

Abstract
The presenilins (PSs) are components of large molecular complexes that contain β-catenin and function as γ-secretase. We report here a striking correlation between amyloid angiopathy and the location of mutation in PS-1 linked Alzheimer’s disease. The amount of amyloid β protein, Aβ42(43), but not Aβ40, deposited in the frontal cortex of the brain is increased in 54 cases of early-onset familial Alzheimer’s disease, encompassing 25 mutations in the presenilin-1 (PS-1) gene, compared to sporadic Alzheimer’s disease. The amount of Aβ40 in PS-1 Alzheimer’s disease varied according to the copy number of ε4 alleles of the Apolipoprotein E gene. Although the amounts of Aβ40 and Aβ42(43) deposited did not correlate with the genetic location of the mutation in a strict linear sense, the histological profile did so vary. Cases with mutations between codon 1 and 200 showed, in frontal cortex, many diffuse plaques, few cored plaques, and mild or moderate amyloid angiopathy. Cases with mutations occurring after codon 200 also showed many diffuse plaques, but the number and size of cored plaques were increased (even when ε4 allele was not present) and these were often clustered around blood vessels severely affected by amyloid angiopathy. Similarly, diverging histological profiles, mainly according to the degree of amyloid angiopathy, were seen in the cerebellum. Mutations in the PS-1 gene may therefore alter the topology of the PS-1 protein so as to favor Aβ formation and deposition, generally, but also to facilitate amyloid angiopathy particularly in cases in which the mutation lies beyond codon 200. Finally we report that the amount of Aβ42(43) deposited in the brain correlated with the amount of this produced in culture by cells bearing the equivalent mutations.
Approximately half of all cases of early onset familial Alzheimer’s disease (AD) are associated with a mutation in a gene located on the long arm of chromosome 14, termed the presenilin-1 (PS-1) gene. More than 70 disease-causing mutations in this gene have so far been identified (http://www.alzforum.org/members/resources/pres_mutations/ps1/pres1table.html), although the precise manner in which these influence the pathological cascade remains uncertain.
In the central nervous system, PS-1 protein is principally located in the perikarya and dendrites of neurones, particularly pyramidal cells of the cerebral cortex and hippocampus and magnocellular basal forebrain neurones, within the Golgi apparatus and smooth endoplasmic reticulum. 1-3 No differences in the distribution or amount of immunocytochemically detectable PS-1 protein 4-6 or its message 7 have been observed between AD (including cases of PS-1 AD) and control patients. Nonetheless, cell lines and transgenic mice bearing PS-1 [and presenilin-2 (PS-2)] mutations produce more amyloid β protein (Aβ), particularly Aβ42(43) than those carrying wild-type PS-1. 8-13 Moreover, fibroblasts from human carriers of PS-1 mutations produce more Aβ42(43) in culture 14 and plasma levels of this are likewise elevated in affected individuals. 14,15 At autopsy, cerebral cortical deposition of Aβ, particularly Aβ42(43) is often high. 16-20 Mutations in the PS-1 gene may therefore confer a metabolic defect that modifies γ-secretase catabolism of APP along pathways that favor the production of Aβ, especially Aβ42(43)
Recent evidence has indicated that both PS-1 and PS-2 are novel membrane-associated aspartyl proteases, 21 consistent with the long-suspected idea that they regulate the catabolic activity of γ-secretase and might even be γ-secretase itself. PS-1 knockout mice show normal α- and β-secretase activity, although the activity of γ-secretase and the production of Aβ1-40 and Aβ1-42 is decreased. 20 Site directed mutagenesis of the aspartate residues of PS-1 reduces γ-secretase catalyzed processing of APP within its transmembrane domain. 21 Immunodepletion of PS-1 from solubilized γ-secretase causes a reduction in γ-secretase activity and solubilized γ-secretase activity co-migrates with PS-1 during gel exclusion chromatography. 22 Crossing linking experiments using inhibitors of γ-secretase, directed to the active site of an aspartyl protease, label PS-1 (and PS-2) 23,24 and strongly suggest that PS-1 (and PS-2) contain the active site of γ-secretase. 23 However, PS-1 (and PS-2) are probably contained in a large functional complex with other proteins. 21,22,24
Studying factors that might influence variability in disease onset, progression, or pathology in AD can be confounded by the heterogeneous basis of the disease, eg, genetic (ApoE ε4 allele) or environmental (head injury) causes. To avoid such difficulties and therefore increase the power of this type of study, we have investigated a large group of patients with AD because of the same definable cause, ie, those with PS-1 mutations. To determine the biological effects of mutations in the PS-1 gene in the human situation, we have characterized the extent and the morphological features of Aβ deposition in 54 cases of PS-1 AD from 25 separate mutations covering the length of PS-1. Deposition of Aβ40 and Aβ42(43) as plaques and amyloid angiopathy, was assessed in frontal cortex and cerebellum and quantified by image analysis, and related to mutation position in PS-1.
Materials and Methods
Formalin-fixed blocks of frontal cortex (Brodmann area 8/9) were obtained from 54 cases, and cerebellar cortex in 48 of these cases, from 28 families with proven chromosome 14-linked AD; 25 separate PS-1 mutations were present in these. Details of these cases are given in Table 1 ▶ . These tissues were kindly donated by colleagues throughout the world for this particular project and no tissue blocks from other regions of cerebral cortex, or noncortical areas, were available to us to study. Tissue blocks from a similar area of frontal cortex were also available from 25 cases of sporadic AD (SAD) with onset of disease before 70 years of age, matching (proportionately) the cases of PS-1 AD for Apolipoprotein E genotype (ApoE). All tissue blocks were similarly and routinely processed into paraffin wax and consecutive sections, cut at a thickness of 6 μm, were mounted onto 3-aminopropyltriethoxysilane (APES)-coated slides.
Table 1.
