

Abstract
HIV-1 uses mononuclear phagocytes (monocytes, tissue macrophages, and dendritic cells) as a vehicle for its own dissemination and as a reservoir for continuous viral replication. The mechanism by which the host immune system clears HIV-1-infected macrophages is not understood. TRAIL may play a role in this process. TRAIL is expressed on the cell membrane of peripheral immune cells and can be cleaved into a soluble, secreted form. The plasma level of TRAIL is increased in HIV-1-infected patients, particularly those with high viral loads. To study the effect of elevated TRAIL on mononuclear phagocytes, we used recombinant human (rh) TRAIL and human monocyte-derived macrophages (MDM) as an in vitro model. Our results demonstrated rhTRAIL-induced apoptosis in HIV-1-infected MDM and inhibited viral replication, while having a reduced effect on uninfected MDM. HIV-1 infection significantly decreased Akt-1 phosphorylation; rhTRAIL exposure further decreased Akt-1 phosphorylation. Infection with a dominant-negative Akt-1 adenovirus potentiated rhTRAIL-induced apoptosis, while constitutively active Akt-1 blocked rhTRAIL-induced apoptosis in HIV-1-infected MDM. From this data we conclude the death ligand TRAIL preferentially provokes apoptosis of HIV-1-infected MDM, and the mechanism is reliant upon the inhibition of Akt-1 phosphorylation. Understanding this mechanism may facilitate the elimination of HIV-1-infected macrophages and lead to new therapeutic avenues for treatment of HIV-1 infection.
Mononuclear phagocytes (MP),3 which include circulating monocytes, tissue macrophages, and dendritic cells, are among the important cell types infected by HIV-1 (1, 2). Infected MP disseminate virus to lymph nodes where CD4+ T lymphocytes become infected, and to various tissues including the lung and CNS where they serve as viral reservoirs, instigating tissue pathology and human disease (3–6). Although both MP and CD4+ T lymphocytes are targets for HIV-1, their responses to infection differ. HIV-1 rapidly depletes CD4+ T cells but infects MP for prolonged periods measured in weeks to months (7). The introduction of antiretroviral therapy (ART) can decrease viral load and improve CD4+ T cell count in infected individuals; however, ART fails to eradicate HIV due to the persistence of viral reservoirs in MP or memory CD4+ T cells. Thus, the final pathways of virus-MP interactions in the ART era remain to be understood.
A critical component to HIV pathogenesis is host-mediated clearance of infected cells and the mechanisms involved. Viral manipulation of death cascades may facilitate evasion of immune response or resistance to treatment through manipulations in cell signaling pathways; however, various factors promote targeted cell death during the infectious processes. Recently, emerging evidence indicates TRAIL may induce cell death in the HIV-1-infected macrophage (8, 9). TRAIL is a member of the TNF superfamily and is closely related to Fas ligand (10, 11). TRAIL interacts with at least five unique receptors found on a variety of cell types. TRAIL receptor one (R1) and receptor two (R2) have death domains and induce cellular apoptosis following ligand binding (12–14). TRAIL-R3 and TRAIL-R4, however, do not possess these domains and instead act as decoy receptors (15, 16). The fifth soluble TRAIL receptor is osteoprotegerin, which appears to be exclusively involved in bone remodeling (17). Previously, we have demonstrated that TRAIL is up-regulated on the cell surface of HIV-infected or immune-activated macrophages (18, 19). Also, TRAIL has been reported to be expressed on the cell membrane of MP, NK cells, and CD4+ T lymphocytes and can be cleaved into a soluble, secreted form (20). The plasma level of TRAIL is increased to nanogram per milliliter ranges in HIV-1-infected patients, particularly those with high viral loads (21).
The signaling cascade for TRAIL in MP is unknown; however, Akt (also known as protein kinase B) phosphorylation is known to be critical for macrophage survival. Inhibition of Akt-1 phosphorylation usually results in macrophage death (22). Akt is a serine/threonine kinase with three human isoforms, Akt-1, Akt-2, and Akt-3. Akt-1 is ubiquitously expressed; Akt-2 and Akt-3 are ~82% identical with Akt-1 and have tissue-specific expression. The mechanism for activation of Akt-1 has yet to be fully characterized, but is correlated with phosphorylation of Thr308 in the catalytic domain and Ser473 at the C terminus (23). Membrane translocation is an essential step in Akt-1 activation, with the addition of an N-terminal myristoylation sequence causing Akt-1 to be constitutively active (24, 25).
In these studies, we used a monocyte-derived macrophage (MDM) primary culture system as a model to test whether TRAIL can clear HIV-1-infected MP in vitro and to determine the related signaling pathway. Our results demonstrated recombinant human (rh) TRAIL preferentially induced cell death and inhibited viral replication in HIV-1-infected MDM. Cell death was dependent upon caspase-3, −8, −9 activation. HIV-1 infection significantly decreased Akt-1 phosphorylation; rhTRAIL exposure further decreased Akt-1 phosphorylation. Dominant-negative Akt-1 potentiated rhTRAIL-induced apoptosis, while constitutively active Akt-1 blocked rhTRAIL-induced apoptosis in HIV-infected MDM. In conclusion, the results of this work provide insight into the apoptotic signaling events of TRAIL-macrophage interaction and demonstrate that TRAIL-induced apoptosis in HIV-1-infected macrophage is reliant in part, upon the inhibition of Akt-1 phosphorylation.
Materials and Methods
Isolation and culturing of primary monocytes
Human monocytes were recovered from PBMC of HIV-1, HIV-2, and hepatitis B virus-seronegative donors after leukapheresis and purified by countercurrent centrifugal elutriation (26). Monocytes were cultured in DMEM (Sigma-Aldrich) supplemented with 10% heat-inactivated human serum, gentamicin (50 μg/ml), ciprofloxacin (10 μg/ml), and M-CSF (1000 U/ml; a gift from Genetics Institute, Cambridge, MA). Monocytes were allowed to differentiate for 7 days at which time they were considered MDM. All human subject studies were performed in full compliance with the University of Nebraska Medical Center and National Institutes of Health (NIH) ethical guidelines.
Infection of MDM
Seven days after plating, MDM were infected with HIV-1 virus at a multiplicity of infection (MOI) of 0.1 virus/target cell (26). Briefly, viral stocks were diluted 1/15 in medium for overnight incubation with MDM. On the second day, medium was removed and substituted with MDM culture medium that was half-exchanged every 2 days (26). Stock virus was screened for mycoplasma and endotoxin using hybridization and Limulus amebocyte lysate assays, respectively.
Measurements of reverse transcriptase (RT) activity
RT activity was determined in triplicate samples of cell culture fluids. For this assay, 10 μl of supernatant was incubated in a reaction mixture of 0.05% Nonidet P-40, 10 μg of poly(A)/ml, 0.25 μg of oligo(dT)/ml, 5 mM DTT, 150 mM KCl, 15 mM MgCl2, and [3H]TTP in Tris-HCl buffer (pH 7.9) for 24 h at 37°C. Radiolabeled nucleotides were precipitated with cold 10% trichloroacetic acid on paper filters in an automatic cell harvester and washed with 95% ethanol. Radioactivity was estimated by liquid scintillation spectroscopy (5).
