal cord. Using a graticule, a randomly selected area within the dorsal funiculi, measuring 100 μm 2 was used to count the number of dead axons within this region. Dead axons were also counted in a further two areas of the same dorsal funiculi region.
Enzyme-Linked Immunosorbent Assay (ELISA): IL-10, IL-4, Transforming Growth Factor-β1, IgG1, IgG2b, and Anti IL-12 Antibodies
The levels of Th2 cytokines IL-10, IL-4 (both purchased as kits from R&D Systems, Abingdon, Oxon, UK), and TGF-β1 (EMAX TGF-β1 ELISA kit, Promega) in the serum from treated rats were analyzed by a sandwich ELISA method (using the manufacturer’s protocol). Briefly, the capture antibody was adsorbed onto 96-well plates overnight at 4°C before washing and adding the test sera and incubating for 2 hours at room temperature. The plates were then washed and incubated with secondary antibody conjugated to biotin for a further 2 hours. After washing, peroxidase was added to develop the color and the plates were read at 450 nm within 30 minutes. IgG (horseradish peroxidase-labeled IgG1 and IgG2b; both from Zymed Labs, CA) isotypes were measured by a similar sandwich ELISA method. The plates were developed with 2,2-azino-bis(3-ethylbenzenthiazoline-6-sulfonic acid) (ABTS)/peroxidase before being read at 405 nm. A similar method was used to determine MBP-specific IgG1, IgG2b, MOG (gift from Dr. Sandra Amor), and anti IL-12 antibodies.
Protein Extraction and tPA ELISA
Protein from snap-frozen spinal cords, weighing 0.1-0.5 g wet weight, were finely chopped and re-suspended in Tris-HCl buffer [100 nmol/L Tris (pH 8.1), 1% Triton X-100, 1 mmol/L PMSF, 10 mg/ml aprotinin] at 100 mg per 1 ml of buffer. 39 Samples were homogenized on ice by sonication (25 seconds), triturated three times through 19- and 21-gauge needles and incubated on ice for 30 minutes. The tissue suspension was then spun at 20,000 × g for 45 minutes at 4°C and the supernatant collected and stored at −70°C. Protein concentrations were determined before ELISA by the Lowry method.
The levels of tPA protein (Biopool, Umea, Sweden) and tPA activity (Technoclone, Vienna, Austria) were determined by ELISA according to the manufacturers instructions. Inhibition of tPA activity with tPA-STOP (American Diagnostica, Greenwich, CT) was performed by incubating protein extracts with 2 nmol/L inhibitor at room temperature for 15 minutes.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 2.01 (GraphPad Inc, San Diego, CA). Unless otherwise stated, sample means were analyzed using one-way analysis of variance with Bonferroni post hoc testing.
Results
IL-12 Induces Repeated Paralytic Relapses after Recovery from Acute EAE
EAE developed in all rats after immunization with MBP, and all animals underwent a first paralytic relapse with clinical scores of 2.5–3 on administration of IL-12 on alternate days following recovery, as described previously. 20 Seven of seven animals injected with IL-12 on recovery from the first relapse underwent a second relapse, reaching peak clinical scores of 3 (Figure 1a) ▶ . Following recovery, administration of IL-12 to these animals induced a second (injections: days 35, 37, and 39), and a third relapse (injections: days 45, 47 and 49), with intervening recovery, reaching peak clinical scores of 2 and 1, respectively (Figure 1a) ▶ . The duration and the onset of the disease following IL-12 injection was similar to the initial IL-12 induced relapse, although the severity was reduced (Figure 1a ▶ : Table a). On recovery from the fourth disease episode, animals did not suffer a further relapse following administration of IL-12.
Figure 1.

IL-12 induces serial paralytic relapses in EAE with diminishing severity. a: IL-12 (3 μg/ml, 0.2 ml i.p.) was injected (arrows) on days 17, 19, 21, days 35, 37, and 39 for second relapse, and days 45, 47, and 49 for third relapse (indicated by arrows). Points represent the mean ± SD of clinical scores. The table represents the: I, incidence; S, severity; and D, duration of disease for each relapse. Values represent median and range. b: Chronic relapse experiment where IL-12 was injected as for the first relapse (days 21, 23, and 25) followed by allowing the animals to recover to clinical score 2 before being injected with IL-12 at a dose of 1.5 μg/ml, 0.2 ml, i.p. every other day (days 35, 37, and 39 followed by every other day until day 63).
In an attempt to induce chronicity after the acute EAE phase, animals were injected as previously to induce the first relapse, allowed to recover to a clinical score 2, whereupon IL-12 was given at half the dose (1.5 μg/rat, 0.2 ml i.p., every other day) for as long as the disease persisted. It was possible to maintain clinical signs of disease for a period of 11 days after which animals recovered and remained resistant to induction of EAE, even with further administration of IL-12 (Figure 1b ▶ and Table b).
