

Abstract
To learn more about the process of amyloid β-protein (Aβ) deposition in the brain, human prefrontal cortices were fractionated by sucrose density gradient centrifugation, and the Aβ content in each fraction was quantified by a two-site enzyme-linked immunosorbent assay. The fractionation protocol revealed two pools of insoluble Aβ. One corresponded to a low-density membrane domain; the other was primarily composed of extracellular Aβ deposits in those cases in which Aβ accumulated to significant levels. Aβ42 levels in the low-density membrane domain were proportional to the extent of total Aβ42 accumulation, which is known to correlate well with overall amyloid burden. In PDAPP mice that form senile plaques and accumulate Aβ in a similar manner to aging humans, Aβ42 accumulation in the low-density membrane domain also increased as Aβ deposition progressed with aging. These observations indicate that the Aβ42 associated with low-density membrane domains is tightly coupled with the process of extracellular Aβ deposition.
Although senile plaques are one of the hallmarks of human brains affected by Alzheimer’s disease (AD), they are also found in the brains of nondemented aged patients. The senile plaque is composed of fibrillar or nonfibrillar aggregates of a small protein (Mr ∼ 4000), called amyloid β-protein (Aβ). This small protein is produced through sequential cleavage from a large transmembrane protein, β-amyloid protein precursor (APP). Two proteases are involved in the generation of Aβ from APP; one is β-secretase, which cleaves the amino terminus of Aβ, and the other is γ-secretase, which cleaves the carboxyl terminus of Aβ. 1 The former has recently been identified as a transmembrane aspartyl protease (β-site APP-cleaving enzyme), 2 and the latter is presumed to be presenilin-1 and presenilin-2. 3,4 As a result of cleavage, two major protein species are formed: Aβ40, ending at Val40, and Aβ42 ending at Ala42.
Although Aβ40 is the major species normally secreted from cells, Aβ42 is the predominant species found in senile plaques, 5 and is the initial species to be deposited in the brain. 5,6 Now that the presenilins are likely to be the long-sought γ-secretase, 3,4 one research direction is straightforward: to elucidate the complicated interplay among enzyme, substrate, and lipid (membrane) environments. The central questions are: where, within the cell, is Aβ produced and how is the production of Aβ, in particular Aβ42, regulated? Any of the pathogenic mutations of presenilins and APP lead to increased production or to an increased proportion of Aβ42. 7 This enzyme-substrate (APP-presenilin) relationship may be modified by a number of interacting proteins, especially presenilin- and APP-binding proteins, and by altered lipid composition of the membrane, especially the contents of cholesterol and sphingomyelin. The chromosome 10 locus, which has just been identified as a major susceptibility gene for AD, 8-10 may produce just such an interacting protein and thus increase the amounts of Aβ.
However, for most AD patients, the plasma Aβ42 levels of whom are not necessarily elevated, causes other than increased Aβ42 production should be sought. One possible cause would be intracellular Aβ trafficking, the abnormalities of which may eventually cause extracellular Aβ deposition. From our previous study, we found that, even in young brains, a substantial fraction of Aβ is insoluble, and that this particular species is a normal metabolite and seems to accumulate with age. 11,12 More than half of the Triton-insoluble Aβ is located in the low-density membrane (LDM) domain of SH-SY5Y human neuroblastoma cells. 13 This domain is rich in glycosphingolipids (especially GM1 ganglioside and sphingomyelin) and cholesterol, and seems to be involved in vesicular trafficking and signal transduction. 14 Furthermore, GM1 ganglioside-bound Aβ is exclusively detected in brains showing diffuse plaques, the earliest stage of senile plaques. 15 Thus, these findings suggest that Aβ deposition is closely related to aberrant trafficking of this specific membrane domain. We followed this line of investigation using sucrose density gradient fractionation to examine many brain specimens from nondemented patients, and found that the extent of Aβ accumulation in the LDM domain is indeed proportional to the extent of Aβ deposition in the extracellular space.
Materials and Methods
Study Participants and Tissue Preparation
The present study was based in part on 20 autopsy cases (16 men and 4 women) from the Gunma Cancer Center (Ohta, Gunma, Japan). All of the patients had malignant neoplasms. The ages at death ranged from 50 to 79 years (seven at 50 to 59 years of age, seven at 60 to 69 years of age, and six at 70 to 79 years of age; postmortem delay, 1 to 13 hours). The other source of eight autopsy cases (six men and two women) was the Tokyo Medical Examiner’s Office (Otsuka, Tokyo, Japan), as described previously. 11,12 Their ages at death ranged from 22 to 48 years (two at 20 to 29 years of age, two at 30 to 39 years of age, and four at 40 to 49 years of age; postmortem delay, 2 to 24 hours). Cases of AD and dementia from other causes were excluded from this series of patients, based on history, medical chart, and neuropathological findings. Those AD cases that were excluded were diagnosed based on both clinical and neuropathological criteria; all cases met the A2 criteria as defined by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association, 16 and were classified type C as defined by the Consortium to Establish a Registry for Alzheimer’s Disease. 17
Cortical blocks were obtained from the prefrontal cortex (Brodmann areas 9 to 11), and stored at −80°C until use. The blocks from adjacent sites and/or from the same locations on the contralateral side were fixed in 10% buffered formalin and processed for histological and immunocytochemical examinations.
The AD brains examined here were kindly provided by Drs. D. J. Selkoe (Harvard Medical School) and C. L. Masters (University of Melbourne). Brains from heterozygous PDAPP transgenic mice, aged 1.6 to 12.3 months, were snap-frozen in 2-methylbutane and stored at −80°C until use. Normal rodent brains were obtained from C57BL/6J mice or Wistar rats at 2 months of age. Rat brains were used freshly or kept at room temperature for 0, 12, or 24 hours before freezing at −80°C.