Western blot analysis
Cell lysates from MDM were prepared with M-PER Mammalian Protein Extraction Buffer (Pierce). Mitochondria and cytosol fraction were prepared from MDM using the mitochondria isolation kit according to the manufacturer’s instruction (Pierce). Protein concentration was determined using the BCA Protein Assay kit (Pierce). Protein (10 μg) was electrophoresed on precast 10% or 4–20% SDS-PAGE (Bio-Rad) and transferred to an Immuno-Blot polyvinylidene difluoride membrane (Bio-Rad). Total Akt-1, phospho-Akt-1, and β-actin proteins were detected using anti-Akt-1 (1/1,000 dilution; Cell Signaling Technology), anti-phospho-Akt-1 at Ser473 (1/500 dilution; Cell Signaling Technology), and anti-β-actin (1/10,000 dilution; Sigma-Aldrich) Abs. Cytochrome c and voltage-dependent anion channel (VDAC) proteins were detected using anti-cytochrome c (1/1,000 dilution; BD Biosciences), and anti-VDAC (1/1,000 dilution; Abcam) Abs. Caspase-8, caspase-9, and caspase-3 proteins were detected using anti-caspase-8 (1/500 dilution; BD Biosciences), anti-caspase-9 (1/500 dilution; R&D Systems), and anti-caspase-3 (1/500 dilution; Cell Signaling Technology) Abs. Membranes were treated overnight with primary Ab at 4°C followed by a HRP-ligand secondary anti-rabbit (1/5,000 dilution; Cell Signaling Technology) or anti-mouse (1/5,000 dilution; Cell Signaling Technology) Ab for 1 h at room temperature. Ag-Ab complexes were visualized by ECL (Amersham Biosciences) and captured with CL-X Posur Film (Pierce). For data quantification, the films were scanned with a CanonScan 9950F scanner; the acquired images were then analyzed on a Macintosh computer using the public domain NIH image program (developed at the U.S. NIH and available on the internet at 〈http://rsb.info.nih.gov/nih-image/〉).
Assessment of cell viability and apoptosis
MDM were plated in poly-D-lysine-coated 48-well plates at a density of 5 × 105 cells/well or 24-well plates with 1.1 × 106 cells/well. Cells were treated with various concentrations of rhTRAIL (R&D Systems) for 24 h in serum-free medium (neuronal basal with 5 mM glutamine, 0.01%BSA, penicillin-streptomycin). Additional cells were pretreated with 20 μM caspase inhibitors or FMK control (R&D Systems) for 2 h, and then treated with rhTRAIL (50 ng/ml) for 24 h. Each condition was tested in triplicate. Cell cultures were maintained for the indicated treatment times. A colorimetric MTT assay was then performed to measure the cell viability as previously described (27). To assess apoptosis, cultures were lysed and assayed by Cell Death Detection ELISA (Roche), following the manufacturer’s instructions (27).
Adenoviral infection
Adenoviral constructs. Replication-defective adenovirus vectors expressing dominant-negative Akt-1 (Ad-DNAkt-1) and constitutively active Akt-1 (Ad-myrAkt-1) were generated as described (28) and provided by K. Walsh (Boston University School of Medicine, Boston, MA). The constitutively active Akt-1 construct has the c-src myristoylation sequence fused in-frame to the N terminus of the HA-Akt-1 (wild-type) coding sequence. The dominant-negative mutant of Akt-1 construct has a hemagglutinin tag at the N terminus and three amino acid substitutions at lysine 179, threonine 308, and serine 473 to alanine. Adenoviral constructs were amplified in 293 cells and purified by ultracentrifugation through a CsCl gradient. MDM were infected with recombinant adenovirus at ~90% confluency in serum-free DMEM for 2 h and then incubated for 48 h in a growth medium before the treatment. Infection efficiency is close to 70% as determined by the GFP expression. At 48-h postinfection with adenovirus, MDM were treated with rhTRAIL (50 ng/ml) for another 24 h. For adenovirus and HIV-1 double infection, MDM were infected with adenovirus for 24 h, then infected with HIV-1; apoptosis assays were performed 3 days after HIV-1 infection.
FACS analysis
Monocytes were cultured for 7 days at a density of 5 × 106 cells/well in 6-well plates in the presence of M-CSF. MDM were then infected with HIV-1 and/or treated with rhTRAIL (50 ng/ml). Twenty-four hours after treatment, cells were resuspended in 3% FBS-PBS and incubated with one of TRAIL receptor Abs (TRAIL-R1 Ab (BD Biosciences; 2 μg/ml), TRAIL-R2 Ab (Chemicon International; 2 μg/ml), TRAIL-R3, -R4 Ab (Santa Cruz Biotechnology; 2 μg/ml), or isotype control IgG (BD Biosciences; 2 μg/ml), and CD14-PE (BD Pharmingen; 2 μg/ml) for 30 min at 4°C. Cells were then washed with 3% FBS/PBS and incubated with anti-goat-FITC (BD Biosciences; 1 μg/ml), or anti-rabbit-FITC (BD Biosciences; 1 μg/ml) for another 30 min at 4°C. Cells were washed with 3% FBS/PBS and resuspended in 4% paraformaldehyde. Expression of cell surface Ags was determined by immunofluorescence flow cytometry and analyzed with CellQuest software. At least 10,000 cells were analyzed per sample.
Statistical tests
Data was analyzed as means ± SD of the mean (SD) unless specified. The data was evaluated statistically by the ANOVA, followed by the Tukey test for paired observations. Significance was considered to be <0.05 unless specified. To account for any donor-specific differences, all experiments were performed with at least three donors. All assays were performed at least three times with triplicate or quadruplicate determinations for each time.