Clinical and Genetic Details on 54 Cases of Alzheimer’s Disease Because of Mutations in PS-1 Gene
| Case | Patient code | PS-1 mutation | Sex | Age at onset | Age at death | Duration | Brain weight | ApoE genotype | Additional clinical features |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 6866 | T116R | M | 35 | 43 | 8 | n.a. | E3/E4 | Nil |
| 2 | 22290 | E120D | M | 53 | 62 | 9 | 1239 | E3/E3 | Language loss |
| 3 | 22466 | E120D | M | 34 | 51 | 17 | 1206 | E3/E3 | Seizures, rigidity, negative cogwheel |
| 4 | A74/93 | E120K | M | 36 | 44 | 8 | 548* | E3/E3 | Nil |
| 5 | A141/93 | ΔE4 truncation | M | 36 | 54 | 18 | 963 | E3/E3 | Myoclonus |
| 6 | A293/94 | M139V | M | 36 | 42 | 6 | 1080 | E3/E4 | Myoclonus, seizures, RTA with loss of consciousness at 28 years |
| 7 | 176/81 | M139V | F | 43 | 53 | 10 | 922 | E3/E4 | Myoclonus, seizures |
| 8 | 734 | M139I | M | 38 | 41 | 3 | 553* | E2/E3 | Seizures |
| 9 | 773 | M139I | M | 38 | 43 | 5 | 1210 | E3/E3 | Nil |
| 10 | A236/91 | I143F | F | 55 | 61 | 6 | 510* | E3/E3 | Nil |
| 11 | 4434 | I143T | F | 34 | 39 | 5 | n.a. | E3/E3 | Myoclonus, epilepsy, toxic hepatitis |
| 12 | 4268 | M146I | M | 43 | 51 | 8 | n.a. | E3/E3 | Anxiety |
| 13 | 95-124 | M146L | M | 45 | 51 | 6 | 840 | E3/E3 | Delusions, myoclonus, hallucinations, Parkinsonism |
| 14 | V1501-92 | M146V | F | 39 | 43 | 4 | n.a. | E3/E3 | Myoclonus, seizures |
| 15 | 178 | H163R | M | 40 | 44 | 4 | 1200 | E3/E3 | Nil |
| 16 | 313 | H163R | F | 43 | 48 | 5 | 370** | E3/E4 | Nil |
| 17 | 3098 | H163R | F | 51 | 62 | 11 | 790 | E3/E3 | Nil |
| 18 | A77 | H163R | M | 50 | 65 | 15 | 1080 | E3/E3 | Language loss, seizures, quadriplegia |
| 19 | A80 | H163R | F | 38 | 62 | 24 | n.a. | E3/E3 | Language loss, clumsiness |
| 20 | 97/19 | S169L | F | 32 | 38 | 6 | 910 | E3/E3 | Myoclonus, seizures |
| 21 | 12829 | G209V | M | 39 | 45 | 6 | 1150 | E3/E3 | Myoclonus, seizures, language loss |
| 22 | 14338 | G209V | M | 39 | 49 | 10 | 940 | E2/E3 | Myoclonus, seizures, language loss |
| 23 | 20399 | G209V | F | 45 | 50 | 3 | 1146 | E3/E3 | Myoclonus, language loss |
| 24 | 22129 | G209V | F | 36 | 55 | 10 | 770 | E3/E3 | Seizures |
| 25 | 22203 | G209V | M | 56 | 40 | 4 | 1179 | E3/E3 | Seizures |
| 26 | X97-63 | M233T | F | 31 | 39 | 8 | n.a. | E3/E4 | Seizures |
| 27 | 305 | A246E | M | 54 | 60 | 6 | 950 | E3/E4 | Nil |
| 28 | 306 | A246E | F | 50 | 56 | 6 | 800 | E3/E4 | Nil |
| 29 | 593 | A246E | M | 55 | 70 | 15 | 1290 | E3/E3 | Nil |
| 30 | 95-9 | A246E | M | – | 65 | – | 880 | E3/E3 | Nil |
| 31 | 40-95 | A246E | F | 43 | 66 | 23 | 800 | E3/E3 | Partial complex seizures since 26 years, language loss |
| 32 | L971 | A246E | F | 52 | 67 | 15 | 850 | E3/E4 | Nil |
| 33 | 183/89 | L250S | F | 56 | 63 | 7 | n.a. | E3/E4 | Myoclonus, language loss, hallucinations, visual agnosia |
| 34 | 14/95 | L250S | F | 49 | 55 | 6 | n.a. | E3/E4 | Myoclonus, ophthalmoplegia, extrapyramidal signs language loss, visual agnosia |
| 35 | 19582 | A260V | F | 38 | 55 | 17 | 323** | E3/E3 | Myoclonus, seizures, language loss |
| 36 | 22239 | A260V | F | 41 | 60 | 19 | 372* | E3/E3 | Bilateral spasticity, language loss |
| 37 | 3650 | P264L | M | 34 | 49 | 15 | 1300 | E3/E3 | Myoclonus, rigidity, language loss |
| 38 | 45643 | R269H | M | 46 | 55 | 9 | 1180 | E4/E4 | Myoclonus, hallucinations |
| 39 | 2968 | E280A | F | 48 | 58 | 10 | 900 | E3/E3 | Seizures, early gait disturbance |
| 40 | 2965 | E280A | M | 54 | 62 | 8 | 900 | E3/E3 | Abnormal reflexes, pyramidal signs |
| 41 | 2761 | E280A | M | 50 | 55 | 5 | 1000 | E3/E3 | Pyramidal signs |
| 42 | 2786 | E280A | F | 39 | 48 | 9 | 1020 | E3/E4 | Myoclonus, seizures, tremor, dystonia |
| 43 | 3396 | E280A | F | 41 | 44 | 3 | 1100 | E3/E3 | Myoclonus, seizures, pyramidal signs |
| 44 | 2970 | E280A | F | 53 | 61 | 8 | 940 | E3/E3 | Nil |
| 45 | 3156 | E280A | F | 55 | 61 | 6 | 1020 | n.a. | Nil |
| 46 | 3153 | E280A | F | 54 | 61 | 7 | 800 | E4/E4 | Nil |
| 47 | 269/81 | E280G | F | 48 | 53 | 5 | 762 | E3/E3 | Seizures |
| 48 | C32 | G384A | M | 36 | 48 | 12 | n.a. | E3/E3 | |
| 49 | 6531 | L286V | M | 53 | 61 | 8 | 1200 | E3/E3 | Nil |
| 50 | L963 | C410Y | F | 45 | 60 | 15 | 840 | n.a. | Parkinsonism |
| 51 | L929 | C410Y | M | 42 | 47 | 5 | 1300 | E3/E3 | Parkinsonism |
| 52 | L93127 | C410Y | F | 56 | 81 | 25 | 850 | E3/E3 | Parkinsonism |
| 53 | L8926 | C410Y | M | 46 | 55 | 9 | 980 | E2/E3 | Parkinsonism |
| 54 | L933 | C410Y | M | 51 | 59 | 8 | 980 | E3/E3 | Myoclonus, hallucinations |
*Denotes weight of half-brain only.
**Denotes weight of half-hemisphere only.
Sections of frontal cortex and cerebellum were immunostained 26 using the end-specific monoclonal antibodies BC05 and BA27, to selectively demonstrate Aβ peptide species Aβ42(43) and Aβ40 respectively within plaques and blood vessel walls. BC05- and BA27-immunostained sections of frontal cortex were subjected to computerized morphometry using an Olympus Image Analysis System (SP500, Model 1500 C2; Olympus Corp., Tokyo, Japan). 26 The numerical density of Aβ40- and Aβ42(43)-containing plaques and the area proportion (amyloid load) of each was determined. The ratio between Aβ40- and Aβ42(43)-containing deposits was calculated on both a numerical density and an area proportion basis.
In histological sections the extent of Aβ40 and Aβ42(43) deposition within plaques (cerebellum only), or as amyloid angiopathy (frontal cortex and cerebellum), was rated semiquantitatively by an experienced neuropathologist (DMAM) according to the following protocol. Rating of plaques was: 1 = few; 2 = moderate; 3 = many; 4 = very many. The presence of subpial or white matter deposits was noted. Rating of amyloid angiopathy was: 1 = few leptomeningeal vessels weakly or patchily stained; 2 = few leptomeningeal vessels, strongly or evenly stained, with mild intracortical vascular involvement; 3 = many leptomeningeal and intracortical vessels patchily or strongly stained; 4 = many leptomeningeal and intracortical vessels strongly or evenly stained, with dyshoric angiopathy.
Results
Relationship Between Aβ40 and Aβ42(43) Deposition and Site of Mutation
There was much variation in the amount of Aβ42(43) deposited as plaques within the frontal cortex between cases of different PS-1 mutation, and sometimes within cases of the same mutation (Figure 1A) ▶ . On average, some mutations were associated with a very heavy Aβ42(43) deposition, some 2 to 3 times the amount of that in others in which deposition was within the usual range (for SAD) 27 with <10% of tissue area occupied. There was no obvious clustering of mutations associated with high, intermediate, or low Aβ deposition at any site(s) (in a strict linear sense) within the PS-1 gene. More importantly, there was no apparent clustering of high- or low-Aβ42(43)-depositing mutations around the active site aspartyl residues at codon 257 and 385, or around codon 298 where PS-1 is cleaved. 28 Nonetheless, present findings clearly imply that some mutations are much more aggressive than others, at least in terms of the extent of tissue deposition of Aβ42(43) they elicit.
Figure 1.