Changes in the Composition of Perivascular Cuffs
The total number of inflammatory cuffs in sections of cervical compared with thoracic, sacral or lumbar regions of the spinal cord showed no significant differences over the time period of relapses and remission (not shown). The degree of inflammation, represented by the number of perivascular cuffs in a 1 cm portion of the cervical spinal cord, was greatest during the first relapse (median, 115) where a maximum cuff score (median, 352) was also recorded (Figure 2a) ▶ . The proportion of macrophages (median, 60%) increased in the first relapse with an associated decrease in the proportion of CD4+ T cells (median, 34%) compared with acute EAE. The greatest proportion of ED1+ macrophages were observed during the first relapse, which correlates well with the clinical severity of the disease. During subsequent relapses, the relative proportion of macrophages and CD4+ T cells within perivascular cuffs returned to those observed in the acute phase while the number of ED1+ activated microglia in the parenchyma increased significantly (P < 0.001).
Figure 2.
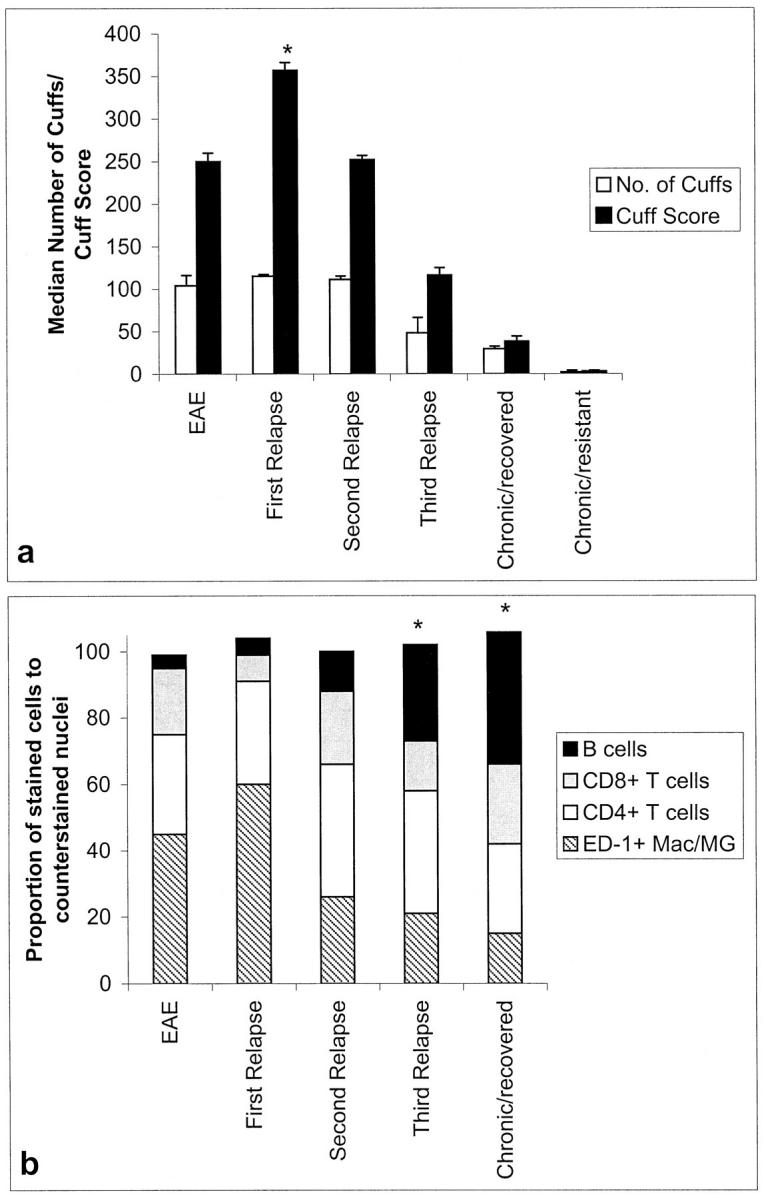
CNS histopathology in IL-12-induced relapses. Graphs show cuff numbers and score (a) and the cellular distribution of the inflammatory cuffs (b). *P < 0.001 versus EAE. Mac, macrophages; MG, microglia.
12% of the total number of cells in inflammatory cuffs of second relapse animals were identified as B cells (P < 0.01 versus acute EAE) compared to the first relapse in which only 4 to 5% was detected (Figure 2b ▶ and Figure 3 ▶ ). In subsequent relapses, the proportion of B cells in lesions increased further reaching a maximum in chronic/recovered animals (animals sacrificed immediately after the chronic bout of disease at day 46) where 40% of cells were B cells (P < 0.03 chronic/recovered versus second relapse). Six days after complete recovery from chronic disease (animals sacrificed day 52) there were no persisting lesions, even though IL-12 was continually administered throughout this period (not shown). The ratio of CD8:CD4+ T cells remained unchanged throughout the course of the relapses (not shown).
Figure 3.