Results
rhTRAIL induces apoptosis in HIV-1-infected macrophage
To evaluate whether TRAIL kills HIV-1-infected macrophage in our primary culture system, MDM were infected with HIV-1ADA for 7 days and then treated with doses of rhTRAIL ranging from 0.01to 300 ng/ml. The percentage of viable cells, as determined by MTT assay, fell significantly in a dose-dependent manner after 24 h of treatment with rhTRAIL in the HIV-1ADA-infected group. Uninfected MDM retained high viability except in the presence of 300 ng/ml rhTRAIL (Fig. 1A). To test whether the cell loss was due to apoptosis, DNA fragmentation ELISA was used in control and HIV-1ADA-infected MDM 3 days after infection. At several time points following treatment, rhTRAIL-mediated DNA fragmentation increased significantly after 4 h of treatment in the HIV-1-infected group (50 ng/ml rhTRAIL) and was sustained through 24 h. In contrast, rhTRAIL induced a moderate increase of DNA fragmentation at 4 h on control MDM and this effect diminished continuously over the time points tested (Fig. 1B). We also tested the apoptotic effects of different concentrations of rhTRAIL (from 0.3 to 300 ng/ml) in control and HIV-1ADA-infected MDM. rhTRAIL induced up to 1.5-fold increase of DNA fragmentation in control MDM in a dose-dependent manner. However, HIV-1 infection potentiated apoptosis in MDM, with rhTRAIL inducing up to a 5-fold increase of DNA fragmentation in HIV-1-infected MDM. Thus, rhTRAIL induces apoptosis on HIV-1ADA-infected MDM, while the effect of rhTRAIL on control MDM is significantly reduced. To further confirm the apoptotic potentiating effect of HIV-1 infection, we used two macrophage-tropic HIV-1 strains HIV-1BAL and HIV-1DJV, and one dual-tropic strain HIV-189.6, all of which dramatically increased the susceptibility of MDM to apoptosis (Fig. 2A). Cells were productively infected by those viral strains as monitored by RT activity assay (Fig. 2B). HIV-1ADA represented the typical HIV-1 infection seen in all tested strains and was used thereafter, referred to as HIV-1 for convenience.
FIGURE 1.
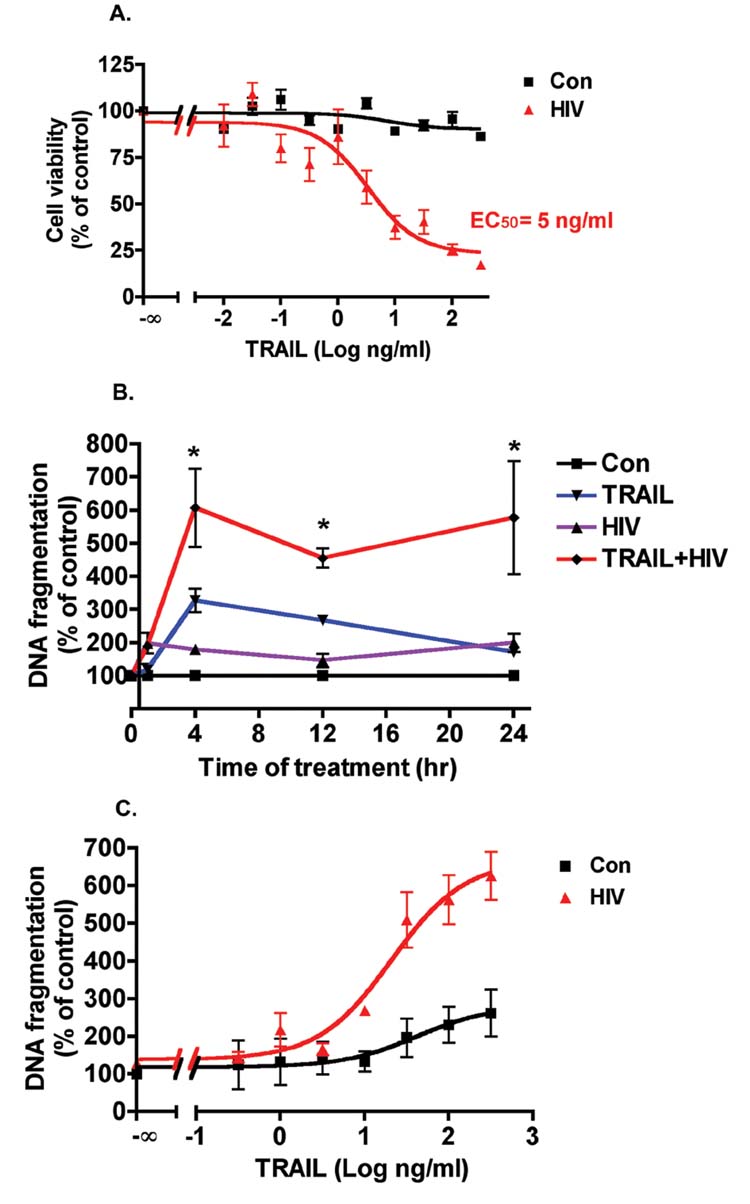
rhTRAIL preferentially induces apoptosis in HIV-1-infected macrophages. A, Human MDM were plated in 24-well plates in triplicate; HIV-1-infected MDM were infected with HIV-1ADA for 7 days. HIV-1-infected and control MDM were then incubated with doses of rhTRAIL ranging from 0.01 to 300 ng/ml for 24 h. Cell viability was determined by MTT assay. Results shown are the averages ± SEM of experiments performed in triplicate (n = 3 donors). Data were analyzed by nonlinear regression and assessed the EC50 for TRAIL in HIV-1-infected MDM as 5 ng/ml. B, Three days after HIV-1ADA infection, control or HIV-1-infected MDM were treated with rhTRAIL (50 ng/ml) at time points indicated. Apoptosis was determined by DNA fragmentation apoptosis ELISA. Results are normalized to each control in percentage and expressed as average ± SD of four experiments performed with different donors.*, A p value of <0.01 comparing HIV or TRAIL with TRAIL + HIV group. C, Human MDM were infected with HIVADA for 3 days, and then control and HIV-1-infected MDM were incubated with various concentrations of rhTRAIL (0.3–300 ng/ml) for 24 h. Apoptosis was determined by DNA fragmentation apoptosis ELISA. Results shown are nonlinear regression curves of averages ± SD of experiments performed in triplicate (n = 3 donors).
FIGURE 2.