Tissue Extraction
Each of the samples was homogenized with a motor-driven Teflon/glass homogenizer in four volumes of Tris-saline [TS: 50 mmol/L Tris-HCl (pH 7.6), 0.15 mol/L NaCl] containing a cocktail of protease inhibitors. Each homogenate was then centrifuged at 540,000 × g for 20 minutes in a TLX centrifuge (Beckman, Palo Alto, CA). The resulting pellet, after being washed once more with TS, was further extracted with 6 mol/L of guanidine-HCl in 50 mmol/L of Tris-HCl (pH 7.6). The homogenate was centrifuged at 265,000 × g for 20 minutes. The supernatant was diluted to 0.5 mol/L guanidine-HCl, and subjected to enzyme-linked immunosorbent assay (ELISA) for TS-insoluble Aβ40 and Aβ42, as described previously. 11,12
Isolation of Detergent-Insoluble LDM Domains
LDM fractions were obtained according to an established protocol with minor modifications. 13 A cortical block from human prefrontal cortex (200 mg) or cerebral tissue from mouse or rat (100 mg) was homogenized in 2 ml of MES-buffered saline (25 mmol/L MES, pH 6.5, 150 mmol/L NaCl) containing 1% Triton X-100, 1 mmol/L phenylmethyl sulfonyl fluoride, 10 μg/ml leupeptin, 1 μg/ml pepstatin, and 10 μg/ml aprotinin. In some cases, appropriate amounts of synthetic Aβ40 or Aβ42 dissolved in dimethyl sulfoxide was added to the buffer just before homogenization. The homogenate was adjusted to 40% sucrose by adding an equal volume of 80% sucrose in MES-buffered saline, placed at the bottom of an ultracentrifuge tube, and overlaid with 4 ml of 35% sucrose and 4 ml of 5% sucrose in MES-buffered saline without Triton X-100. The discontinuous gradient was centrifuged at 39,000 rpm for 20 hours in an SW 41 rotor (Beckman) at 4°C. An interface at 5/35% sucrose (fraction 2) and each of the layers composed of 5, 35, and 40% sucrose (fractions 1, 3, and 4, respectively) were collected, diluted threefold with MES-buffered saline, and centrifuged. The resultant pellets and the pellet (fraction 5) derived from an original sucrose gradient centrifugation were extracted with 6 mol/L guanidine-HCl and subjected to ELISA.
Separation of the LDM fraction from myelin was performed as described previously. 18 Each mouse brain tissue sample (∼90 mg) was homogenized in 250 mmol/L sucrose in 3 mmol/L imidazol, pH 7.4, with a Dounce homogenizer, and the homogenate was centrifuged at 1000 × g for 10 minutes. The postnuclear supernatant was adjusted to 40.6% sucrose, 3 mmol/L imidazol, pH 7.4, and placed at the bottom of a tube. This was overlaid sequentially with 35 and 25% sucrose in 3 mmol/L imidazol, pH 7.4, and the homogenization buffer. The gradient was centrifuged at 37,000 rpm for 60 minutes on an SW 50.1 rotor (Beckman) at 4°C. The three interfaces as well as all of the layers were collected from the top, and the suspensions were centrifuged after dilution with TS. The resultant pellets and the pellet derived from an original sucrose gradient centrifugation were subjected to ELISA, as described above.
Antibodies
The antibodies used for ELISA were BAN50 (raised against Aβ1-16; the epitope is located in Aβ1-10), BNT77 (raised against Aβ11-28; the epitope is located in Aβ11-16), BA27 (raised against Aβ1-40; specific for Aβ40), and BC05 (raised against Aβ35-43; specific for Aβ42). The specificities of these antibodies were described in detail previously. 19 Antibodies 4G8 (specific for Aβ17-24) and 6E10 (raised against Aβ1-17) were obtained from Senetek PLC (Napa, CA). Polyclonal antibodies against APP were raised against the synthetic peptides, APP666-695 (cytoplasmic domain). Other antibodies used in this study were those to tau, HT7, and AT8 (Innogenetics, Zwijndrecht, Belgium); flotillin and calnexin (Transduction Laboratories, Lexington, KY); BiP (Grp78; StressGen, Victoria, British Columbia, Canada); TGN46 (a gift of Dr. Vas Ponnambalam, University of Dundee, Dundee, Scotland); human lysosome-associated membrane protein 2 (Developmental Studies Hybridoma Bank, Iowa City, IA); myelin basic protein (MBP; Biomeda, Foster City, CA, and Oncogene, Cambridge, MA); and myelin proteolipid protein (Cosmo Bio, Tokyo, Japan).
ELISA
The two-site ELISA for Aβ quantification consisted of a combination of three monoclonal antibodies, BNT77 or BAN50, BA27, and BC05. Antibody BNT77 or BAN50 was coated as a capture antibody on a multiwell plate (Immunoplate I; Nunc, Roskilde, Denmark), and BA27 or BC05 was used as a detection antibody after conjugation with horseradish peroxidase.
Because BC05 weakly cross-reacts with APP, 13 the TS-insoluble Aβ42 levels, when less than ∼5 pmol/g, must be corrected for their true levels. 12
Western Blotting
The proteins were separated by using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidenedifluoride membrane (Immobilon; Nihon Millipore Ltd., Yonezawa, Japan), followed by labeling with various antibodies. For the detection of Aβ, the pellet from fraction 2 was delipidated with chloroform/methanol as described previously. 12 The residue was extracted with formic acid, and the extract was cleared by brief centrifugation. An aliquot of the supernatant was dried with a Speed Vac (Savant Instruments, Farmingdale, NY) and solubilized with Laemmli’s sample buffer containing 4 mol/L urea. These samples were run on a 16.5% Tris/tricine gel, and separated proteins were transferred onto a nitrocellulose membrane (pore size, 0.22 μm; Nihon Millipore Ltd., Yonezawa, Japan), which was dipped in boiled phosphate-buffered saline to enhance the sensitivity. The bound antibodies were detected by enhanced chemiluminescence (Amersham Pharmacia, Buckingham, UK).