Mean percentage area of frontal cortex occupied by Aβ42(43) (A) and Aβ40 (B) in 54 cases of PS-1 AD, plotted against mutation site.
Deposition of Aβ40 was likewise highly variable, although cases showing a heavier deposition tended to be those in which the mutation was located after codon 200 (Figure 1B) ▶ . There was no correlation between the amounts of Aβ40 and Aβ42(43) within plaques across the 54 cases.
Comparisons with Sporadic AD
The area proportion of tissue occupied by Aβ42(43)-containing plaques was overall significantly (P < 0.001) higher in PS-1 AD compared to SAD (13.6 ± 5.9% versus 7.6 ± 2.3%, respectively). However, the area occupied by Aβ40 in PS-1 AD did not differ overall from that in SAD (2.5 ± 3.7 versus 2.2 ± 2.2, respectively). Such data extend previous studies based on small series of patients or on individual cases. 16-20 They are also consistent with experimental studies using cell lines and transgenic mice, 8-13 or fibroblasts from human carriers 14 showing that the PS-1 mutations result in an increased production of Aβ42(43) but not Aβ40, compared to wild-type PS-1.
Relationship between Aβ Deposition and ApoE Genotype
We stratified the PS-1 and SAD cases according to ApoE genotype, grouping into ε4 and non-ε4 allele bearers. The area proportion of tissue occupied by Aβ42(43) was significantly greater in PS-1 AD, compared to SAD, for both non-ε4 (13.2 ± 6.5 versus 8.1 ± 2.9, respectively; P < 0.01) and ε4 (14.9 ± 4.8 versus 7.2 ± 1.6, respectively; P < 0.001) allele bearers. Area measures for Aβ40 did not however differ from those of SAD, either for non-ε4 (1.8 ± 2.9% versus 1.1 ± 0.9%, respectively) or ε4 (4.1 ± 5.3% versus 3.2 ± 2.7%, respectively) allele bearers. Nevertheless, as we 27 and others 29 have reported previously for SAD, the area proportion of tissue occupied by Aβ40, but not Aβ42(43) was greater (P < 0.05) in cases of PS-1 AD in the presence of ε4 allele (with ε4 4.1 ± 5.3% versus 1.8 ± 2.9% without ε4). Such data imply that the presence of E4 protein isoform in the brain in PS-1 AD may, as in SAD, lower the threshold to fibrillization of Aβ40, thereby promoting its deposition on pre-existing Aβ42(43)-containing plaques.
Relationship between Aβ Deposition and Aβ Production
We correlated the mean amount of Aβ42(43) deposited in the frontal cortex in 19 of the PS-1 mutations with the amount of Aβ1-42 secreted in culture by Green monkey kidney cells (COS-1 cells) bearing the same mutations. This latter data 30 was obtained with permission from Professor T. Iwatsubo, University of Tokyo. We found a significant (r = 0.789, P < 0.001) correlation between the amount of tissue deposition in human brain and secretion of Aβ in culture for each PS-1 mutation (Figure 2) ▶ . This indicates that variations in the amount of Aβ deposited in the tissue reflect differences in the level of production of Aβ. Tissue deposition of Aβ can therefore be viewed as an index of tissue secretion. Moreover, these results reinforce the utility of cell lines for investigating the effects of PS-1 mutations in vivo.
Figure 2.

Correlation between the mean percentage area of frontal cortex occupied by Aβ42(43) from patients with various PS-1 mutations and the amount of Aβ42 produced by cell lines bearing the equivalent mutations. The cell line Aβ42 value is expressed as a ratio of the total Aβ produced to normalize against variations in expression. Mutations compared are E120K, M139I, M139V, I143F, I143T, M146L, M146V, H163R, A246E, L250S, A260V, P264L, E280G, L286V, G384A, and C410Y.
Correlations with Amyloid Angiopathy
As has been observed previously, 16-19 amyloid angiopathy was particularly prominent within the frontal cortex of many of the present cases, this involving both intraparenchymal, as well as leptomeningeal, vessels with a dyshoric change being commonplace (Figure 3, c and d) ▶ . Indeed, a particularly heavy Aβ40 deposition, in the form of large plaques clustered around affected vessels, was seen in cases with severe amyloid angiopathy, especially when dyshoric angiopathy was present (Figure 3d) ▶ . Variations in plaque Aβ40 (but not Aβ42(43) deposition) correlated (Rs = 0.672; P < 0.001) with the rating for amyloid angiopathy. These observations suggest a possible hematogenous source for at least some of the Aβ40, with plasma Aβ40 leaking across damaged blood vessels and seeding on local pre-existing Aβ42(43)-containing deposits. Although some cases with severe amyloid angiopathy were bearers of ApoE ε4 allele, there was no correlation between possession of this and the extent of amyloid angiopathy across the 54 cases (data not shown).
Figure 3.

Type 1 and type 2 histologies in frontal cortex in PS-1 AD. In type 1 histology (for example, case 4 with E120K mutation), there are numerous deposits of Aβ42(43) in the form of both diffuse and cored plaques (a). Aβ40 deposits are sparse and amyloid angiopathy is mild (c). In type 2 histology (for example, case 49 with G384A mutation), there are again numerous deposits of Aβ42(43) in the form of both diffuse and cored plaques (b), but Aβ40 deposits are likewise frequent (d) and amyloid angiopathy is severe (b and d). a and b: BC05 immunoperoxidase-hematoxylin. c and d: BA27 immunoperoxidase-hematoxylin.
The distribution of amyloid angiopathy with respect to mutation site revealed an uneven distribution (Figure 4) ▶ with most of those cases showing heavier pathology (ie, grade 2 and above) (27 of 34) occurring after codon 200. This differed significantly (chi-square = 15.4; P < 0.001) from that proportion (5 of 20) of cases with heavy pathology occurring before codon 200.
Figure 4.

Variations in extent of amyloid angiopathy in 54 cases of PS-1 AD, plotted against mutation site. Note, I143F, M146L, and S169L have a rating of zero.
Histological Patterns of Aβ Deposition
We then examined the overall pattern of plaque and vessel Aβ deposition in the 54 cases of PS-1 linked AD. It was strikingly clear that there were two distinct topographical profiles, defined as type 1 and type 2.
Type 1
In this pattern of pathology BC05 (Aβ42(43)) immunostaining revealed many diffuse and cored plaques, fairly evenly distributed along the whole of the gyral length (Figure 3a) ▶ . Usually, more plaques were present in cortical layers II and upper III, where diffuse deposits predominated, than in layers V and VI where cored plaques were more frequent. Cored plaques were also more frequent at the depths of the cortical sulci (Figure 3a) ▶ . Relatively few subpial deposits were seen and only occasional white matter plaques were present. In BA27 (Aβ40) immunostaining either a few, or a moderate number of, small cored or compacted deposits were fairly evenly spread along cortical laminae (usually in layers II and III) but often concentrated at the depths of the cortical sulci (Figure 3b) ▶ . A few subpial deposits were stained and occasional white matter plaques were present. In both BC05 and BA27 immunostaining, amyloid angiopathy was mild or moderate (or sometimes absent) and confined mostly to leptomeningeal arteries, with only occasional intraparenchymal arteries being involved.
Type 2
In this pattern of pathology, BC05 immunostaining revealed a similar number and distribution of diffuse and compacted or cored plaques as in type 1 pathology, except these tended to be very large and concentrated around amyloidotic arteries, especially at the depths of sulci (Figure 3c) ▶ . In BA27 immunostaining many of the diffuse plaques were weakly immunoreactive, although often there were many, very large, cored plaques clustered, as large conglomerates, around the amyloidotic arteries (Figure 3d) ▶ . Amyloid angiopathy was nearly always severe, involving both leptomeningeal and intraparenchymal arteries, and dyshoric change was frequent (Figure 3, c and d) ▶ .