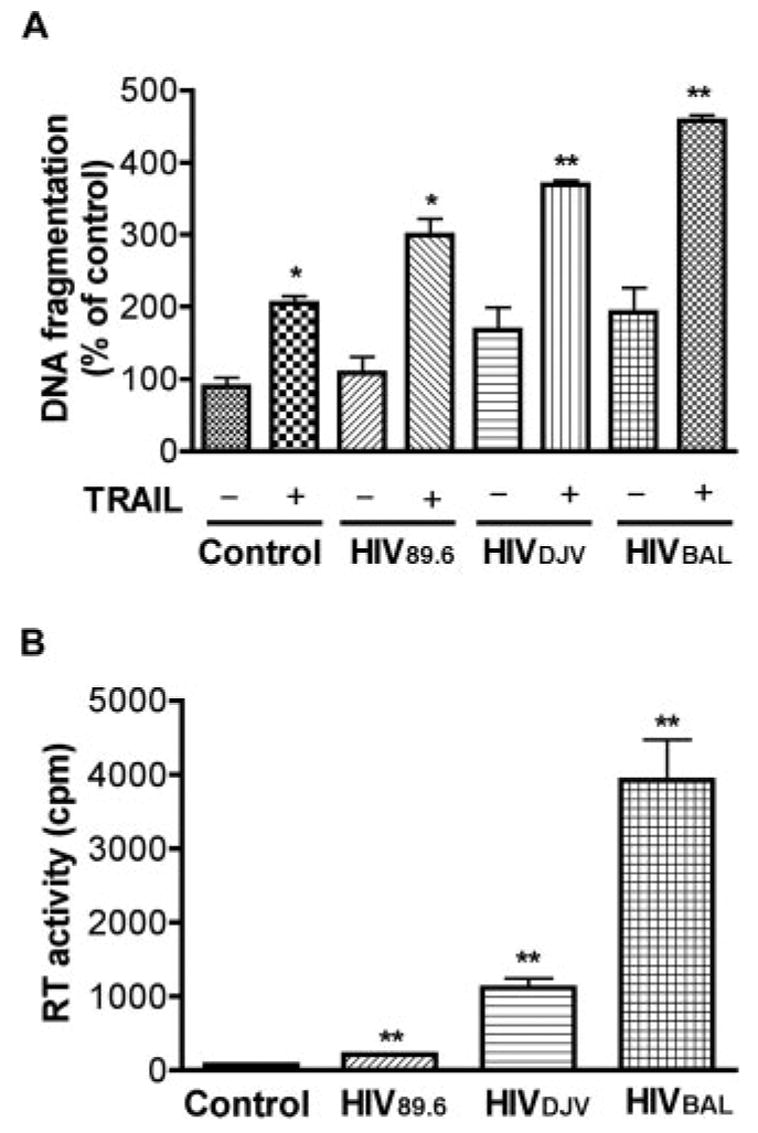
rhTRAIL induces apoptosis in human macrophage infected with different HIV-1 viral strains. A, Human MDM were infected with HIVBAL, HIVDJV, and HIV89.6 for 3 days, then incubated with or without TRAIL (50 ng/ml) for 24 h. Apoptosis was determined by DNA fragmentation apoptosis ELISA.*, p < 0.05 compared with control. #, p < 0.05 compared with rhTRAIL-treated uninfected MDM. ##, p < 0.01 compared with rhTRAIL-treated uninfected MDM. B, The infection levels of different HIV-1 strains were monitored by RT activity. **, p < 0.01 compared with control. Results shown are representative experiments performed with three different donors.
rhTRAIL inhibits HIV-1 replication in MDM
To test whether rhTRAIL-mediated apoptosis results in inhibition of HIV-1 replication in HIV-1-infected MDM, we infected MDM with HIV-1 for 3 days and then treated with rhTRAIL at doses ranging from 0.01 to 300 ng/ml. RT activity assay was used to measure HIV-1 RT activities in the culture supernatants. After rhTRAIL treatment for 24 h, RT activities decreased dramatically in a dose-dependent manner. Pharmacological analysis showed the EC50 for rhTRAIL in the inhibition of RT activity was 10 ng/ml (Fig. 3A). We used serial HIV-1 dilutions (from 1/5000 to 1/15) to infect MDM and then treated those cells with rhTRAIL (50 ng/ml) for 24 h. rhTRAIL significantly reduced the RT activity in the HIV-1-infected group (Fig. 3B). AZT, a drug that inhibits HIV-1 replication by interfering with RT, was used as positive control (Fig. 3B).
FIGURE 3.
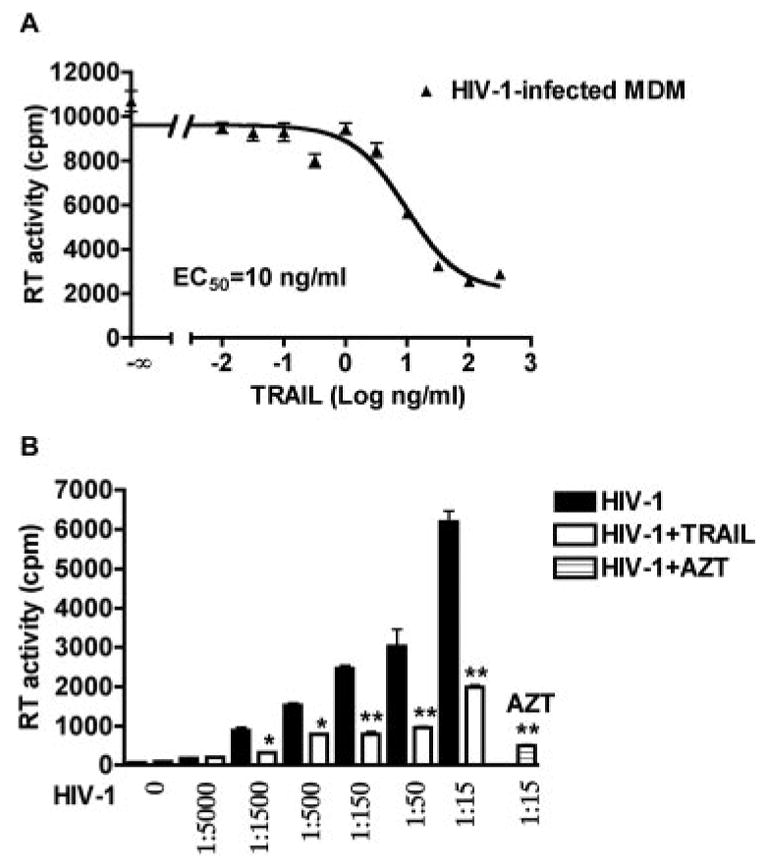
rhTRAIL reduces HIV-1 viral production from MDM-infected cultures. A, Human MDM were infected with HIVADA for 3 days, and then incubated with various concentration of rhTRAIL (0.3–300 ng/ml) for 24 h. RT activity was determined by RT activity assay. Results shown are the representative nonlinear regression curves of averages ± SD of experiments performed in three different donors. B, MDM were infected with seven different HIV-1 dilutions, 0 denotes uninfected control cells. A 1/15 dilution of HIV-1 virus used here equaled a MOI of 0.1 virus/target cell. ■, HIV-1 infection levels as determined by RT activity assay; □, HIV-1-infected with rhTRAIL (50 ng/ml) treatment for 24 h; ▤ HIV-1-infected with AZT (5 μM) treatment for 24 h. *, p < 0.05 compared with corresponding HIV-1-infected group. **, p < 0.01 compared with corresponding HIV-1-infected group.
TRAIL receptor regulation by HIV-1 infection and/or rhTRAIL treatment
Having demonstrated that rhTRAIL kills HIV-1-infected MDM and inhibits HIV-1 replication, we went on to explore the mechanism by which rhTRAIL mediated apoptosis in HIV-1-infected MDM. Due to the proapoptotic role of TRAIL-R1, -R2 and the protective role of TRAIL-R3, -R4, an obvious mechanism would involve TRAIL receptor regulation. To determine the regulation of TRAIL receptors, we performed FACS analysis on HIV-1-infected and uninfected MDM. Three days after infection, TRAIL-R1-R4 were all present in the MDM. As shown in Fig. 4, the fluorescent intensities of receptor staining in FACS are visibly higher than the isotype control. HIV-1 infection or rhTRAIL treatment did not significantly change the cell surface protein level of TRAIL receptors in all three donors (Table I). Further, the transcriptional level of TRAIL receptors was assayed by real-time PCR. After normalization with the internal control GAPDH, no significant difference between the mRNA levels for TRAIL-R1-R4 was observed with HIV-1-infected MDM, 3 days after infection (data no shown). Because cell surface receptors remain unchanged, we presume the apoptotic mechanism must exist downstream of the receptors.