Other Methods
Immunocytochemical examinations for senile plaques and neurofibrillary tangles were performed using 4G8 or polyclonal antibodies to Aβ1-28, and HT7 or AT8, respectively. The density of senile plaques was assessed semiquantitatively as follows: 20 −, none; +/−, only one or two focal areas of an entire tissue section showing few senile plaques (focal presence); +, senile plaques in several areas, with the mean density of <1 per mm2; ++, senile plaques in limited cortical layers with a density between 1 and 10 per mm2; +++, senile plaques in most cortical layers with a density being >10 per mm2. The density of neurofibrillary tangles was also assessed semiquantitatively as follows: −, none; +/−, less than one per entire section; +, a few per entire section; ++, less than one per mm2; +++, more than one per mm2. The apoE genotype was determined by polymerase chain reaction as described previously. 11
Results
The Presence of Aβ40 and Aβ42 in the Detergent-Insoluble LDM Fraction from Normal Human Brains
The distinct membrane domains are easily isolated by their density in sucrose layers in the presence of Triton X-100. In SY5Y cells, approximately one half of the Triton-insoluble Aβ40 and Aβ42 is associated with LDM domains. 13 To learn more about the origin of brain insoluble Aβ, human brain homogenates were fractionated by sucrose gradient centrifugation. Resident proteins of subcellular organelles were localized among the fractions with several specific markers (Figure 1) ▶ . The LDM domain was recovered in fraction 2, into which flotillin was exclusively fractionated. However, fraction 2 was found to be significantly contaminated with myelin, as shown by the presence of a dense creamy layer and immunoreactivity with MBP (Figure 1) ▶ . Endoplasmic reticulum as represented by BiP (GRP78) and calnexin, Golgi complex by TGN46, and lysosomes by lysosome-associated membrane protein 2 were fractionated mainly in fraction 4 (Figure 1) ▶ . Regarding plasma membrane markers, Na/K ATPase was recovered in fractions 2 and 4, whereas integrin β was only found in fraction 4 (data not shown). With human brain specimens, a substantial amount of APP was fractionated into the pellet (Figure 1 ▶ , fraction 5) that contrasted with that found for mouse APP. We do not know the exact reason, but this Triton insolubility may be caused during the postmortem period. It should be noted that there were very low levels of APP in the LDM fraction (Figure 1 ▶ , fraction 2).
Figure 1.
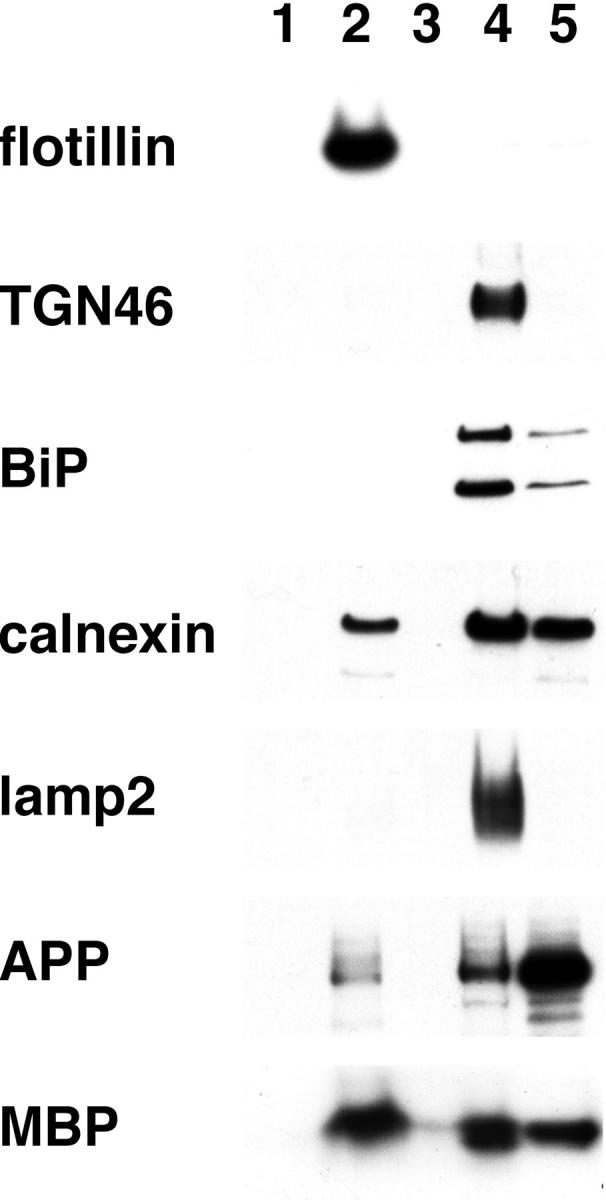
Localization of subcellular organelle markers after fractionation by sucrose density gradient centrifugation of human brain homogenates. Flotillin is a marker for LDM domain, TGN46 for Golgi complex, BiP (lower band) and calnexin for endoplasmic reticulum, lyosome-associated membrane protein 2 for lysosomes, and MBP for myelin. Equal volumes of aliquots from each fraction (Fr 2:Fr 4:Fr 5 = 1:4:1 in volume) were subjected to Western blotting using the specific antibodies. It should be noted that APP is the most abundant in fraction 4, if relative volume is taken into account.