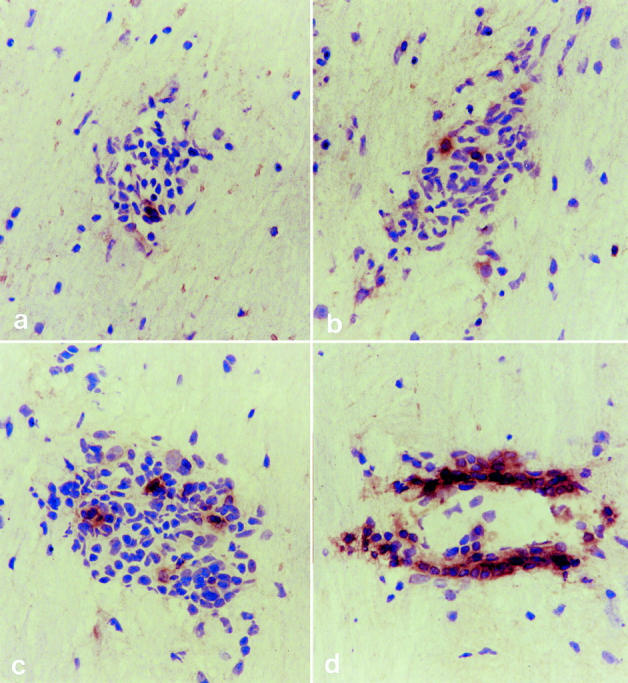
B cell localization in inflammatory cuffs in cervical regions of the spinal cord in acute EAE (a), first relapse (b), second relapse (c), and third relapse animals (d). Original magnification, ×1000.
IL-12 Did Not Induce the Th2 Cytokines IL-10 and IL-4 but Induced TGF-β1 in Serum
A significant increase in circulating TGF-β1 levels was detected in the serum of EAE animals when compared to controls (P < 0.01 for control versus EAE, unpaired, two-tailed Student’s t-test) (Figure 4) ▶ , which remained elevated during the relapses but did not reach statistical significance (analysis of variance, P > 0.05). There was no detection of the classical Th2 mediators IL-10 and IL-4 in serum samples (not shown). Additionally, only low levels of anti IL-12 antibodies were detected by ELISA, which varied over time but had no association with clinical signs of disease (not shown).
Figure 4.

Biologically active TGF-β levels increase during EAE and relapses. ***P < 0.01.
Altered Isotype Levels after IL-12-Induced Relapses
Isotype specific IgG results showed a biphasic response for IgG1 and IgG2b (Figure 5, a and b ▶ , respectively). Anti MBP-specific IgG1 levels increased during the first relapse, with a slight drop in levels during the second relapse stage. There was a significant rise in IgG1 levels in third relapse and chronic/recovered animals compared to EAE (P < 0.001), which was not observed in chronic/resistant rats. Anti MBP-specific IgG2b levels, however, increased significantly in first relapse compared to EAE animals (P < 0.05). There was a further increase in anti MBP-specific IgG2b during the second relapse compared to first relapse (P < 0.05), after which levels returned to control levels. No myelin oligodendrocyte glycoprotein (MOG)-specific antibodies could be detected (not shown).
Figure 5.

IgG1 and IgG2b isotype responses showed a general increase in levels of MBP-specific IgG1 (*P < 0.001 EAE versus first relapse, +P < 0.001 first relapse versus third relapse, #P < 0.001 first relapse versus chronic/recovered) (a) and MBP-specific IgG2b (*P < 0.05 EAE versus first relapse, #P < 0.05 first relapse versus second relapse) (b) after IL-12-induced relapses.
Axonal Death Is Observed in Third Relapse Animals
Semithin sections stained with toluidine blue showed a significant degree of axonal death, which was present throughout the dorsal funiculi regions of both the cervical and lumbar portions of the spinal cord in third relapse animals (Figure 6b) ▶ . Axons in the damaged area tended to be of smaller diameter compared to those of an identical region in control untreated animals (Figure 6a) ▶ . The number of dead axons in the cervical (mean, 154 ± 3) and the lumbar (156 ± 3) regions of the spinal cord in third relapse animals were very similar. No axonal death was observed in cervical or lumbar portions of the spinal cord from animals with earlier relapses.
Figure 6.
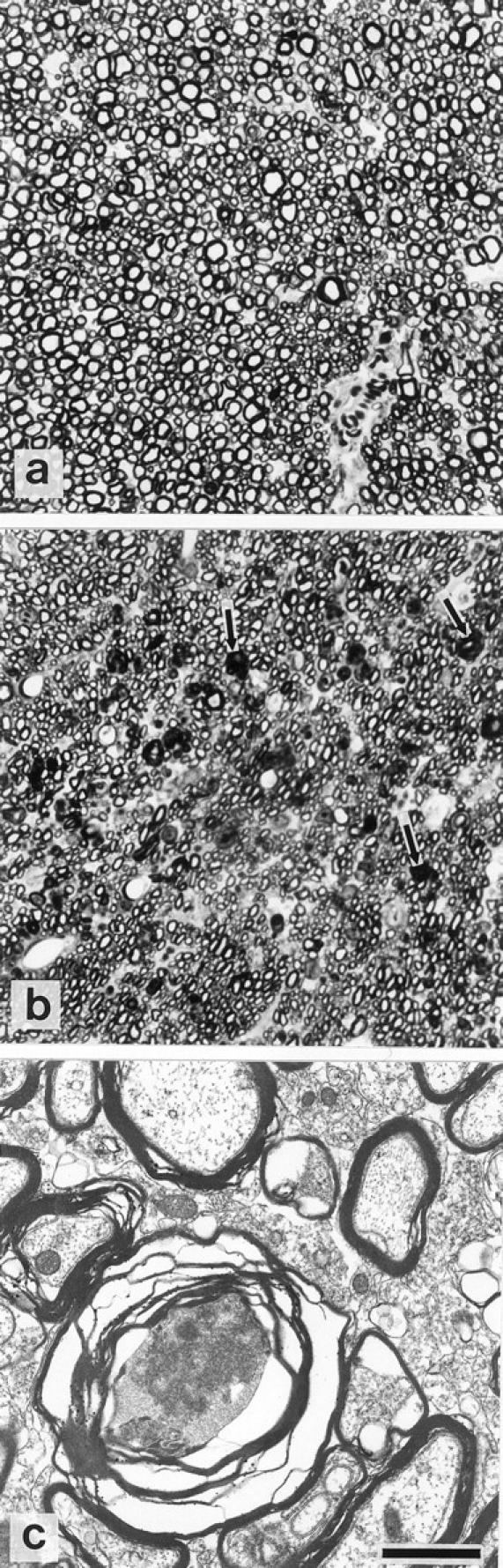
Axonal death in the dorsal funiculi region of the spinal cord during the third relapse (b: representative cervical region shown) indicated by arrows. Note the smaller diameter of axons in the damaged region (b) compared to the identical region in untreated control animals (a). c: EM showing degenerating axon surrounded by disrupted myelin. Original magnification in a and b, ×1000; bar in c = 1 μm.