FIGURE 4.

Regulation of TRAIL receptors by HIV-1 infection and TRAIL treatment. A–D, Cell surface expression of TRAIL receptor R1–4 was detected by FACS. Three days post-HIV-1 infection, control or HIV-1 infected MDM were treated with or without rhTRAIL (50 ng/ml) for 24 h. Cells were dual-immunostained for the monocyte Ag, CD14 (CD14-PE) and TRAIL receptors 1–4 (two-step staining; primary Abs, anti-TRAIL receptor 1–4 or isotype control IgG1; secondary Ab, anti-goat-FITC or anti-rabbit-FITC). Cells were gated by CD14 and fluorescent intensities of different TRAIL receptors are presented as color lines in histograms. Results are representative of four separate experiments performed with three donors.
Table I.
Regulation of TRAIL receptors by HIV-1 infection and TRAIL treatmenta
| Control | TRAIL | HIV | TRAIL ± HIV | |
|---|---|---|---|---|
| TRAIL R1 | 54.91 ± 9.15 | 54.12 ± 6.65 | 50.37 ± 7.548 | 50.25 ± 8.51 |
| TRAIL R2 | 59.9 ± 17.1 | 58.62 ± 15.56 | 56.12 ± 18.36 | 61.28 ± 13.48 |
| TRAIL R3 | 48.82 ± 10.88 | 39.68 ± 8.11 | 42.86 ± 7.73 | 55.53 ± 15.13 |
| TRAIL R4 | 64.99 ± 18.38 | 50.7 ± 11.59 | 51.7 ± 11.34 | 38.91 ± 7.4 |
Data are mean fluorescent intensities ± SEM of different TRAIL receptors assessed by FACS (see Fig. 4). Data analyzed by ANOVA showed no statistically significant differences between any of the treatment groups for any TRAIL receptors.
TRAIL mediates cytochrome c release and the caspase cascade in HIV-1-infected MDM
Two major signaling pathways lead to apoptosis: the extrinsic pathway that leads to caspase-8 activation, and the intrinsic mitochondrial pathway that results in cytochrome c release and caspase-9 activation (29). TRAIL-mediated signaling events were suggested to be classical death receptor-initiated signaling cascade that involve both the intrinsic and extrinsic pathway (30). To elucidate the signaling cascade of TRAIL in HIV-1-infected MDM, we first investigated the involvement of the mitochondrial pathway. Our data showed rhTRAIL induced the release of cytochrome c from the mitochondria. After 4 h of rhTRAIL treatment, release of cytochrome c from MDM is enhanced after infection by HIV-1 (Fig. 5A). Further, when HIV-1-infected MDM were treated with increasing concentrations of rhTRAIL (0.05–500 ng/ml) along with control, rhTRAIL provoked caspase-8, −3, and −9 activation in HIV-1-infected MDM in a dose-dependent manner but had minimal effect on uninfected MDM (Fig. 5B). It is notable that cytochrome c and active caspase-9 were also present in the control cells, an observation likely due to the serum-free conditions used for the treatment of MDM. When using 100 ng/ml of the soluble receptors R1 or R2 to act as decoys during rhTRAIL treatment, the effects of rhTRAIL were blocked (Fig. 5C), confirming apoptosis in HIV-1-infected MDM was mediated by TRAIL. Further, rhTRAIL-mediated apoptosis in HIV-1-infected MDM was blocked by the caspase-8 (Z-IETD-FMK, 20 μM) or caspase-3 (Z-DEVD-FMK, 20 μM) inhibitor, and partially blocked by the caspase-9 (Z-LEHD-FMK, 20 μM) inhibitor (Fig. 5D). These data illustrate the requirement of the caspases in TRAIL-mediated apoptosis of infected MDM. Also, the effects of partial blockade by the caspase-9 inhibitor indicate apoptotic pathways bypassing caspase-9 exist. In summary, our data support that TRAIL-mediated apoptosis on infected MDM involves the classical death receptor-initiated signaling cascade.
FIGURE 5.
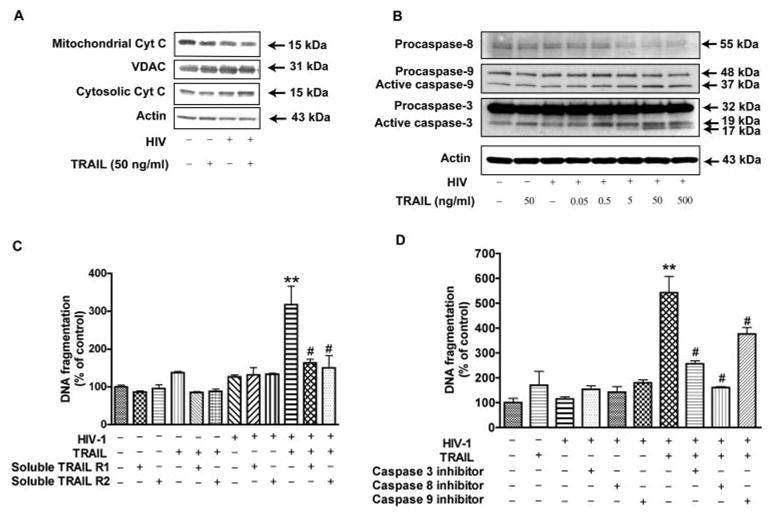
TRAIL mediates cytochrome c release and the caspase cascade in HIV-1-infected MDM. A, HIV-1-infected or control MDM were treated for 4 h in the presence or absence of 50 ng/ml rhTRAIL. Cells were subjected to subcellular fractionation; the cytosolic and the mitochondrial fractions were separated by SDS-PAGE and blotted for immunodetection of cytochrome c. VDAC and β-actin were used as loading controls for the mitochondrial and cytosolic fractions, respectively. B, MDM were treated for 24 h in the presence or absence of 50 ng/ml rhTRAIL; HIV-1-infected MDM were treated with increasing concentrations of rhTRAIL (0.05–500 ng/ml). Cells were then harvested and cell lysates were subjected to SDS-PAGE and immunoblotting for the caspase-8, −9, or −3. The Ab against caspase-8 detects inactive form (55 kDa). The anti-caspase-9 Ab detects inactive form (48 kDa), and cleaved intermediate (37 kDa). The anti-caspase-3 Ab detects inactive form (32 kDa), and cleaved active from (19 and 17 kDa). Actin was used as the loading control. C–D, Uninfected and infected human MDM were pretreated with or without soluble TRAIL receptor 1 and 2 for TRAIL (100 ng/ml, C), inhibitors (20 μM, D) to caspase-3, −8, or −9 or a negative control for 2 h. C, Two soluble receptors TRAIL R1, R2 (100 g/ml) were shown, with or without TRAIL treatment and HIV-1 infection. D, Caspase inhibitors were shown with or without TRAIL and HIV-1 infection, 20 μM of negative control for caspase inhibitors were added. Cells were lysed and apoptosis was detected by DNA fragmentation apoptosis ELISA. Data is presented as mean ± SEM. **, p < 0.01 compared with control or HIV-1. #, p < 0.01 compared with rhTRAIL-treated and HIV-1-infected cells. &, p < 0.05 compared with rhTRAIL-treated HIV-1-infected cells. Data are representative of three donors.