In normal cases, defined as having TS-insoluble Aβ42 levels <5 pmol/g, 12 the amounts of Aβ40 and Aβ42 in the LDM fraction were 46 to 77% (mean, 58%) and 25 to 62% (mean, 40%) of the total Triton-insoluble Aβ40 and Aβ42 (sum of the insoluble Aβ in each fraction), respectively. Thus, more than one half of the total Triton-insoluble Aβ40 and more than one third of the total Triton-insoluble Aβ42 were located in the LDM fraction (Figure 2) ▶ . In each LDM fraction from normal cases, the level of Aβ40 was always higher (usually more than twofold) than that of Aβ42 except one (case 9) (Figure 2) ▶ . The levels of Aβ40 in fraction 5 were lower than those in fraction 2. Seemingly, those of Aβ42 in fraction 5 were similar to or slightly higher than those in fraction 2. However, their levels in fraction 5 should be considered overestimates, owning to the presence of a large amount of APP (Figure 2) ▶ . 12,13
Figure 2.

Quantification of Aβ40 and Aβ42 in each sucrose density gradient fraction from the autopsied cases. After sucrose density gradient centrifugation, each fraction was diluted and again spun down. Each resulting pellet was extracted with 6 mol/L of guanidine hydrochloride, and the extract, after being diluted, was subjected to ELISA. Cases are placed in an order of increasing levels of TS-insoluble Aβ42 (see Table 1 ▶ for actual ELISA levels). Normal cases, defined as having TS-insoluble Aβ42 levels <5 pmol/g, are indicated by an asterisk. Open and filled bars represent the levels of Aβ40 and Aβ42, respectively.
The Extent of Aβ Deposition in Human Brains Correlates Well with the Levels of Aβ in the LDM Fraction
During the process of Aβ accumulation (TS-insoluble Aβ42 >5 pmol/g), 12 the levels of Aβ42 associated with the LDM fraction seemed to increase preferentially: Aβ42 levels become higher than Aβ40 levels in the LDM fraction (Figure 2) ▶ . The levels of Aβ42 in fraction 5 increased in a concomitant manner, and were much higher than those of Aβ40 in the same fraction (Figure 2) ▶ . In these cases, presumably, fraction 5 consists of a small amount of intracellular Triton-insoluble Aβ, 12 and a larger amount of extracellularly deposited Aβ. An increase in the levels of Aβ40 in LDM fraction appeared to start only after significant accumulation of Aβ42. The same tendency was again observed in fraction 5. Thus, increases in the Aβ42 level in the LDM fraction and fraction 5 predominated over those in the Aβ40 levels in the corresponding fractions.
We examined the relationship between the levels of Aβ40 or Aβ42 in the LDM fraction (Figure 3, A and B ▶ ; x axis) and the TS-insoluble Aβ42 levels that represent Aβ deposits (Figure 3, A and B ▶ ; y axis). The Aβ42 levels in the LDM fraction stayed less than ∼0.5 pmol/g, when TS-insoluble Aβ42 levels were less than ∼5 pmol/g and thereafter the former rises in proportion to the latter. Similarly, the levels of Aβ42 in fraction 5 rose as the levels of TS-insoluble Aβ42 increased (data not shown). Regarding Aβ40, its levels in both LDM fraction and fraction 5 gradually increased with the levels of TS-insoluble Aβ42 (Figure 3A ▶ , data not shown). The slope for the rate of Aβ40 increase was much smaller than that for the Aβ42 increase.
Figure 3.

Relationship between Aβ40 (A) or Aβ42 levels (B) in the LDM domain (y axis) and TS-insoluble Aβ42 levels (x axis). Open and filled circles represent the levels of Aβ40 and Aβ42, respectively.
As expected, when the Aβ40 and Aβ42 levels in LDM fraction were plotted against Aβ40 and Aβ42 levels in fraction 5, respectively, high correlations were found again (data not shown). Five AD brains were similarly examined for the levels of Aβ40 and Aβ42 in both fractions. All AD brains exhibited high Aβ42 levels in both fractions, but these values overlapped with those obtained in nondemented patients (data not shown). In contrast, the Aβ40 levels in LDM fraction and fraction 5 were far greater than those for nondemented patients (data not shown).
Table 1 ▶ shows the relationship between biochemical parameters and immunocytochemical results. When the levels of TS-insoluble Aβ42 were <5 pmol/g (Table 1) ▶ , none or only negligible levels of senile plaques were observed. When Aβ42 levels were more than ∼100 pmol/g, Aβ42-positive plaques were constantly observed except for case 30. This case was unusual in that, despite a large accumulation of Aβ42 quantified by ELISA, there were no senile plaques (Figure 2) ▶ .
Table 1.