The distribution of type 1 and type 2 histologies within frontal cortex of individual cases, with respect to mutation site, is given in Figure 5 ▶ . Sixteen of 21 cases with type 1 histology occurred before codon 200 whereas 29 of 33 cases with type 2 histology occurred beyond codon 200. These distributions of pathological subtype either side of codon 200 were, in both instances, highly significantly different (chi-square = 22.6, P < 0.001).
Figure 5.

Distribution of cases of PS-1 AD with type 1 or type 2 histologies within frontal cortex (bottom) and cerebellum (top), according to mutation site.
However, whereas in the majority of instances the type 1 and type 2 profiles did not co-exist within cases of those mutations where multiple (family) members were represented, there was some overlap in others. For example, in the case of H163R mutation, one member showed type 1 histology, whereas four members showed type 2. However, these five cases comprised of two members of an American pedigree (Table 1) ▶ and three from a Japanese pedigree. All three members of the Japanese pedigree had type 2 histology, whereas one of the American cases had type 1 histology whereas the other, a bearer of ApoE ε4 allele, had type 2 histology. Similarly, of the six bearers of A246E mutation, three had type 1 histology and were all bearers of ApoE ε3/ε3 genotype whereas the other three were bearers of ApoE ε4 allele and had type 2 histology. In contrast, of the five bearers of the G209V mutation, one had type 1 histology and four had type 2 histology, yet all bore the ApoE ε3/ε3 genotype.
Comparisons between Type 1 and Type 2 Histologies
Comparing the mean age at onset (±SD) between type 1 (n = 21; 41.5 ± 7.7 years) and type 2 (n = 32; 46.7 ± 6.6 years) histologies, we found that type 1 cases had a disease onset (P = 0.012) on average 5 years before type 2 cases. A similar trend was also observed for disease duration with type 1 cases (n = 21; 7.9 ± 4.6) being 2.4 years shorter (P = 0.081) than type 2 cases (n = 32; 10.3 ± 5.6 years). Furthermore, there was a near significant difference (P = 0.067) in the mean percentage area occupied by Aβ42(43) (type 1, n = 21, 15.3 ± 6.5%; type 2, n = 33, 12.4 ± 5.4%). The opposite trend was observed with the mean percentage area occupied by Aβ40 with type 2 (n = 33, 2.9 ± 3.4%) having significantly more (P = 0.009) than type 1 (n = 21, 1.8 ± 4.1%) cases. Moreover, 5 of 21 type 1 cases had Aβ40 levels >2% (1 of 21 >3%), whereas 11 of 33 cases with type 2 histology had Aβ40 levels >2% (9 of 33 >3%). Thus although only 1 of 21 cases with type 1 histology had high (ie, >3%) levels of Aβ40, 9 of 33 cases with type 2 histology had Aβ40 levels above this (chi-square = 4.3; P < 0.05). Thus there was an overall tendency for Aβ40 deposition to be greater, and to occur more frequently in type 2 histology cases, although this association was far from perfect. The differences in Aβ40 and Aβ42(43) deposition between type 1 and 2 histologies were independent of ApoE gene effects as similar numbers of ε4 allele bearers were present in each group (type 1, 6 of 21; type 2, 8 of 31; chi-square = 0.05; P > 0.05). Indeed, among those cases with type 2 histology, the average rating for amyloid angiopathy in bearers of ε4 allele (2.5) was similar to that in nonbearers (2.54).
Changes in the Cerebellum
We next examined the patterns of histological change within the cerebellum of 48 of the 54 cases where this region was also available. Aβ deposition, both as plaques and as amyloid angiopathy, was seen in all cases, although only trace amounts of amyloid angiopathy were seen in two cases. Again, two distinct histological patterns of Aβ deposition were seen.
Type 1 Histology
In BC05-immunostained sections either a moderate number of, or many, diffuse plaques were seen in the molecular layer, some of which contained areas where the amyloid was more compacted (Figure 6a) ▶ . Cored plaques, similar to those typically seen in the cerebral cortex, were also occasionally seen in the molecular layer. In most instances a few or a moderate number of cored, or more often irregular-shaped, coarse amyloid deposits were usually present in the Purkinje cell layer or granule cell layer (Figure 6a) ▶ . Amyloid angiopathy was mild, moderate, or severe, sometimes affecting intracortical, as well as leptomeningeal, vessels (Figure 6a) ▶ and occasionally with dyshoric change.
Figure 6.
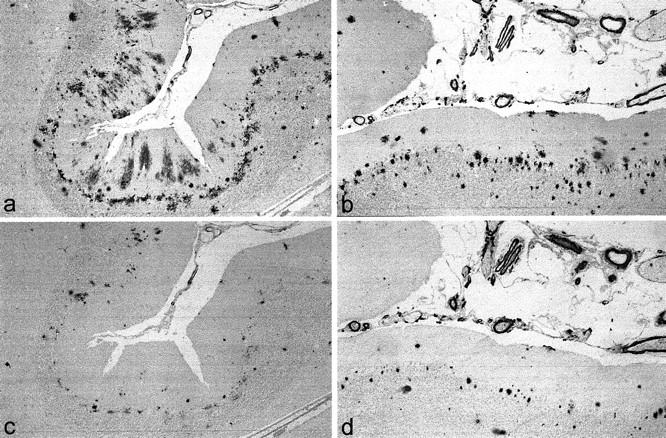
Type 1 and type 2 histologies in cerebellum in PS-1 AD. In type 1 histology (for example, case 12 with M146I mutation), there are numerous deposits of Aβ42(43) in the molecular layer in the form of diffuse plaques and (in some instances, as illustrated here) cored deposits in the Purkinje cell layer (a). Aβ40 deposits are sparse and confined to the cored deposits, and amyloid angiopathy is mild (c). In type 2 histology (for example, case 48 with L286V mutation), there are numerous deposits of Aβ40 and Aβ42(43) but only in the form of cored plaques, chiefly within the Purkinje cell layer, and amyloid angiopathy is severe (b and d). a and b: BC05 immunoperoxidase-hematoxylin. c and d: BA27 immunoperoxidase-hematoxylin.
In BA27-immunostained sections, the diffuse plaques in the molecular layer were nearly always unstained with only rare patches of immunoreaction in some of the more compacted regions (Figure 6b) ▶ . The cored plaques in the Purkinje and granule cell layers were nearly always BA27 immunoreactive, usually strongly so (Figure 6b) ▶ . Amyloid angiopathy distribution was as seen in BC05 immunoreaction, although usually more strongly staining (Figure 6b) ▶ .
Type 2 Histology
In BC05-immunostained sections there were usually fewer diffuse plaques (than in type 1 histology) (Figure 6c) ▶ and often none at all. Well-defined cored plaques were always present in all layers and in many instances were much more numerous than in cases with type 1 histology, especially within the granule cell layer. Coarse, irregular deposits were more common than the discrete cored plaques in some instances and these were particularly numerous within Purkinje and granule cell layers (Figure 6c) ▶ .
In BA27-immunostained sections only rare patches of immunoreaction were seen in compacted regions of the diffuse plaques (Figure 6d) ▶ . However, the cored and coarse deposits in the molecular, Purkinje (Figure 6d) ▶ , and granule cell layers were always strongly immunoreactive. Amyloid angiopathy was generally more severe (Figure 6, c and d) ▶ . In most instances intracortical, as well as leptomeningeal, vessels were affected, sometimes with dyshoric change.