HIV-1 infection decreases phosphorylation of Akt-1
The fact that rhTRAIL did not induce caspase-9 activation as compared with HIV-1-infected cells (Fig. 5) indicates an inhibition of caspase-9 in control MDM that was lost upon HIV-1 infection. Because caspase-9 is a well-known target of the Akt-1 pathway, we evaluated the involvement of Akt-1 in TRAIL-induced apoptosis. Following HIV-1 infection, Akt-1 phosphorylation was decreased (Fig. 6A). The infection levels of different HIV-1 dilutions were monitored by RT activity assay (Fig. 6C). Quantification indicated a progressive loss of Akt-1 phosphorylation as HIV-1 infection levels increased (Fig. 6A). This data demonstrates HIV-1 infection decreases phosphorylation of Akt-1 in a dose-dependent manner. Interestingly, TRAIL exposure further decreased Akt-1 phosphorylation on HIV-1-infected MDM (Fig. 6B). Together, these data indicate an inverse correlation between Akt-1 level and apoptosis in macrophage. TRAIL-induced apoptosis may involve inhibition of Akt-1 phosphorylation.
FIGURE 6.
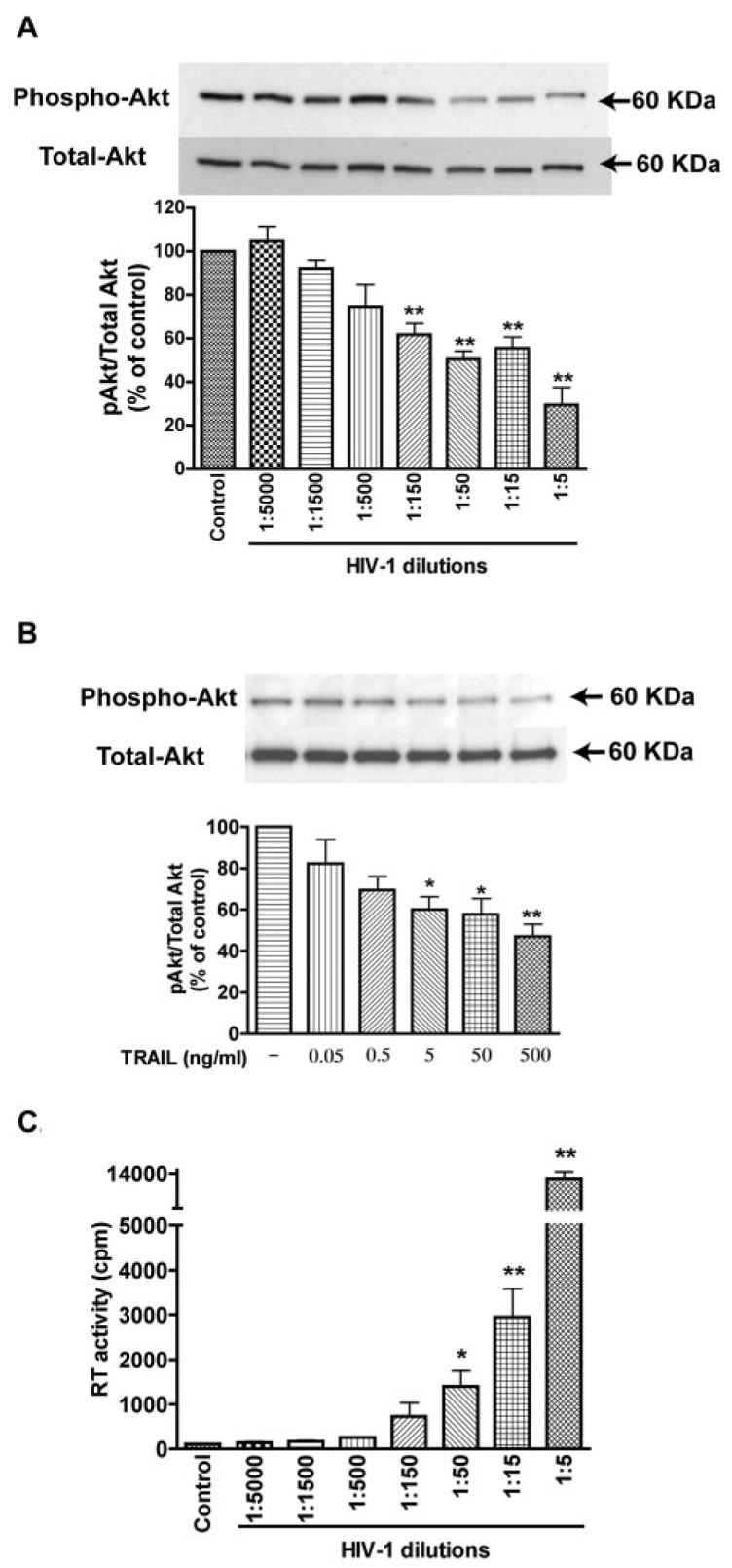
HIV-1 infection decreases phosphorylation of Akt-1. A, MDM were infected with seven different HIV-1 dilutions; control denotes uninfected cells. Cell lysates were collected 3 days after infection and subjected to immunoblotting for phospho-Akt (Ser473), or total Akt. Results were quantified as a ratio of phospho-Akt to total Akt and then normalized to control. Average ± SD of three independent experiments were shown. The infection levels of different HIV-1 dilutions were monitored by RT activity (C). B, HIV-1-infected MDM (3 days after infection, 1/15 dilution in A) were treated with increasing concentrations of TRAIL (0.05–500 ng/ml). Cell lysates were collected and subjected to immunoblotting for phospho- or total Akt. Normalized ratio of phospho-Akt to total Akt was shown. Results shown are average ± SD of three independent experiments. *, p < 0.05 compared with control. **, p < 0.01 compared with control.