Summary of Cases Examined
| ID | Age (years) | Gender | Apo E | Senile plaques | CAA | NFT | TS-insoluble Aβ40/Aβ42 (pmol/g) | LDM-Aβ40/Aβ42 (pmol/g) |
|---|---|---|---|---|---|---|---|---|
| 95848* | 48 | M | 3 /3 | − | − | − | 1.74 /0.28 | 0.80 /0.32 |
| 23* | 53 | M | 3 /4 | − | − | − | 1.44 /0.38 | 0.41 /0.23 |
| 97971* | 44 | M | 3 /3 | n.d. | n.d. | n.d. | 2.25 /0.54 | 0.67 /0.12 |
| 95348* | 40 | M | 3 /3 | n.d. | n.d. | n.d. | 1.30 /0.56 | 0.62 /0.19 |
| 90235* | 22 | M | 3 /4 | − | − | − | 2.90 /0.60 | 0.91 /0.24 |
| 99579* | 37 | M | 3 /3 | n.d. | n.d. | n.d. | 3.00 /1.14 | 0.33 /0.17 |
| 96393* | 28 | F | 3 /3 | − | − | − | 3.34 /1.14 | 0.44 /0.06 |
| 96078* | 33 | F | 3 /3 | − | − | +/− | 3.60 /1.14 | 0.56 /0.19 |
| 51* | 63 | F | 3 /3 | − | − | − | 4.18 /1.40 | 0.94 /0.43 |
| 28* | 68 | M | 3 /3 | − | − | − | 2.88 /2.40 | 0.48 /0.29 |
| 41* | 57 | M | 3 /3 | +/− | − | − | 2.40 /3.24 | 1.19 /0.11 |
| 9* | 59 | M | 3 /3 | +/− | − | − | 1.92 /4.20 | 0.54 /0.64 |
| 27 | 65 | M | 3 /4 | − | − | − | 3.03 /10.7 | 0.53 /0.53 |
| 45 | 69 | M | 3 /4 | − | + | − | 5.25 /20.0 | 0.98 /1.31 |
| 16 | 57 | F | 3 /3 | − | + | − | 5.30 /47.2 | 0.62 /0.57 |
| 33 | 59 | M | 3 /4 | − | − | − | 4.18 /53.6 | 1.23 /7.33 |
| 47 | 64 | M | 3 /4 | − | − | − | 8.26 /67.5 | 0.99 /0.45 |
| 4 | 79 | F | 3 /4 | + | − | − | 14.9 /95.3 | 2.94 /6.86 |
| 96249 | 46 | M | 3 /4 | n.d. | n.d. | n.d. | 6.20 /113.1 | 1.73 /8.92 |
| 8 | 60 | M | 3 /3 | + | − | − | 12.7 /211.1 | 2.39 /80.8 |
| 7 | 68 | M | 3 /4 | ++ | − | − | 19.5 /211.4 | 1.02 /4.36 |
| 30 | 50 | M | 4 /4 | − | + | − | 17.7 /376.5 | 1.27 /5.63 |
| 43 | 79 | M | 3 /4 | ++ | − | − | 19.1 /658.0 | 1.30 /196.5 |
| 11 | 58 | M | 3 /3 | ++ | − | − | 11.7 /1300.6 | 1.26 /232.3 |
| 15 | 71 | M | 3 /4 | +++ | − | + | 682.7 /4862.2 | 8.46 /1197.7 |
| 55 | 74 | M | 3 /4 | +++ | + | + | 26.1 /5152.0 | 3.60 /849.7 |
| 34 | 79 | M | 3 /3 | ++ | − | − | 19.7 /6379.6 | 1.81 /1333.1 |
| 19 | 75 | F | 3 /3 | +++ | − | ++ | 25.3 /8903.5 | 5.27 /462.0 |
CAA, cerebral amyloid angiopathy; NFT, neurofibrillary tangle; n.d., not determined.
Cases 15, 55, and 19, in which NFTs are observed, may fall into Braak stages III, III, and IV by AT8 immunostaining, respectively.
*Normal cases, defined as having TS-insoluble Aβ42 levels below 5 pmol/g.
PDAPP Mice Show Increasing Aβ42 Accumulation in LDM Fractions as Aβ Deposition Progresses During Aging
Posthumous use of human materials always causes problems for analysis; there may be some significant postmortem alterations, and compartmentalization of Aβ may be altered. Thus, we examined PDAPP mice overexpressing APPV717F (∼10-fold endogenous mouse APP), 21 which are known to form senile plaques and accumulate Aβ in a manner similar to that in humans. 22 In PDAPP mice, the levels of Aβ42 dramatically increase in the hippocampus and cortex after 8 to 10 months of age. 22 The structural alterations surrounding mature plaques are very similar to those found in AD brains; apparently degenerating neuronal processes, reactive astrocytes, and activated microglia are involved in the formation of the lesions. 23
We used the above protocol to fractionate the mouse brain homogenates. Up to the age of 3.5 months, approximately one half of the total Triton-insoluble Aβ40 and one third of Aβ42 resided in the LDM fraction (Figure 4) ▶ . Once Aβ deposition progresses, as reflected by the levels of Aβ42 in fraction 5, the levels of Aβ42 in LDM fraction preferentially increase. The predominance of Aβ42 over Aβ40 in the LDM fraction and in fraction 5, as observed in human brains, became apparent after 8.4 months of age. It should be noted that in these mouse brains APP was recovered exclusively in fraction 4 instead of fraction 5 (data not shown). Overall, the profiles are similar to those found in human brains (Figure 2) ▶ . Thus, although marked overproduction of mutant APP and therefore of Aβ40 and Aβ42 were found in the PDAPP mouse brains, which differs from that for human brains, both species showed a similar profile in terms of Aβ accumulation in the LDM fraction and in Aβ deposition.
Figure 4.

Fractionation of brain homogenates of PDAPP mouse brains. Brains of PDAPP mice aged 1.6 to 12.3 months were fractionated by sucrose density gradient centrifugation. Each fraction was spun down and the resulting pellet was extracted with guanidine hydrochloride. The extract was subjected to ELISA. Open and filled bars represent the levels of Aβ40 and Aβ42, respectively. The figures at the right in the last three columns indicate levels of Aβ42 (pmol/g).
Aβ in LDM Fraction Represents Mostly the Aβ Associated with LDM Domains
The present protocol is used most often to purify LDM domains from cultured cells, but has not been applied to brain fractionation except for two reports. 24,25 In the case of human brains, a dense creamy layer is always observed to contaminate the LDM fraction: this is most likely myelin, as suggested by labeling with antibodies to MBP. In PDAPP mice, although such creamy layers were less obvious, the LDM fraction also contained the myelin marker. Thus, we cannot exclude the possibility that significant amounts of Aβ are absorbed in the myelin and recovered in the LDM fraction. This is possible because Aβ, especially in its aggregated form, seems to have a relatively high affinity for lipid, and especially cholesterol. 26,27 We attempted several detergents, but were unable to selectively solubilize myelin. We then used an entirely different protocol for fractionating PDAPP mouse brain homogenates, which was originally developed to isolate endosomes. 18 By using this protocol, myelin markers and LDM markers were substantially separated (Figure 5) ▶ . Aβ was not associated with myelin markers, indicating that Aβ is not bound to myelin. Thus, we believe that the Aβ in the LDM fraction represents Aβ tightly associated with LDM domains in the brain (Figure 5) ▶ .