The distribution of cerebellar type 1 and type 2 histologies within individual cases with respect to mutation site is given in Figure 5 ▶ . Fourteen of 24 cases with type 1 histology occurred before codon 200 whereas 22 of 24 cases with type 2 histology occurred beyond codon 200. These distributions of histological subtype either side of codon 200 were again, in both instances, highly significantly different (chi-square = 13.5; P < 0.001).
Correlations between type 1 and type 2 histologies in frontal cortex and cerebellum (Figure 5) ▶ in the 48 cases in which both regions were available for study revealed a coincidence of histological type in 42 of the cases. There was only one case (case 12 with M146I mutation) with type 1 histology in the frontal cortex that did not also have type 1 histology in the cerebellum. Conversely, there were five cases (two with G209V mutation and three with E280A mutation) with type 2 histology in the frontal cortex that did not also have type 2 histology in the cerebellum.
Discussion
In this present study we have shown that the histopathological profile of Alzheimer-type changes in cases of PS-1 AD is not uniform, with two distinct phenotypes being present. The type 1 pattern of histology in frontal cortex and cerebellum is entirely typical of that usually seen in sporadic AD, 26 whereas the type 2 histology is unusual and is distinguished mainly from type 1 histology by the excessive amyloid angiopathy both in the cerebral cortex and cerebellum. Although average plaque Aβ40 levels, and proportion of cases with high plaque Aβ40, were greater in type 2 cases, this is not a good discriminant of histological subtype as the severity of amyloid angiopathy. Many type 2 cases with severe amyloid angiopathy show low (normal) levels of plaque Aβ40.
It is common, indeed usual, to find marked anatomical variations in the distribution and severity of the characteristic pathological changes of AD (ie, amyloid plaques, amyloid angiopathy, and neurofibrillary tangles) throughout the brain. However, because the histological distinctions seen in the frontal cortex also occur in the cerebellum of the same cases, we feel that this pattern of pathology would be similarly mirrored in other topographical brain regions. Unfortunately, no other brain regions were available to us for this present study, and clearly this is an issue that will require further study.
A severe involvement of the cerebellum in PS-1 AD by Aβ deposition (plaque formation) has been noted on previous occasions in respect of particular mutations (eg, E280A, 17 I143T, 31 M139V, 16,32,33 G209V 16,33,34 ). Findings reported here are consistent with these previous descriptions, although this present study has extended the range of PS-1 mutations examined, allowing comparisons to be made between many more of the mutations associated with this form of AD. In sporadic AD, cored and coarse Aβ deposits in the cerebellum, of the kind described here in PS-1 AD, are rare. 35-38 However, the high presence of these is not specific to PS-1 AD and such deposits can be found in the brains of elderly patients with Down’s syndrome 35-37,39-41 and AD because of APP717 mutation. 42 The observation 17 of an especially high PS-1 mRNA expression in PS-1 AD (E280A) compared to sporadic AD may have relevance to the unusually severe cerebellar pathology seen in this particular mutation, and perhaps others. However, it may just be that PS-1 mutations, like the APP717 mutations and the trisomy of Down’s syndrome, have such a devastating effect on the brain that the pathology progresses more severely into peripherally affected regions, like the cerebellum, when compared to sporadic AD.
Broadly speaking, the two histological phenotypes in cerebral cortex and cerebellum are associated with mutations grouping toward opposite ends of the PS-1 gene structure. The type 1 histological profile is generally seen in cases with mutations extending up to codon 200, with type 2 profiles following codon 200. However, some overlap does occur. For example, in G209V, A246E, and E280A mutations, there are cases with type 1 or type 2 histology. Although the eight cases with E280A mutations were spread between several families, all cases with A246E or G209V mutations were from the same kindreds. Furthermore, the M146I mutation is associated with type 2 histology, when other mutations at the same codon (M146L and M146V) produce type 1 histology. This implies that there may perhaps be other (genetic) modifiers that act in concert with PS-1 to determine the histological phenotype, although possession of ApoE ε4 allele does not seem to be instrumental in this respect.
There are other mutations in PS-1 gene also associated with AD phenotype. In at least four pedigrees 43-48 there is a functional deletion of exon 9 (ΔE9) that occurs either because of missense mutations that result in splice acceptor site changes 44-46 or as a result of a 4.5-kb deletion 47,48 within exon 9. Although these mutational events lie beyond codon 200 in the PS-1 gene, they are not associated with the type 2 histology, as above. These exon 9 deletion cases are characterized histologically by very large, rounded plaques within the frontal cortex, known as “cotton wool” plaques. 43,48,49 These are composed of both Aβ40 and Aβ42(43) and are relatively free from neuritic changes and glial cell components, and usually devoid of a compact amyloid core. 49 In the cerebellum the plaques are almost entirely of a compact type, again composed of Aβ40 and Aβ42(43) with only a few diffuse Aβ42(43)-containing plaques. 49 The amount of Aβ40, and the ratio between Aβ40 and Aβ42(43) in frontal cortex, is higher than that seen in other cases of AD because of different PS-1 mutations, or in cases of sporadic AD, of similar ApoE genotype. 49 Amyloid angiopathy in frontal cortex and cerebellum is highly variable. 49 This kind of histological picture was not seen in any of the PS-1 cases studied here, even in those (eg, E280A, L286V) in which the mutation is located close to the splice acceptor site at codons 289/290. Furthermore, these exon 9 deletion cases often present clinically with a spastic paraparesis. 43,48 To our knowledge, none of the PS-1 cases studied here displayed such clinical features. Hence, the exon 9 deletion cases would seem to present a separate distinct histological (and clinical) subtype, despite the mutational events that cause such changes being located after codon 200.
How these tissue variations in Aβ40 and Aβ42(43) deposition in the form of plaques, or the presence and extent of amyloid angiopathy, producing two clearly distinguishable histological profiles might relate to PS-1 protein structure or mutant PS-1 protein topology is unclear. Possession of ApoE ε4 allele, which in the cerebral cortex is associated with the formation of cored plaques and the deposition of Aβ40 in sporadic AD, 27,29 does not seem to be implicated. ApoE ε4 allele frequency was similar in cases with type 1 (12.5%) and type 2 (9.4%) histologies, neither differing from population control data. Others 50 have reported the ε4 allele frequency to be normal in PS-1 AD.
However, because the type 1 profile has more Aβ42(43) and is associated with an earlier age at onset and shorter disease duration, these observations have important implications for the amyloid cascade hypothesis. 51 The data imply that increasing Aβ42(43) deposition accelerates the disease process. Mutations favoring a heavy Aβ42(43) deposition are spread (in a strict linear sense) throughout the entire protein length without displaying any obvious clustering. Indeed, mutations associated with a heavy deposition can occur at, or adjacent to, the same codon (eg, codon 139) as the lighter depositing ones. It is therefore possible that even subtle differences in amino acid substitution at any one codon might translate into more dramatic alterations in the topology (folding characteristics) of the PS-1 protein and γ-secretase activity. It is possible that the variations in mutational effect we have observed here result from more aggressive mutations (ie, in terms of Aβ42(43) deposition) spatially clustering around the active site of PS-1. Furthermore, the observation that some mutations are more aggressive than others has clear implications for the transgenic modeling of PS-1 linked AD. Transgenic mice containing PS-1 mutations, used in previous studies 8-11 have mutations that are relatively mild in their Aβ42(43) promoting ability compared to some of the others reported here.