Inhibition of Akt-1 phosphorylation sensitizes MDM to TRAIL-induced apoptosis
PI3K is an upstream kinase for Akt-1, with inhibition of PI3K resulting in decreased Akt-1 phosphorylation. To investigate the effect of Akt-1 inhibition in MDM, we used LY294002, a cell-permeable, potent, and specific PI3K inhibitor. MDM were treated with different doses of LY294002 ranging from 30 nM to 30 μM. Results show LY294002 induced DNA fragmentation of MDM in a dose-dependent manner with an EC50 of 1 μM (Fig. 7A). Exposure to as little as 100 nM LY294002 for 24 h significantly inhibited Akt-1 phosphorylation, but did not change the level of total Akt-1 (Fig. 7B). To test whether Akt-1 inhibition could potentiate the effect of TRAIL, uninfected MDM were pre-treated with LY294002 and then incubated with rhTRAIL for 24 h. As shown in Fig. 7C, rhTRAIL caused only a moderate increase of DNA fragmentation in cultured MDM. When pre-treated with LY294002, rhTRAIL induced significantly higher levels of DNA fragmentation in MDM. These observations demonstrate in macrophage the PI3K/Akt-1 pathway is necessary for protection against TRAIL-induced apoptosis.
FIGURE 7.
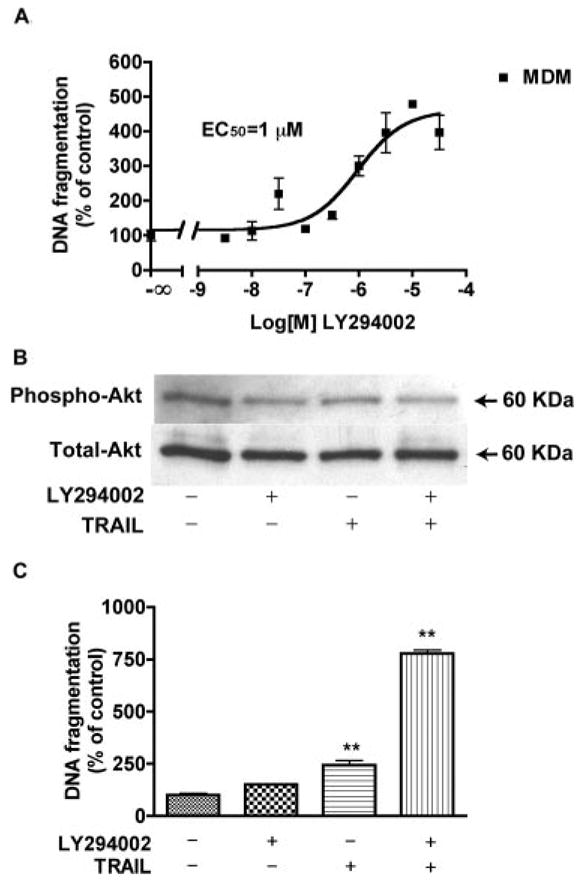
LY294002 pretreatment potentiates TRAIL-induced MDM apoptosis. A, Human MDM were incubated with various concentrations of LY294002 (30 nM - 30 μM) for 24 h. Apoptosis is determined by DNA fragmentation apoptosis ELISA. Results were analyzed by nonlinear regression and curve fit that assess the EC50 for LY294002 in MDM as 1 μM. B, MDM were pretreated with 0.1 μM LY294002 for 6 h, then incubated with or without 50 ng/ml rhTRAIL for 24 h. Cell lysates were collected and subjected to immunoblotting for phospho-Akt (Ser473), or total Akt. C, With the same treatment of B, apoptosis of MDM was determined by DNA fragmentation apoptosis ELISA. Results are expressed as average ± SD. **, A p value of <0.01 comparing TRAIL + LY294002 group with TRAIL or LY294002 alone, or comparing TRAIL with control. Experiments are representative of three replicate assays done with three different donors.
Akt-1 is required for macrophage survival following TRAIL treatment
To specifically target Akt-1, we used an adenovirus delivery system to deliver a dominant-negative Akt-1 to MDM in vitro. As seen in Fig. 8B, infection with adenovirus expressing only GFP (Ad-GFP) had minimal toxicity to cultured MDM. However, using the same delivery system expressing dominant-negative Akt-1 (Ad-DN-Akt), MDM viability decreased as the MOI was increased. We chose the MOI of 1000 as the viral titer where no significant loss of cells was seen at both Ad-GFP- and Ad-DN-Akt-transfected groups. At this viral concentration, infection efficiency was ~70% (Fig. 8A). Also, the DN-Akt overexpression was examined by immunoblotting as shown in Fig. 8C, virus-encoded DN-Akt was 3-fold higher than vector control. When MDM expressing Ad-GFP were exposed to 50 ng/ml rhTRAIL, DNA fragmentation was significantly escalated. This elevated DNA fragmentation is greater than the effect of TRAIL on untransfected MDM, where DNA fragmentation increased up to 1-fold. These observations suggest that adenovirus, like other reported viruses (31–33), may sensitize cells to TRAIL-induced apoptosis. More importantly, when MDM expressing Ad-DN-Akt were treated with rhTRAIL (50 ng/ml), there was a 13-fold increase in apoptosis as seen using DNA fragmentation ELISA ( p < 0.001, Fig. 8D). These observations support the necessity of Akt-1 in protecting macrophages from TRAIL-induced apoptosis.
FIGURE 8.
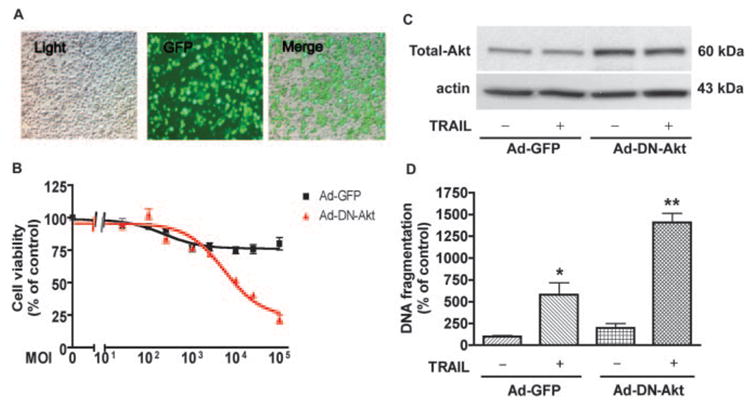
Dominant-negative Akt-1 potentiates TRAIL-induced apoptosis in uninfected macrophages. A, Human MDM were infected with adenovirus vector containing a GFP reporter system. Light panel, Pictures taken under light microscopy; GFP panel, same field taken under green fluorescence. Merge panel, light and GFP panels merged. B, MDM were infected with various MOI of viral particles for Ad-GFP and Ad-DN-Akt. Forty-eight hours after infection, cell viabilities were determined by MTT assay; results were normalized as percentage of untransfected control. C, Akt overexpression was monitored by immunoblotting using anti-Akt-1 Abs (Total Akt). D, In the MOI of 1000, infected MDM were incubated with or without 50 ng/ml rhTRAIL for another 24 h. Apoptosis was determined by DNA fragmentation apoptosis ELISA. *, A p value of <0.01 comparing to control; **, a p value of <0.01 comparing to control TRAIL-treated vector control or Ad-DN-Akt transfected alone.