Figure 5.

Separation of myelin from Aβ by another sucrose density gradient centrifugation protocol. After sucrose density gradient centrifugation, resulting fractions (Fr 1–7 and pellet) were subjected to Western blotting for localization of proteolipid protein and flotillin. Remaining fractions were spun down and the pellets were extracted. The extracts were subjected to ELISA for Aβ40 and Aβ42 levels. Open and filled bars represent the levels of Aβ40 and Aβ42, respectively.
Another concern is that subcellular fractionation is usually established using fresh tissues and the use of frozen tissue may have led to some aberrant distributions of subcellular organelles, and the Triton-insoluble Aβ may have been mislocalized. In general, cytosolic proteins in human brains tend to become insoluble. To examine this possibility, we fractionated fresh and frozen rat brains with various postmortem intervals according to the protocol, and obtained exactly the same locations of several markers specific for subcellular organelles. This strongly suggests that at least subcellular organelles of interest from frozen brains are fractionated in the same manner as has been found in fresh brains.
One more concern is that monomeric and/or oligomeric Aβ, generated through mechanical disruption of Aβ deposits by homogenization, could have been redistributed among membranous compartments, and have led to high levels of Aβ in the LDM fraction. To examine this possibility, a small (3 pmol/g wet tissue) or a large amount (500 pmol/g wet tissue) of exogenous Aβ40 or Aβ42 was added to homogenate of normal mouse brain, each of which was similarly fractionated by sucrose density gradient centrifugation. Most of the Aβ40 in both cases and most of Aβ42, when a small amount was added, was recovered in fraction 4 (more accurately, soluble part of fraction 4). By contrast, if a large amount was added, most of the Aβ42 was recovered in the fraction 5 (pellet; data not shown). This result strongly suggests that the presence of large amounts of Aβ40 and Aβ42 in LDM fraction is neither derived from soluble Aβ that should have been recovered in fraction 4, nor from deposited Aβ fibrils that should have been recovered in fraction 5.
The presence of Aβ40 and Aβ42 in the LDM fraction from human brains was confirmed by Western blotting using Aβ monoclonal antibodies (Figure 6) ▶ . The blot clearly showed that sodium dodecyl sulfate-stable Aβ dimers were invariably present in the LDM fraction from human specimens. In PDAPP mice, Aβ40 dimers are constantly observed, whereas Aβ42 dimers were seen only in one half of the mice. In addition, the uppermost Aβ-immunoreactive band was by far the predominant species in PDAPP mice, whereas two or three bands were always detected in human specimens (Figure 6) ▶ . Possibly, the presence of truncated Aβ species may depend on the duration of Aβ accumulation in vivo (decades in humans 11,12 versus months in mice 22 ).
Figure 6.

Western blotting of LDM fractions from PDAPP mouse brains and human brains. LDM fractions were prepared from two PDAPP mice aged 3.5 months (lanes 1 and 2) and from two aged 10.3 months (lanes 3 and 4), and from the brains of a nondemented patient aged 46 years (lane 5) and an AD patient (lane 6). The Aβ40/Aβ42 levels (pmol/g) in LDM fraction by ELISA were 2.0/1.5, 0.6/0.9, 2.2/8.6, 2.2/7.6, 1.7/8.9, and 5.0/102.7, respectively. In the left lanes 25 pg of Aβ40 and 200 pg of Aβ42 were loaded. For an unknown reason, the highest BA27 reactivity was observed in the LDM fraction prepared from one PDAPP mouse aged 10.3 months (lane 4). Arrowheads at the left indicate the positions of Aβ monomers and dimers.
Discussion
We have shown here firstly that, in both human and PDAPP mouse brains, substantial fractions of Aβ40 and Aβ42 reside in the LDM domains, with the levels of the former being approximately twofold higher than those of the latter before Aβ42 accumulation. Secondly, progression of Aβ42 deposition invariably accompanies a predominant accumulation of Aβ42 in LDM domains, with the Aβ40 levels being relatively stable. 12 Finally, the extents of Aβ40 and Aβ42 accumulation in LDM domains correlate well with the extent of Aβ42 deposition as represented by the levels of TS-insoluble Aβ42 or by the levels of Aβ42 in fraction 5 (data not shown). The levels of Aβ42 in turn correlate well with the amyloid burden, 11 which is the Aβ-positive area relative to the total area of the tissue section. We carefully analyzed several cases in which the observed deposition of Aβ42 was minimal, but could not determine which of the fractions (LDM fraction or fraction 5) accumulated Aβ42 first. This suggests that the increases in the Aβ levels in these two compartments occur at the same time, and may be tightly coupled. In normal brains, Aβ40 predominated over Aβ42 in LDM domains. This was also the case in fraction 5 and TS-insoluble Aβ. 12 Furthermore, it is notable that the Aβ accumulation in LDM domains has the same characteristics as seen in Aβ deposition in the extracellular space: predominance of Aβ42, and a delayed, and smaller extent of, Aβ40 accumulation, when Aβ deposition has commenced (Figure 3, A and B) ▶ . Thus, the Aβ associated with LDM domains accurately reflects the characteristics and extents of Aβ deposition in the extracellular space. This strongly suggests that the former is closely related to Aβ deposition in the extracellular space.
Most interestingly, mutant PS2, but not wild-type PS2, transgenic mice show an age-dependent accumulation of Aβ42 in LDM domains (1/4 to 1/5 the Aβ42 levels in LDM domains of PDAPP mice). 28 Altogether, human brains, PDAPP mouse brains, and mutant PS2 transgenic mouse brains share the same abnormalities in the brain just before and during the process of Aβ deposition or during aging: a predominant increase in Aβ42 associated with LDM domains. This suggests that LDM domains may contribute to a common pathway leading to extracellular Aβ deposition. This feature is common to brains affected by mutations of APP and presenilins, and to normal human brains.