The molecular basis for the mutational clustering (ie, downstream of codon 200) for amyloid angiopathy is difficult to explain, and why this should differ from that pattern favoring Aβ42(43) deposition as plaques is unclear. However, it is known that PS-1 is involved in Notch signaling. 52,53 Transgenic mice lacking PS-1 or Notch, or mice homozygous for a processing-deficient allele of Notch 1, have altered vascularization of the yolk sac. 54 Several other lines of evidence tie Notch signaling with vascular development. The Notch ligand Dll4 and Notch 4 are expressed in the vascular endothelium. 55,56 Furthermore, mice deficient for both Notch 1 and Notch 4 have severe defects in angiogenic vascular remodeling, indicating an essential role for Notch signaling in vascular morphogenesis and remodeling. 57 Moreover, mutations in Notch 3 result in CADASIL, a disease with a nonamyloid angiopathy. 58 Collectively, these observations raise the possibility that PS-1 mutations associated with AD may act through two separate, although complimentary, mechanisms. Firstly, they increase the amount of Aβ42(43) produced and deposited, and secondly, they effect Notch signaling resulting in a breakdown of the vascular epithelium. This, in turn, may result in plasma Aβ (especially Aβ40) leaking into brain tissue, seeding on pre-existing local deposits of Aβ42(43), and producing the large plaques we have seen around such vessels and promoting the amyloid angiopathy itself. It should be noted that the proposed functionally important residues (ie, the aspartyl residues of the active site and the PS-1 cleavage site) are all located in the region of PS-1 where mutations are associated with severe amyloid angiopathy (ie, in type 2 histology cases). Nevertheless, whatever the ultimate reasons for the differing histological profiles in PS-1 AD, they do not contribute to disease progression because the type 2 cases are seemingly less aggressive, at least in terms of time of onset of illness and its duration.
The PS-1 gene consists of 11 exons and encodes a primary 2.7-kb transcript that is translated into a 467-amino acid, 43- to 50-kd, holoprotein. 59,60 The holoprotein is normally processed into 17-kd C-terminal fragments and 27- to 28-kd N-terminal fragments, cleavage occurring at, or around, amino acid 298 (ie, within the proximal part of the hydrophilic loop). 28 Both C-terminal fragments and N-terminal fragments of PS-1 form part of a larger, stable complex that contains β-catenin. 61-65 It is clear that very little holoprotein is normally present within the cell, this being rapidly cleaved by a proposed protease, termed presenilase. 28 It has recently been shown that full-length PS-1 is a zymogen and requires cleavage to activate γ-secretase activity. 23 However, it has also been demonstrated that the delta 9 deletion mutated PS-1 (which cannot be cleaved) also possesses γ-secretase activity. 23 It remains to be established whether any of the point mutations reported here also confer γ-secretase activity on full length PS-1. Other recent work 66 implies that PS-1 mutations may affect the cleavage of APP at its amino terminus, thereby modulating the activity of β-secretase.
In summary, therefore, it seems that in PS-1-linked AD there are two distinct histological profiles and that these result from the location of the particular mutation. Cases of type 1 histology show a greater Aβ42(43) deposition and have an earlier age at onset and shorter disease duration. The extent of Aβ deposition across cases does not relate to the mutational location in a strict linear sense, but cases showing high levels of deposition may cluster around (and be a marker of) the putative active site of PS-1. However, of great potential importance are the findings that the extent of amyloid angiopathy is related to mutational position, and this might involve a PS-1-mediated dysfunction of Notch signaling. Finally, we demonstrate that altered Aβ processing in a cell culture model of mutant PS-1 correlates with the relevant pathological changes in humans.
Acknowledgments
We thank all of the members of the Familial Alzheimer’s Disease Pathology Study Group and their colleagues who assisted in the collection of the pathological specimens and provided supporting clinical and genetic information.
Footnotes
Address reprint requests to David M. A. Mann, Clinical Neuroscience Research Group, Department of Medicine, Stopford Building, University of Manchester, Oxford Rd., Manchester M13 9PT, United Kingdom. E-mail david.mann@man.ac.uk.
The Familial Alzheimer’s Disease Pathology Study Group Members are Juan Arango, Tom Bird, Christine Van Broeckhoven, William Brooks, Rosemary Brown, Nigel Cairns, Patrick Cras, David Ellison, Matti Haltia, Kunio Ii, Arne Jorgensen, Jillian Krill, Peter Lantos, Carol Lippa, Ralph Martins, David Nochlin, Daniel Pollen, Carlyn Rosenberg, Martin Rossor, and Takeshi Tabira.
References
- 1.Nishimura M, Yu G, St George-Hyslop PH: Biology of presenilins as causative molecules for Alzheimer’s disease. Clin Genet 1999, 55:219-225 [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ: The cell biology of beta-amyloid precursor protein and presenilins in Alzheimer’s disease. Trends Cell Biol 1998, 8:447-453 [DOI] [PubMed] [Google Scholar]
- 3.Sisodia SS, Kim SH, Thinakaran G: Function and dysfunction of the presenilins. Am J Hum Genet 1999, 65:7-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busciglio G, Hartmann H, Lorenzo A, Wong C, Baumann K, Sommer B, Staufenbiel M, Yankner BA: Neuronal localisation of presenilin-1 and association with amyloid plaques and neurofibrillary tangles in Alzheimer’s disease. J Neurosci 1997, 17:5101-5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levery AI, Jenkins N, Copeland N, Price DL, Sidodia SS: Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med 1997, 3:756-760 [DOI] [PubMed] [Google Scholar]
- 6.Levey AI, Heilman CJ, Lah JJ, Nash NR, Rees HD, Wakai M, Mirra SS, Rye DB, Nochlin D, Bird TD, Mufson EJ: Presenilin-1 protein expression in familial and sporadic Alzheimer’s disease. Ann Neurol 1997, 41:742-753 [DOI] [PubMed] [Google Scholar]
- 7.Elder GA, Tezapsidis N, Carter J, Shioi J, Bouras C, Li H-C, Johnson JM, Efthimipopolous S, Friedrich VL, Robakis NK: Identification and neuron specific expression of the S182/presenilin 1 protein in human and rodent brains. Exp Neurol 1996, 145:308-320 [DOI] [PubMed] [Google Scholar]
- 8.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada C-M, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS: Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron 1996, 17:1005-1013 [DOI] [PubMed] [Google Scholar]
- 9.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, Hyslop PSG, Selkoe DJ: Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med 1997, 3:67-72 [DOI] [PubMed] [Google Scholar]
- 10.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-Tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S: Increased amyloid-β42(43) in brains of mice expressing mutant presenilin. Nature 1996, 383:710-713 [DOI] [PubMed] [Google Scholar]