Ultrastructural analysis by electron microscopy highlighted the axonal/myelin changes occurring during the death of affected axons in the spinal cord of third relapse animals (6c). However, macrophages with myelin fragments were not convincingly detected in EM sections although they were detected by immunocytochemistry.
Myelin and iNOS Positive Macrophages and Microglia Are Present in Perivascular Cuffs and Parenchyma of Spinal Cord in IL-12-Induced Second and Third Relapse
Double staining of MBP and ED-1 in serial sections showed that very little MBP was present in ED1+ macrophages in the inflammatory cuff of EAE or first relapse animals. However, numerous ED1+ cells (Figure 7a) ▶ with MBP immunostaining were observed (Figure 7b) ▶ in perivascular cuffs in second and third relapse animals together with staining of microglia in both white matter (WM) and gray matter (GM) regions (Figure 7d) ▶ of the spinal cord. Likewise, no QD9 or EP peptide positive (not shown) immunostaining was detected in acute EAE or first relapse animals (not shown), however, QD-9 and EP immunostaining in first, second and third relapse animals mirrored that of MBP immunostaining (not shown). Control untreated animals showed a normal pattern of MBP immunostaining of myelin (not shown). Although the cell numbers decreased, the proportion of cuffs positive for MBP increased significantly following relapses such that by the third relapse all cuffs were MBP positive (Figure 7f) ▶ .
Figure 7.
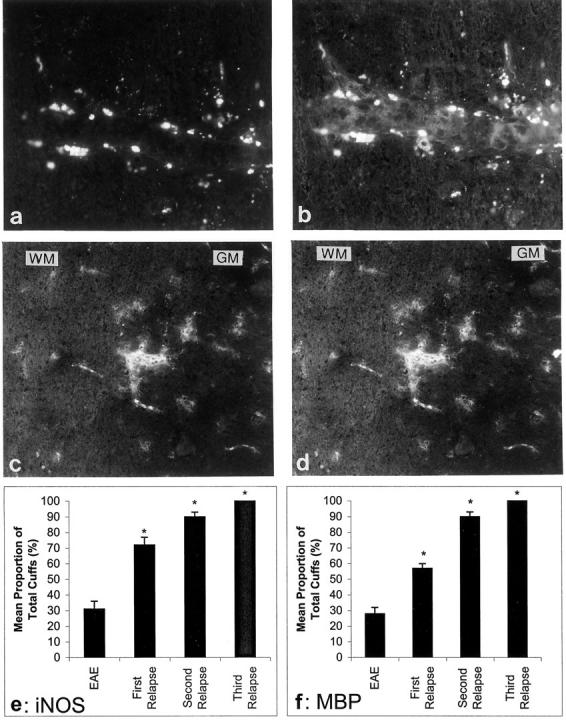
Representative MBP and iNOS immunostaining localized in macrophages in the spinal cord of Lewis rats. Double staining on serial sections with ED-1 (a) and MBP (b). c: iNOS immunostaining was closely linked to the pattern of MBP (serial sections) (d). *P < 0.001 versus EAE. Original magnifications, ×500. Graphs to show the proportion of inflammatory cuffs which were positive for iNOS (e) and MBP (f).
iNOS localization in acute EAE and first relapse animals was mainly confined to the inflammatory cuffs while the proportion of iNOS+ cuffs increased during the second and third relapses and there was a spread of iNOS+ immunostaining to individual cells in the parenchyma of both WM and GM as well as intense positive staining in inflammatory cuffs (Figure 7c) ▶ . Similar to MBP, there was an increase in the proportion of cuffs which were positive for iNOS and by the third relapse, all inflammatory cuffs were positive (Figure 7e) ▶ . Chronically treated animals showed a similar profile to third relapse animals (not shown).