An investigation based on the assumption that presenilins are γ-secretases, has led to the suggestion that the Golgi complex is the intracellular production site for Aβ40 and Aβ42. 29 The complex of N- and C-terminal fragments of presenilin, which is considered to be an active γ-secretase, co-fractionates with Golgi markers. 29 LDM domains are considered to occur in the Golgi region as well, and thus, it would be reasonable to speculate that a fraction of the generated Aβ is incorporated into the LDM domain occurring there, and is delivered constitutively to the plasma membrane. It may be that generated Aβ42 is partitioned preferentially into LDM domains in the Golgi complex, whereas most Aβ40 may be on the route to the secretion pathway. This is quite possible because Aβ42 is longer than Aβ40 by two hydrophobic residues, Ileu and Ala, and should have a higher affinity for the lipid bilayer. Mutant APP and PS2 generate more Aβ42, which thus may replace Aβ40 and result in an increase in Aβ42 in the LDM domains.
Currently we do not know how LDM-accumulated Aβ42 is related to Aβ42 deposition in the extracellular space. Because of the shared characteristics of the two compartments, one can postulate that extracellular Aβ deposits and LDM-Aβ are in dynamic equilibrium. In fact, senile plaques appear to be in a dynamic process of deposition and dissolution of Aβ, especially Aβ42. 30 For example, vaccination enhances the latter process and finally leads to a reduced amount of senile plaques. 31 This dynamic process may be mediated by lipoproteins in the brain that can bind and carry monomeric or oligomeric Aβ. 32 This view may be consistent with the coexistence of Aβ and ApoE in senile plaques. 33 For analogy, one can consider the case of cholesterol transfer between the plasma membrane and high-density lipoprotein (reverse cholesterol transport). 34 If this analogy is true, then the accumulation of Aβ in LDM domains of brain cells indeed reflects the extent of Aβ deposition in the extracellular space. Another possibility, which does not necessarily exclude the above hypothesis, is the shedding of the LDM domains into the extracellular space, which in turn may act as seeds for the formation of Aβ fibrils. 15
We do not know what triggers the vicious cycle for progressive Aβ deposition 11,12 and what determines the levels of Aβ42 in LDM domains. The levels of Aβ42 in LDM domains seem to be a determining factor for extracellular Aβ deposition. In brains affected by mutations to the genes for APP or presenilins, the Aβ42 levels in LDM domains are higher, even from the neonatal period. In most patients, the Aβ42 levels in LDM can become high only after reaching a critical age. 11,12 A protease(s) associated with LDM or in the brain parenchyma may have an important role in release of Aβ42 from the cell and its clearance from the parenchyma, respectively. A decrease in the activities of such a protease by its down-regulation or up-regulation of endogenous inhibitors would cause an accumulation of Aβ42 in LDM domains and in the parenchyma, and lead to some functional disturbances of the cell. One such candidate protease is neprilysin, which has recently been identified as a major metalloprotease for Aβ degradation in the brain. 35
Acknowledgments
We thank Ms. J. Saishoji and Ms. N. Naoi for tissue preparation, and Ms. M. Anzai for preparation of the manuscript.
Footnotes
Address correspondence to Yasuo lhara, M.D., Department of Neuropathology, Faculty of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: .
Supported in part by Research Grants for Longevity Sciences from the Ministry of Health and Welfare, Japan and from the Sasakawa Health Science Foundation, Japan.
N. O. and M. M. K. contributed equally to this work.
References
- 1.Selkoe DJ: The cell biology of β-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol 1998, 8:447-453 [DOI] [PubMed] [Google Scholar]
- 2.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M: β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286:735-741 [DOI] [PubMed] [Google Scholar]
- 3.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ: Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and β-secretase activity. Nature 1999, 398:513-517 [DOI] [PubMed] [Google Scholar]
- 4.Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ: Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000, 405:689-694 [DOI] [PubMed] [Google Scholar]
- 5.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y: Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ-monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13:45-53 [DOI] [PubMed] [Google Scholar]
- 6.Iwatsubo T, Mann DMA, Odaka A, Suzuki N, Ihara Y: Amyloid β-protein (Aβ) deposition: Aβ42(43) precedes Aβ40 in Down’s syndrome. Ann Neurol 1995, 37:294-299 [DOI] [PubMed] [Google Scholar]
- 7.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S: Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 1996, 2:864-870 [DOI] [PubMed] [Google Scholar]
- 8.Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE: Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science 2000, 290:2302-2303 [DOI] [PubMed] [Google Scholar]
- 9.Myers A, Holmans P, Marshall H, Kwon J, Meyer D, Ramic D, Shears S, Booth J, DeVrieze FW, Crook R, Hamshere M, Abraham R, Tunstall N, Rice F, Carty S, Lillystone S, Kehoe P, Rudrasingham V, Jones L, Lovestone S, Perez-Tur J, Williams J, Owen MJ, Hardy J, Goate AM: Susceptibility locus for Alzheimer’s disease on chromosome 10. Science 2000, 290:2304-2305 [DOI] [PubMed] [Google Scholar]
- 10.Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG: Linkage of plasma Aβ42 to a quantitative locus on chromosome 10 in late-onset Alzheimer’s disease pedigrees. Science 2000, 290:2303-2304 [DOI] [PubMed] [Google Scholar]