- 11.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada C-M, Eckman C, Younkin S, Hsiao K, Duff K: Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 1998, 4:97-100 [DOI] [PubMed] [Google Scholar]
- 12.Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ: Enhanced production and oligomerization of the 42-residue amyloid β-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem 1997, 272:7977-7982 [DOI] [PubMed] [Google Scholar]
- 13.Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J, Grünberg J, Haass C, Iwatsubo T, Obata K: The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci USA 1997, 94:2025-2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S: Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 1996, 2:864-869 [DOI] [PubMed] [Google Scholar]
- 15.Martins RN, Turner BA, Carroll RT, Sweeney D, Kim KS, Wisniewski HM, Blass JP, Gibson GE, Gandy S: High levels of amyloid-beta protein from S182 (Glu246) familial Alzheimer’s cells. Neuroreport 1995, 7:217-220 [PubMed] [Google Scholar]
- 16.Mann DMA, Iwatsubo T, Cairns NJ, Lantos PL, Nochlin D, Sumi SM, Bird TD, Poorkaj P, Hardy J, Hutton M, Prihar G, Crook R, Rossor MN, Haltia M: Amyloid β protein (Aβ) deposition in chromosome 14-linked Alzheimer’s disease: predominance of Aβ42(43). Ann Neurol 1996, 40:149-156 [DOI] [PubMed] [Google Scholar]
- 17.Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Martinez A, Madrigal L, Hincapie L, Arango LJC, Anthony DG, Koo EH, Goate AM, Selkoe DJ, Arango VJC: The E280A presenilin 1 Alzheimer mutation produces increased Aβ42 deposition and severe cerebellar pathology. Nat Med 1996, 2:1146-1150 [DOI] [PubMed] [Google Scholar]
- 18.Ishii K, Ii K, Hasegawa T, Shoji S, Doi A, Mori H: Increased Aβ42(43)-plaque deposition in early-onset familial Alzheimer’s disease brains with the deletion of exon 9 and the missense point mutation (H163R) in the PS-1 gene. Neurosci Lett 1997, 228:17-20 [DOI] [PubMed] [Google Scholar]
- 19.Gómez-Isla T, Wasco W, Pettingell WP, Gurubhagavatula S, Schmidt SD, Jondro PD, McNamara M, Rodes LA, DiBlasi T, Growdon WB, Seubert P, Schenk D, Growdon JH, Hyman BT, Tanzi RE: A novel presenilin-1 mutation increases β-amyloid and neurofibrillary changes. Ann Neurol 1997, 41:809-813 [DOI] [PubMed] [Google Scholar]
- 20.Romero I, Jorgensen P, Bolwig G, Fraser PE, Rogaeva E, Mann D, Havsager A-M, Jorgensen AL: A presenilin-1 Thr116Asn substitution in a family with early onset Alzheimer’s disease. Neuroreport 1999, 10:2255-2260 [DOI] [PubMed] [Google Scholar]
- 21.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ: Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398:513-517 [DOI] [PubMed] [Google Scholar]
- 22.De Strooper B, Saftig B, Craesserts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuwen F: Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391:387-390 [DOI] [PubMed] [Google Scholar]
- 23.Li Y-M, Lai M-T, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi X-P, Yin K-C, Shafer JA, Gardell SJ: Presenilin-1 is linked to γ-secretase activity in the detergent solubilized state. Proc Natl Acad Sci USA 2000, 97:6138-6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y-M, Xu M, Lai M-T, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelli JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi X-P, Yin K-C, Shafer JA, Gardell SJ: Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 2000, 405:689-693 [DOI] [PubMed] [Google Scholar]
- 25.Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski BL, Donkor IO, Selkoe DJ: Peptidomimetic probes and molecular modelling suggests that Alzheimer’s γ-secretase is an intramembrane-cleaving aspartyl protease. Biochemistry 2000, 38:4720-4727 [DOI] [PubMed] [Google Scholar]
- 26.Iwatsubo T, Odaka N, Suzuki N, Mizusawa H, Nukina N, Ihara Y: Visualization of Aβ42(43)-positive and Aβ40-positive senile plaques with specific Aβ monoclonal antibodies: evidence that an initially deposited species is Aβ1–42(43). Neuron 1994, 13:45-53 [DOI] [PubMed] [Google Scholar]
- 27.Mann DMA, Iwatsubo T, Pickering-Brown SM, Owen F, Saido TC, Perry RH: Preferential deposition of amyloid β protein (Aβ) in the form Aβ40 in Alzheimer’s disease is associated with a gene dosage effect of the apolipoprotein E E4 allele. Neurosci Lett 1997, 221:81-84 [DOI] [PubMed] [Google Scholar]
- 28.Podlisny MB, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, Diehl T, Levesque G, Fraser P, Haass C, Koo EHM, Seubert P, St George-Hyslop P, Teplow DB, Selkoe DJ: Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol Dis 1997, 3:325-337 [DOI] [PubMed] [Google Scholar]
- 29.Gearing M, Mori H, Mirra SS: Aβ peptide length and apolipoprotein E genotype in Alzheimer’s disease. Ann Neurol 1996, 39:395-399 [DOI] [PubMed] [Google Scholar]
- 30.Murayama O, Tomita T, Nihonmatsu N, Murayama M, Sun X, Honda T, Iwatsubo T, Takashima A: Enhancement of amyloid β 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer’s disease. Neurosci Lett 1999, 265:61-63 [DOI] [PubMed] [Google Scholar]
- 31.Martin JJ, Gheuens J, Bruyland M, Cras P, Vandenberghe A, Masters CL, Beyreuther K, Dom R, Ceuterick C, Lübke U, Van Heuverswijn H, De Winster G, Van Broeckhoven C: Early-onset Alzheimer’s disease in 2 large Belgian families. Neurology 1991, 41:62-68 [DOI] [PubMed] [Google Scholar]
- 32.Haltia M, Viitanen M, Sulkava R, Ala-Hurula V, Poyhonen M, Goldfarb L, Brown P, Levy E, Houlden H, Crook R, Goate A, Clark R, Korenblat K, Pandit S, Keller HD, Lilius L, Liu L, Axelman K, Forsell L, Winblad B, Lannfelt L, Hardy J: Chromosome 14-encoded Alzheimer’s disease: genetic and clinicopathological description. Ann Neurol 1994, 36:362-367 [DOI] [PubMed] [Google Scholar]
- 33.Mann DMA, Iwatsubo T, Snowden JS: Atypical amyloid (Aβ) deposition in the cerebellum in Alzheimer’s disease: an immunohistochemical study using end-specific Aβ monoclonal antibodies. Acta Neuropathol 1996, 91:647-653 [DOI] [PubMed] [Google Scholar]
- 34.Lampe TH, Bird TD, Nochlin D, Nemens E, Risse SC, Sumi SM, Koerker R, Leaird B, Wier M, Raskind MA: Phenotype of chromosome 14-linked familial Alzheimer’s disease in a large kindred. Ann Neurol 1994, 36:368-378 [DOI] [PubMed] [Google Scholar]
- 35.Cole G, Williams P, Alldrick D, Singrao S: Amyloid plaques in the cerebellum in Alzheimer’s disease. Clin Neuropathol 1989, 4:188-191 [PubMed] [Google Scholar]
- 36.Cole G, Neal JW, Singrao SK, Jasani B, Newman GR: The distribution of amyloid plaques in the cerebellum and brain stem in Down’s syndrome and Alzheimer’s disease: a light microscopical analysis. Acta Neuropathol 1993, 85:542-552 [DOI] [PubMed] [Google Scholar]
- 37.Mann DMA, Jones D, Prinja D, Purkiss MS: The prevalence of amyloid (A4) protein deposits within the cerebral and cerebellar cortex in Down’s syndrome and Alzheimer’s disease. Acta Neuropathol 1990, 80:318-327 [DOI] [PubMed] [Google Scholar]
- 38.Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J: Spectrum of morphological appearance of amyloid deposits in Alzheimer’s disease. Acta Neuropathol 1989, 78:337-347 [DOI] [PubMed] [Google Scholar]
- 39.MacKenzie IRA, McKelvie PA, Beyreuther K, Masters CL: βA4 amyloid protein deposition in the cerebellum in Alzheimer’s disease and Down’s syndrome. Dementia 1991, 2:237-242 [Google Scholar]