Increased tPA Immunostaining, Protein, and Activity in Second and Third Relapse Animals and tPA Localization on Axons
tPA immunostaining followed a similar trend to iNOS, such that sections from third relapse animals showed extensive staining in macrophages/activated microglia in inflammatory cuffs, as well as microglial cells in GM and WM (Figure 8b) ▶ . There was a similar pattern of tPA immunostaining observed in sections from second relapse animals (not shown). tPA immunostaining was also localized on axons in both WM and GM in sections from second (not shown) and third relapses (arrows). However, immunostaining of tPA in sections of spinal cords from first relapse animals was cell-associated and mainly confined to inflammatory cuffs in WM and GM as well microglia in GM and the surrounding parenchyma without any localization of tPA on axons (Figure 8a) ▶ . Serial sections showed that immunostaining for plasminogen activator inhibitor-1 was absent during the relapses and only apparent during the chronic/recovered and chronic/resistant phases (not shown).
Figure 8.
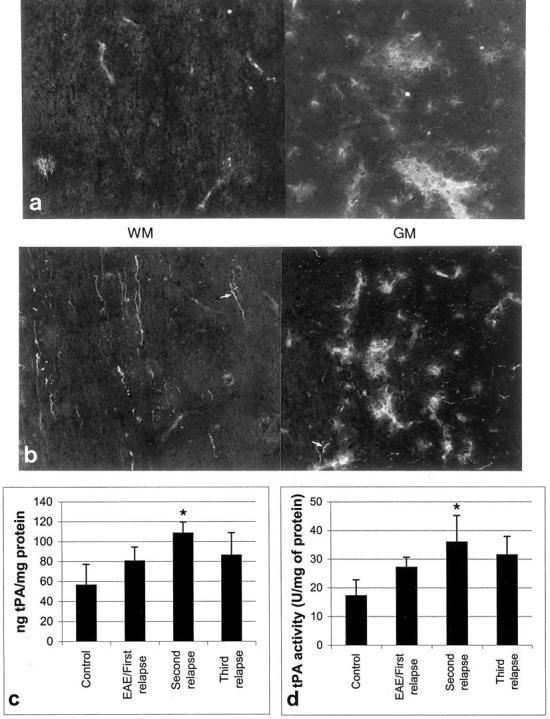
A composite photomicrograph of neighboring frames to show tPA expression in macrophages of inflammatory cuffs in spinal cord WM and GM in first relapse (a) and in axons of third relapse (b) animals. Original magnifications, ×500. Total tPA protein levels (c) as well as tPA activity (d) increased during the second relapse (*P < 0.001 versus control).
The levels of tPA and activity increased with repeated relapses so that during the second relapse the mean amount of tPA protein was 108 ng/mg of protein (Figure 8c) ▶ and the mean activity was 36 U/mg of tPA (Figure 8d) ▶ .
Discussion
This study has shown that a series of three relapses of EAE with decreasing clinical severity can be induced by IL-12 in otherwise disease-resistant Lewis rats. An increase in the number and score of inflammatory cuffs in both white and gray matter of the spinal cord was observed during the first relapse, thereafter decreasing, with the composition of the inflammatory cuff changing to a significantly increased proportion of B cells. The proportion of MBP-positive macrophages increased during the serial relapse as did the serum levels of MBP-specific IgG1 and IgG2b. Axonal death was observed in the dorsal funiculi regions of both the cervical and the lumbar portions of the spinal cord although macrophage stripping of myelin sheaths could not be convincingly detected by EM. iNOS and tPA expression was increased in macrophages in inflammatory cuffs and in microglia outside the lesions in both gray matter and white matter of the spinal cord together with tPA being localized on axons in both these regions in second and third relapse animals.
The rapid recurrence of the clinical signs of EAE following IL-12 administration implies that IL-12 reactivates residual inflammatory T cells and macrophages in the CNS initiating an encephalitogenic Th1-cell response from the pool of already sensitized MBP-specific T cells. 20 The prominence of macrophages in the spinal cord of first relapse animals is consistent with the severity of the disease and higher morbidity in animals administered IL-12. The decrease in clinical and pathological severity of EAE with subsequent relapses suggests that IL-12 effector functions may be suppressed by the presence of detectable levels of antibodies against IL-12. Although it was not determined whether the antibodies against IL-12 were neutralizing, it has been previously reported that antibodies to IL-12 can neutralize the effects of exogenously added IL-12. 19,40 Furthermore, the induction of TGF-β may have contributed to the reduction of disease severity following relapses due to the fact that TGF-β has been shown to be an immunosuppressive cytokine that inhibits IL-12-mediated responses in natural killer and T cells. 41,42 Indeed, systemic administration of TGF-β is able to prevent the development of EAE. 43,44 Another possibility is that the shift in the composition of the inflammatory cuff from a T cell to a B cell predominant population in successive relapses may induce humoral immunity with increasing B cell numbers which could represent a regulatory role. Disease symptoms of EAE are not completely resolved in a B-cell deficient mouse 45 while mice lacking B cells have sustained chronic disease with evidence of demyelination. 46
The increased proportion of B cells also coincides with increased MBP-specific IgG1 and IgG2b isotype responses in serum and MBP positive macrophages in perivascular cuffs of the spinal cords, indicative of myelin phagocytosis. During the second and third relapse microglia/macrophages in areas distant from the cuff in both white and gray matter as well as in cuffs, were positive for MBP, indicating processing of myelin without apparent disruption of the lamellar structure. Similar observations of activated microglia with internalized myelin fragments have been reported in white matter in MS, 47,48 and are considered one of the earliest detectable events in lesion development. 49 Gay et al 49 showed that microglial-bound, fixed complexes of IgG and complement with evidence of complement activation were a hallmark of the putative primary lesion. Moreover, confirmation of myelin proteolysis was to be found in the detection of macrophage associated peptide fragments. The absence of anti-MOG antibodies in serum may explain the observed limitations on demyelination, because these complement-fixing demyelinating antibodies, unlike those directed against MBP, target MOG expressed on the surface of myelin.