- 11.Funato H, Yoshimura M, Kusui K, Tamaoka A, Ishikawa K, Ohkoshi N, Namekata K, Okeda R, Ihara Y: Quantitation of amyloid β-protein (Aβ) in the cortex during aging and in Alzheimer’s disease. Am J Pathol 1998, 152:1633-1640 [PMC free article] [PubMed] [Google Scholar]
- 12.Morishima-Kawashima M, Oshima N, Ogata H, Yamaguchi H, Yoshimura M, Sugihara S, lhara Y: Effect of apolipoprotein E allele ε4 on the initial phase of amyloid β-protein accumulation in the human brain. Am J Pathol 2000, 157:2093-2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morishima-Kawashima M, Ihara Y: The presence of amyloid β-protein in the detergent-insoluble membrane compartment of human neuroblastoma cells. Biochemistry 1998, 37:15247-15253 [DOI] [PubMed] [Google Scholar]
- 14.Simons K, Ikonen E: Functional rafts in cell membranes. Nature 1997, 387:569-572 [DOI] [PubMed] [Google Scholar]
- 15.Yanagisawa K, Odaka A, Suzuki N, Ihara Y: GM1 ganglioside-bound amyloid β-protein (Aβ): a possible form of preamyloid in Alzheimer’s disease. Nat Med 1995, 1:1062-1066 [DOI] [PubMed] [Google Scholar]
- 16.Tierney MC, Fisher H, Lewis AJ, Zorzitto ML, Snow WG, Reid DW, Nieuwstraten P: The NINCDS-ADRDA Work group criteria for the clinical diagnosis of probable Alzheimer’s disease: clinicopathological study of 57 cases. Neurology 1988, 38:356-364 [DOI] [PubMed] [Google Scholar]
- 17.Mirra SS, Heyman A, McKee D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg LT, Ball MJ, Bierer LM, Claasen D, Hansen L, Hart M, Hedreen J, Henderson V, Hyman BT, Joachim C, Markesbery W, Martinez AJ, McKee A, Miller C, Moossy J, Nochlin D, Perl D, Petito C, Rao GR, Schelper RL, Slager U, Terry R: The consortium to establish a registry for Alzheimer’s disease (CERAD). : Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 42:479-486 [DOI] [PubMed] [Google Scholar]
- 18.van der Goot FG: Separation of early steps in endocytic membrane transport. Electrophoresis 1997, 18:2689-2693 [DOI] [PubMed] [Google Scholar]
- 19.Shinkai Y, Yoshimura M, Morishima-Kawashima M, Ito Y, Shimada H, Yanagisawa K, Ihara Y: Amyloid β-protein deposition in the leptomeninges and cerebral cortex. Ann Neurol 1997, 42:899-908 [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi H, Sugihara S, Ogawa A, Ihara Y: Alzheimer β amyloid deposition, which is hastened by ApoE ε4 gene, precedes neurofibrillary pathology in the frontal association cortex of non-demented senior subjects. J Neuropathol Exp Neurol (in press) [DOI] [PubMed]
- 21.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J: Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373:523-527 [DOI] [PubMed] [Google Scholar]
- 22.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L: Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA 1997, 94:1550-1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D: Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer’s disease. J Neurosci 1996, 16:5795-5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S-J, Liyanage U, Bickel PE, Xia W, Lansbury PT, Jr, Kosik KS: A detergent-insoluble membrane compartment contains Aβ in vivo. Nat Med 1998, 4:730-734 [DOI] [PubMed] [Google Scholar]
- 25.Sawamura N, Morishima-Kawashima M, Waki H, Kobayashi K, Kuramochi T, Ito M, Oyama F, Ando S, Ihara Y: Mutant-presenilin 2-transgenic mice: a large increase in the levels of Aβ42 is presumably associated with the low-density membrane domain that contains decreased levels of glycerophospholipids and sphingomyelin. J Biol Chem 2000, 275:27901-27908 [DOI] [PubMed] [Google Scholar]
- 26.Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG: Lipid binding to amyloid β-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J Neurochem 1997, 69:1746-1752 [DOI] [PubMed] [Google Scholar]
- 27.Yamazaki T, Chang T-Y, Haass C, Ihara Y: Accumulation and aggregation of amyloid β-protein in late endosomes of Niemann-Pick type C cells. J Biol Chem (in press) [DOI] [PubMed]
- 28.Oyama F, Sawamura N, Kobayashi K, Morishima-Kawashima M, Kuramochi T, Ito M, Tomita T, Maruyama K, Saido TC, Iwatsubo T, Capell A, Walter J, Grunberg J, Ueyama Y, Haass C, Ihara Y: Mutant presenilin 2 transgenic mouse: effect on an age-dependent increase of amyloid β-protein 42 in the brain. J Neurochem 1998, 71:313-322 [DOI] [PubMed] [Google Scholar]
- 29.Xia W, Ray WJ, Ostaszewski BL, Rahmati T, Kimberly WT, Wolfe MS, Zhang J, Goate AM, Selkoe DJ: Presenilin complexes with the C-terminal fragments of amyloid precursor protein at the sites of amyloid β-protein generation. Proc Natl Acad Sci USA 2000, 97:9299-9304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hyman BT, Marzloff K, Arriagada PV: The lack of accumulation of senile plaques or amyloid burden in Alzheimer’s disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol 1993, 52:594-600 [DOI] [PubMed] [Google Scholar]
- 31.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P: Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400:173-177 [DOI] [PubMed] [Google Scholar]
- 32.Fagan AM, Younkin LH, Morris JC, Fryer JD, Cole TG, Younkin SG, Holtzman DM: Differences in the Aβ40/Aβ42 ratio associated with cerebrospinal fluid lipoproteins as a function of apolipoprotein E genotype. Ann Neurol 2000, 48:201-210 [PubMed] [Google Scholar]
- 33.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K: Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res 1991, 541:163-166 [DOI] [PubMed] [Google Scholar]
- 34.Yokoyama S: Release of cellular cholesterol: molecular mechanism for cholesterol homeostasis in cells and in the body. Biochim Biophys Acta 2000, 1529:231-244 [DOI] [PubMed] [Google Scholar]
- 35.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC: Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 2000, 6:143-150 [DOI] [PubMed] [Google Scholar]