- 40.Mann DMA, Younis N, Jones D, Stoddart RW: The time course of pathological events concerned with plaque formation in Down’s syndrome with particular reference to the involvement of microglial cells. Neurodegeneration 1992, 1:201-215 [Google Scholar]
- 41.Mann DMA, Iwatsubo T: Diffuse plaques in the cerebellum and corpus striatum in Down’s Syndrome contain amyloid β protein (Aβ) only in the form of Aβ42(43). Neurodegeneration 1996, 5:115-120 [DOI] [PubMed] [Google Scholar]
- 42.Mann DMA, Iwatsubo T, Ihara Y, Cairns NJ, Lantos PL, Bogdanovic N, Lannfelt L, Winblad B, Maat-Schieman MLC, Rossor MN: Predominant deposition of amyloid-β42(43) in plaques in cases of Alzheimer’s disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am J Pathol 1996, 148:1257-1266 [PMC free article] [PubMed] [Google Scholar]
- 43.Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, Farrer M, Hutton M, Lincoln S, Hardy J, Gwinn K, Somer NM, Paetau A, Kalimo H, Ylikovski R, Poyhonen M, Kucera S, Haltia M: A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med 1998, 4:452-455 [DOI] [PubMed] [Google Scholar]
- 44.Kwok JBJ, Taddei K, Hallupp M, Fisher C, Brooks WS, Broe GA, Hardy J, Fulham MJ, Nicholson GA, Stell R, St George Hyslop PH, Fraser PE, Kakulas B, Clarnette R, Relkin N, Gandy SE, Schofield PR, Martins RN: Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzheimer’s disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. Neuroreport 1997, 8:1537-1542 [DOI] [PubMed] [Google Scholar]
- 45.Perez-Tur J, Froelich S, Prihar G, Crook R, Baker M, Duff K, Wragg M, Busfield F, Lendon C, Clark RF, Roques P, Fuldner RA, Johnston J, Cowburn R, Forsell C, Axelman K, Lilius L, Houlden H, Karran E, Roberts GW, Rossor M, Adams MD, Hardy J, Goate A, Lannfelt L, Hutton M: Identification of a mutation in Alzheimer’s disease which destroys a splice acceptor site in the presenilin-1 gene. Neuroreport 1996, 7:204-207 [Google Scholar]
- 46.Prihar G, Verkkoniemi A, Perez-Tur J, Crook R, Lincoln S, Houlden H, Somer M, Paetau A, Kalimo H, Grover A, Myllykangas L, Hutton M, Hardy J, Haltia M: Alzheimer disease PS-1 exon 9 deletion defined. Nat Med 1999, 5:1090. [DOI] [PubMed] [Google Scholar]
- 47.Sato S, Kamino K, Miki T, Doi A, Ii K, St George Hyslop PH, Ogihara T, Sakaki Y: Splicing mutation of presenilin 1 gene for early-onset familial Alzheimer’s disease. Hum Mutat 1998, Suppl:S91–S94 [DOI] [PubMed]
- 48.Verkkoniemi A, Somer M, Rinne JO, Myllykangas L, Crook R, Hardy J, Viitanen M, Kalimo H, Haltia M: Variant Alzheimer’s disease with spastic paraparesis. Clinical characterisation. Neurology 2000, 54:1103-1109 [DOI] [PubMed] [Google Scholar]
- 49.Mann DMA, Takeuchi A, Sato S, Cairns NJ, Lantos PL, Rossor MN, Haltia M, Kalimo H, Iwatsubo T: Cases of Alzheimer’s disease due to deletion of exon 9 of the presenilin-1 gene show an unusual but characteristic β amyloid pathology known as “cotton wool” plaques. Neuropathol Appl Neurobiol (in press) [DOI] [PubMed]
- 50.Van Broeckhoven C, Backhovens H, Cruts M, Martin JJ, Crook R, Houlden H, Hardy J: APOE genotype does not modulate age of onset in families with chromosome 14 encoded Alzheimer’s disease. Neurosci Lett 1994, 169:179-180 [DOI] [PubMed] [Google Scholar]
- 51.Hardy J: Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci 1997, 20:154-159 [DOI] [PubMed] [Google Scholar]
- 52.Berezovska O, Xia MQ, Hyman BT: Notch is expressed in adult brain, and is coexpressed with presenilin-1 and is altered in Alzheimer’s disease. J Neuropathol Exp Neurol 1998, 57:738-745 [DOI] [PubMed] [Google Scholar]
- 53.Capell A, Steiner H, Romig H, Keck S, Baader M, Grim MG, Baumeister H: Presenilin-1 differentially facilitates endoproteolysis of beta-amyloid and Notch. Nat Cell Biol 2000, 2:205-211 [DOI] [PubMed] [Google Scholar]
- 54.Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA: Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature 2000, 405:966-970 [DOI] [PubMed] [Google Scholar]
- 55.Shirayoshi Y, Yuasa Y, Suzuki T, Sugaya K, Kawase E, Ikemura T: Proto-oncogene of int-3, a mouse Notch homologue, is expressed in endothelial cells during early embryogenesis. Genes Cells 1997, 2:213-224 [DOI] [PubMed] [Google Scholar]
- 56.Shutter JR, Scully S, Fan W, Richards WG, Kitajewski J, Deblandre S, Stark KL: Dll4, a Notch ligand expressed in arterial endothelium. Genes Dev 2000, 14:1313-1318 [PMC free article] [PubMed] [Google Scholar]
- 57.Krebs LT: Notch signalling is essential for vascular morphogenesis in mice. Genes Dev 2000, 14:1343-1352 [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas NJ, Morris CM, Scaravilli F, Johansson J, Rossor M, De Lar H, Nicoll J, Blank C, Coulthard A, Bushby K, Ince PG, Burn D, Kalaria RJ: Hereditary vascular dementia linked to Notch 3 mutations. CADASIL. Ann NY Acad Sci 2000, 903:293-298 [DOI] [PubMed] [Google Scholar]
- 59.Clark RF, Hutton M, Fuldner RA, Froelich S, Karran B, Talbot C, Crook R, Lendon C, Prihar G, He C, Korenblat K, Martinez A, Wragg M, Busfield F, Behrens MI, Myers A, Norton J, Morris J, Mehta N, Pearson C, Lincoln S, Baker M, Duff K, Zehr C, Perez-Tur J, Houlden H, Ruiz A, Ossa F, Lopera M, Arcos L, Madrigal L, Collinge J, Humphreys C, Ashworth T, Sarner S, Fox N, Harvey R, Kennedy A, Roques P, Cline RT, Philips CA, Venter JC, Forsell L, Axelman L, Lilius J, Johnston R, Cowburn M, Vitanen M, Winblad B, Kosik K, Haltia M, Poyhonen M, Dickson D, Mann D, Neary D, Snowden J, Lantos P, Lannfelt L, Rossor M, Roberts GW, Adams MD, Hardy J, Goate A: The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet 1995, 11:219-222 [DOI] [PubMed] [Google Scholar]
- 60.Sherrington G, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH: Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375:755-760 [DOI] [PubMed] [Google Scholar]
- 61.Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C: The proteolytic fragments of the Alzheimer’s disease-associated presenilin-1 form heterodimers and occur as a 100–150-kDa molecular mass complex. J Biol Chem 1998, 273:3205-3211 [DOI] [PubMed] [Google Scholar]
- 62.Murayama M, Tanaka S, Palacino J, Murayama O, Honda T, Sun X, Yasutake K, Nihonmatsu N, Wolozin B, Takashima A: Direct association of presenilin-1 with beta-catenin. FEBS Lett 1998, 433:73-77 [DOI] [PubMed] [Google Scholar]
- 63.Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva R, Xu D, Liang Y, Duthie M, St George-Hyslop PH, Fraser PE: The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains β-catenin. J Biol Chem 1998, 273:16470-16475 [DOI] [PubMed] [Google Scholar]
- 64.Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA: Destabilization of β-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 1998, 395:698-702 [DOI] [PubMed] [Google Scholar]
- 65.Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS: Presenilin 1 interaction in the brain with a novel member of the Armadillo family. Neuroreport 1997, 8:1489-1494 [DOI] [PubMed] [Google Scholar]
- 66.Russo C, Schettini G, Saido TC, Hulette C, Lippa C, Lannfelt L, Ghetti B, Gambetti P, Tabaton M, Teller JK: Presenilin-1 mutations in Alzheimer’s disease. Nature 2000, 405:531-532 [DOI] [PubMed] [Google Scholar]