Dead axons were noted in sections of spinal cords during the third relapse phase. In the region of axonal death, surrounding axons were noticeably smaller suggesting some degree of stripping/degradation of the myelin sheath. This is in accordance with reports in MS where axonal damage has been shown to be ongoing during inflammation and precedes the demyelination process. 50 The axonal death observed in our experiments could be a result of damage caused by exposure to neurotoxic agents such as NO. Indeed, NO in serum has been shown to parallel the course of EAE 51 and inhibitors of the enzyme ameliorate the disease, 52 suggesting its involvement in clinical symptoms of EAE. Because iNOS and MBP were co-localized in macrophages and microglia in the third relapse, it may be that anti-MBP antibodies also contribute to axonal damage in the spinal cord that appears with repeated episodes of inflammation.
A further candidate for the observed axonal damage may be tPA due to the fact that it has been shown to activate microglia and promote neuronal degeneration. 53 Increased levels and activity of tPA have been shown to correlate with neuronal degeneration caused by the generation of excitotoxins in the hippocampus of mice. 54-56 Because there was increased tPA protein and increased activity of this tPA in second and third relapses of our experiments, it is quite possible that this may have contributed to the axonal death observed in third relapse animals. Furthermore, the increased amounts of tPA may have caused activation of resident microglia in both white and gray matter of the spinal cord, leading to increased phagocytic activity, indicated by the presence of ingested myelin in microglia from these regions. Conversely, the presence of tPA, may in fact indicate a degree of axonal protection, as it was recently reported that tPA/plasmin-mediated fibrinolysis limits axonal degeneration and demyelination after sciatic nerve injury in mice. 57
In conclusion, our results show that IL-12 can precipitate repeated relapses in otherwise resistant Lewis rats, leading to the appearance of B cells. The axonal/myelin changes seen in IL-12 induced relapses may be due to a number of factors including humoral immune antibody-mediated damage to axons, neurotoxic/excitotoxic mediators, and/or proteolytic enzymes, such as tPA, which are produced by macrophages and other inflammatory cells.
Acknowledgments
We thank Professor D.N. Landon and Mr. B. C. Young (Institute of Neurology) for the semithin sections and for performing EM. We would also like to thank Dr. P. McGeer for providing the QD-9 and EP antibodies.
Footnotes
Address reprint requests to Dr. Zubair Ahmed, Neuroinflammation Group, Miriam Marks Department of Neurochemistry, Institute of Neurology, University College London, 1 Wakefield Street, London, WC1N 1PJ, UK. E-mail: z.ahmed@ion.ucl.ac.uk.
Supported by the Brain Research Trust, United Kingdom.
References
- 1.Lassman H, Zimprich F, Rossier K, Vass K: Inflammation in the central nervous system: basic mechanisms and immunological concepts. Rev Neurol 1991, 147:763-781 [PubMed] [Google Scholar]
- 2.Ben-Nun A, Cohen I: Experimental autoimmune encephalomyelitis (EAE) mediated by T cell lines: process of selection of lines and characterization of the cells. J Immunol 1982, 129:303-308 [PubMed] [Google Scholar]
- 3.Huitinga I, Van Roojien N, de Groot CJA, Uitdehaag BMJ, Dijkstra CD: Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med 1990, 172:1025-1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powell MB, Mitchell D, Lederman J, Buchmeier J, Zamvil SS, Graham M, Ruddle NH, Steinman L: Lymphotoxin production by myelin basic protein specific T cell clones correlate with encephalitogenicity. Int Immunol 1990, 2:539-544 [DOI] [PubMed] [Google Scholar]
- 5.Sedgwick JD, McPhee JHM, Puklawec M: Isolation of encephalitogenic CD4+ T cell clones in the rat: cloning methodology and IFN-γ secretion. J Immunol Methods 1989, 143:3492-3497 [DOI] [PubMed] [Google Scholar]
- 6.Mossman TR, Coffman RL: Th1 and Th2 cells: different patterns of lymphokine secretion leads to different functional properties. Annu Rev Immunol 1989, 7:145-173 [DOI] [PubMed] [Google Scholar]
- 7.Kennedy MK, Torrance DS, Picha KS, Mohler KM: Analysis of cytokine mRNA expression in the central nervous system of mice with experimental allergic encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol 1992, 149:2496-2505 [PubMed] [Google Scholar]
- 8.Smith T, Schmied M, Hewson AK, Lassman H, Cuzner ML: Apoptosis of T cells and macrophages in the central nervous system of intact and adrenolectomized Lewis rats during experimental allergic encephalomyelitis. J Autoimmun 1996, 9:167-174 [DOI] [PubMed] [Google Scholar]
- 9.Khoury SJ, Hancock WW, Weiner HL: Oral tolerance to myelin basic protein and natural recovery from experimental allergic encephalomyelitis are associated with downregulation of inflammatory cytokines and differential upregulation of transforming growth factor beta, interleukin 4, and prostaglandin E expression in the brain. J Exp Med 1992, 176:1355-1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM: Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 1993, 260:547-549 [DOI] [PubMed] [Google Scholar]
- 11.Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, Wysocka M, Trinchieri G, Murphy KM, O’Garra A: Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol 1995, 154:5071-5079 [PubMed] [Google Scholar]
- 12.Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P: The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science 1994, 263:235-237 [DOI] [PubMed] [Google Scholar]
- 13.Heinzel FP, Schoenhaut DS, Rerko RM, Rosser LE, Gately MK: Recombinant interleukin-12 cures mice infected with Leishmania major. J Exp Med 1993, 177:1505-1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schijns VE, Haagmans BL, Horzinek MC: IL-12 stimulates an antiviral type 1 cytokine response but lacks adjuvant activity in IFN-γ-receptor-deficient mice. J Immunol 1995, 155:2525-2532 [PubMed] [Google Scholar]
- 15.Heesen C, Sieverding F, Schoser BG, Hadji B, Kunze K: Interleukin-12 is detectable in sera of patients with multiple sclerosis-association with chronic progressive disease course? Eur J Neurol 1999, 6:591-596 [DOI] [PubMed] [Google Scholar]
- 16.Comabella M, Balashov K, Issazadeh S, Smith D, Weiner HL, Khoury SJ: Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J Clin Invest 1998, 102:671-678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balashov KE, Smith DR, Khoury SJ, Hafler DA, Weiner HL: Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci USA 1997, 94:599-603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Boxel-Dezaire AHH, Hoff SCJ, van Ooosten BW, Verweij CL, Drager AM, Ader J, van Houwelingen JC, Barkhof F, Polman CH, Nagelkerken L: Decreased interleukin-10 and increased interleukin-12 p40 mRNA are associated with disease activity and characterize different stages in multiple sclerosis. Ann Neurol 1999, 45:695-703 [DOI] [PubMed] [Google Scholar]
- 19.Leonard JP, Waldburger KE, Goldman SJ: Prevention of experimental allergic encephalomyelitis by antibodies against IL-12. J Exp Med 1995, 181:381-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith T, Hewson AK, Kingsley CI, Leonard JP, Cuzner ML: Interleukin-12 induces relapse in experimental allergic encephalomyelitis in the Lewis rat. Am J Pathol 1997, 150:1909-1917 [PMC free article] [PubMed] [Google Scholar]
- 21.Cuzner ML, Opdenakker G: Plasminogen activators and matrix metalloproteases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol 1999, 94:1-14 [DOI] [PubMed] [Google Scholar]
- 22.Pender MP: The pathophysiology of myelin basic protein-induced acute experimental allergic encephalomyelitis in the Lewis rat. J Neurol Sci 1988, 86:277-289 [DOI] [PubMed] [Google Scholar]
- 23.Glynn P, Linnington C: Cellular and molecular mechanisms of autoimmune demyelination in the central nervous system. Crit Rev Neurobiol 1989, 4:367-385 [PubMed] [Google Scholar]
- 24.Prineas JW: The neuropathology of multiple sclerosis. Koetsier JC eds. Handbook of Clinical Neurology. 1985, :pp 213-257 Elsevier Press, The Netherlands [Google Scholar]
- 25.Lassmann H, Suchanek G, Kitz K, Stemberger H, Schwerer B, Bernheimer H: Antibodies in the pathogenesis of demyelination in chronic relapsing EAE (cr-EAE). Prog Clin Biol Res 1984, 146:165-170 [PubMed] [Google Scholar]
- 26.Baker D, O’Neill JK, Gschmeissner SE, Wilcox CE, Butter C, Turk JL: Induction of chronic relapsing experimental allergic encephalomyelitis in Biozzi mice. J Neuroimmunol 1990, 28:261-270 [DOI] [PubMed] [Google Scholar]
- 27.Gualandris A, Jones TE, Strickland S, Tsirka SE: Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurosci 1996, 16:2220-2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida S, Shiosaka S: Plasticity-related serine proteases in the brain (review). Int J Mol Med 1999, 3:405-409 [DOI] [PubMed] [Google Scholar]
- 29.Tsirka SE, Gualandris A, Amaral DG, Strickland S: Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature 1995, 377:340-344 [DOI] [PubMed] [Google Scholar]
- 30.Tsirka SE, Rogove AD, Strickland S: Neuronal cell death and tPA. Nature 1996, 384:123-124 [DOI] [PubMed] [Google Scholar]
- 31.Cuzner ML, Gveric D, Strand C, Loughlin AJ, Paemen L, Opdenakker G, Newcombe J: The expression of tissue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol 1996, 55:1194-1204 [DOI] [PubMed] [Google Scholar]
- 32.Bo L, Dawson TM, Wesselingh S: Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann Neurol 1994, 36:778-786 [DOI] [PubMed] [Google Scholar]
- 33.Boullerne A, Petry K, Meynard M, Geffard M: Indirect evidence for nitric oxide involvement in multiple sclerosis by characterization of circulating antibodies directed against conjugated S-nitrosocysteine. J Neuroimmunol 1995, 60:117-124 [DOI] [PubMed] [Google Scholar]
- 34.Smith KJ, Kapoor R, Felts PA: Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol 1999, 9:69-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH: Mechanism of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci 1993, 13:2651-2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson VL, Brahmbhatt HP, Mong JA, Dawson TM: Expression of inducible nitric oxide synthase causes delayed neurotoxicity in primary mixed neuronal-glial cortical cultures. Neuropharmacology 1994, 33:1425-1430 [DOI] [PubMed] [Google Scholar]
- 37.Tamatani M, Ogawa S, Niitsu Y, Toyhama M: Involvement of Bcl-2 family and caspase-3-like protease in NO-mediated neuronal apoptosis. J Neurochem 1998, 71:1588-1596 [DOI] [PubMed] [Google Scholar]
- 38.Matsuo A, Akiguchi I, Lee GC, McGeer EG, McGeer PL, Kimuru J: Myelin degradation in multiple system atrophy detected by unique antibodies. Am J Pathol 1988, 153:735-744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Padro T, Quax PA, van den Hoogen CM, Roholl P, Verheijen JH, Ameis JJ: Tissue-type plasminogen-activator and its inhibitor in rat aorta—effect of endotoxin. Arterioscler Thromb 1994, 14:1459-1465 [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez-Scarano F, Baltuch G: Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci 1999, 22:219-240 [DOI] [PubMed] [Google Scholar]
- 41.Bright J, Sriram S: TGF-β inhibits IL-12-induced activation of Jak-STAT pathway in T lymphocytes. J Immunol 1998, 161:1772-1777 [PubMed] [Google Scholar]
- 42.Schmitt E, Hoehn P, Huels C, Goedert S, Palm N, Rude E, Germann T: T helper type 1 development in naïve CD4+ T cells requires the coordinate action of interleukin-12 and IFN-γ and is inhibited by TGF-β. Eur J Immunol 1994, 24:793-798 [DOI] [PubMed] [Google Scholar]
- 43.Johns L, Flanders KC, Ranges GE, Sriram S: Successful treatment of experimental allergic encephalomyelitis with TGF-β1. J Immunol 1991, 147:1792-1796 [PubMed] [Google Scholar]
- 44.Racke MS, Sriram S, Carlino J, Cannella B, Raine C, McFarlin D: Long-term treatment of chronic relapsing experimental allergic encephalomyelitis by TGF-β2. J Neuroimmunol 1993, 46:175-183 [DOI] [PubMed] [Google Scholar]
- 45.Wolf SD, Dittel BN, Hardardottir F, Janeway CA, Jr: Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med 1996, 184:2271-2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hjelmstrom P, Juedes AE, Fjell J, Ruddle NH: Cutting edge: B cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J Immunol 1998, 161:4480-4483 [PubMed] [Google Scholar]
- 47.Li H, Newcombe J, Cuzner ML: Characterisation and distribution of phagocytic macrophages in MS plaques. Neuropathol Appl Neurobiol 1993, 19:214-223 [DOI] [PubMed] [Google Scholar]
- 48.Cuzner ML, Smith T: Immune responses in the central nervous system in inflammatory demyelinating diseases. Rothwell NJ eds. Immune Responses in the Nervous System. 1995, :pp 117-142 BIOS Scientific Publishers Ltd., Oxford [Google Scholar]
- 49.Gay FW, Drye TJ, Dick GWA, Esiri MM: The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis: identification and characterization of the primary demyelinating lesion. Brain 1997, 120:1461-1483 [DOI] [PubMed] [Google Scholar]
- 50.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L: Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998, 338:278-285 [DOI] [PubMed] [Google Scholar]
- 51.Mitrovic B, Ignarro LJ, Montestruque S, Smoll A, Merrill JE: Nitric oxide as a potential pathological mechanism in demyelination: its different effects on primary glial cells in vitro. Neuroscience 1994, 61:575-585 [DOI] [PubMed] [Google Scholar]
- 52.Okuda Y, Nakatsuji Y, Fujimura H, Esumi H, Ogura T, Yanagihara T, Sakoda S: Expression of the inducible isoform of nitric oxide synthase in the central nervous system of mice correlates with the severity of actively induced experimental allergic encephalomyelitis. J Neuroimmunol 1995, 62:103-112 [DOI] [PubMed] [Google Scholar]
- 53.Rogove AD, Siao C, Keyt B, Strickland S, Tsirke SE: Activation of microglia reveals a non-proteolytic cytokine function for tissue plasminogen activator in the central nervous system. J Cell Sci 1999, 112:4007-4016 [DOI] [PubMed] [Google Scholar]
- 54.Nagai N, Urano T, Endo A, Takahashi H, Takada Y, Takada A: Neuronal degeneration and a decrease in laminin-like immunoreactivity is associated with elevated tissue-type plasminogen activator in the rat hippocampus after kainic acid injection. Neurosci Res 1999, 33:147-154 [DOI] [PubMed] [Google Scholar]
- 55.Rogove AD, Tsirke SE: Neurotoxic response by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol 1998, 8:19-25 [DOI] [PubMed] [Google Scholar]
- 56.Tsirke SE: Clinical implications of the involvement of tPA in neuronal cell death. J Mol Med 1997, 75:341-347 [DOI] [PubMed] [Google Scholar]
- 57.Akassoglou K, Kombrinck KW, Degen JL, Strickland S: Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol 2000, 149:1157-1166 [DOI] [PMC free article] [PubMed] [Google Scholar]