To further examine the role of Akt-1, we tested whether over-expression of constitutively active Akt-1 blocks TRAIL-induced apoptosis in HIV-1-infected MDM. The infection efficiency for HIV-1-infected MDM was ~70% (Fig. 9A). The control vector in the experiment was adenovirus encoding GFP but not c-src myristoylated Akt-1. The overexpression of myr-Akt-1 was also detected by immunoblotting against total Akt-1 (Fig. 9B). After infection with constitutively active Akt-1, the effect of rhTRAIL in HIV-1-infected MDM was partially blocked ( p < 0.05) as compared with the same treatment in the vector group or Ad-myr-Akt infection alone (Fig. 9C). These results indicate that inhibition of Akt-1 phosphorylation is required for TRAIL-induced apoptosis in HIV-1-infected MDM. Together, these observations demonstrate that the mechanism of TRAIL-mediated apoptosis on MDM is reliant upon the inhibition of Akt-1 phosphorylation.
FIGURE 9.
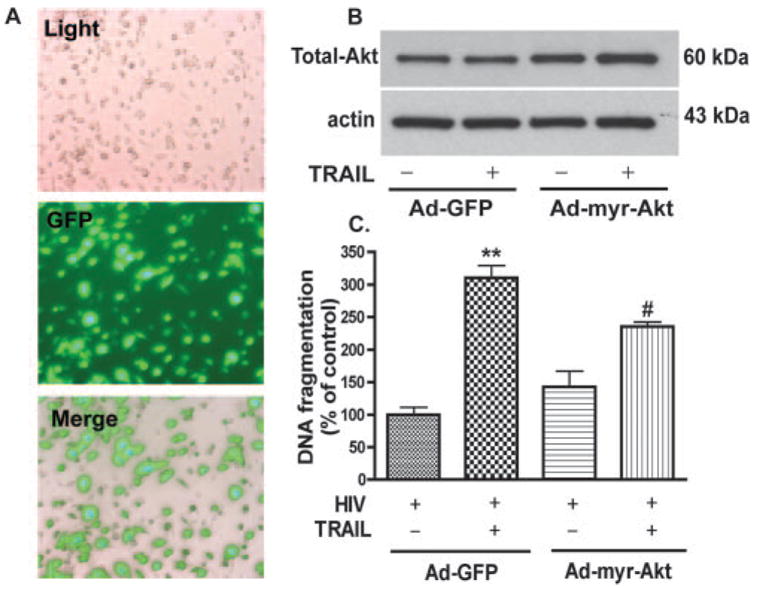
Constitutively active Akt-1 blocks TRAIL-induced MDM apoptosis. Human MDM were infected with adenovirus vector containing GFP reporter system and constitutively active Akt-1 (Ad-myr-Akt) for 24 h and then infected with HIV-1. A, Pictures taken at 48-h postinfection of HIV-1-infected MDM. Light panel, Pictures taken under light microscopy; GFP panel, same field taken under green fluorescence. Merge panel, Light and GFP panels merged. Transfection rate, as demonstrated by GFP production, is ~70%. B, Akt overexpression was monitored by immunoblotting using anti-Akt-1 Abs (total Akt). C, At 72 h post-HIV-1 infection, apoptosis levels were assayed by DNA fragmentation apoptosis ELISA. Results shown are from one donor run in triplicate ± SD. **, p < 0.01 as compared with control. #, p < 0.05 compared with rhTRAIL-treated cells. Data are representative of three donors.
Discussion
Eliminating HIV reservoirs and combating long-term viral persistence is critical to the treatment of AIDS (34–36). Our study aimed to determine whether TRAIL could clear HIV-1-infected MP in vitro. The pharmacological studies demonstrate rhTRAIL mediates apoptosis in HIV-1-infected MDM, whereas cell death observed in uninfected cells is present but at significantly lower levels. rhTRAIL also induced a remarkable and dose-dependent decrease of viral production. Moreover, our results demonstrate rhTRAIL induces activation of caspases-8, −9, and −3 in HIV-1-infected MDM, while caspase inhibition blocks apoptosis. HIV-1 infection significantly decreased Akt-1 phosphorylation; rhTRAIL exposure further decreased Akt-1 phosphorylation. Infection with a dominant-negative Akt-1 adenovirus potentiated rhTRAIL-induced apoptosis, while constitutively active Akt-1 blocked rhTRAIL-induced apoptosis in HIV-infected MDM. These observations suggest that the death ligand TRAIL preferentially provokes apoptosis of HIV-1-infected MDM, a mechanism reliant upon the inhibition of Akt-1 phosphorylation.
The physiological relevance of this observation is significant because of elevated endogenous levels of soluble TRAIL in plasma after HIV-1 infection (21). Further, it has been demonstrated that TRAIL is up-regulated on the cell surface of HIV-infected or immune-activated macrophages (19, 37). The function of TRAIL up-regulation during HIV-1 infection remains unclear, however, this process could mediate innate control of HIV-1 virus in MP as reservoirs. It has been reported that treatment with leucine zipper human TRAIL induces apoptosis of HIV-infected MDM in vitro as well as PBLs from HIV-infected patients. In addition, leucine zipper human TRAIL-treated cells produce less viral RNA and p24 Ag than untreated controls (8). Our new data also supported this notion showing rhTRAIL treatment induced a significant and dose-dependent decrease of viral production. However, the fact that HIV-1 persists even in the presence of elevated soluble TRAIL indicates the efficacy of TRAIL-mediated apoptosis is insufficient to clear the entire infected cell population. This may be due to the sequestration of virus in sites with little or no TRAIL, an inadequate concentration of soluble TRAIL to achieve a fully therapeutic level, or the conformation of soluble TRAIL may not be in the most active state (38). Thus, determining the mechanisms by which TRAIL mediates its physiologic effects may help us to better understand the pathogenesis of HIV-1 infection.
TRAIL is up-regulated during HIV-1 infection (21) and HIV-1 associated dementia (HAD) (19). Under normal conditions, TRAIL is generally not found in the CNS (39). However, recent studies have demonstrated that during disease states such as HAD (19, 40), TRAIL is present and may play an important role in disease pathogenesis. Monocytes stimulated with IFN-γ were shown to increase both the release of soluble TRAIL and the expression of membrane-bound TRAIL (20). Previous work has shown TRAIL up-regulation on HIV-infected and immune-activated macrophages (19), cells that are instrumental in the disease pathogenesis of HAD (41–44). Understanding the mechanism by which TRAIL mediates MP death and its pathways may allow the discovery of new therapeutic avenues for HIV-1 infection.
Signaling pathways involved in the generation of TRAIL-induced apoptosis have been extensively investigated (45–48). Regulation of apoptosis induced by TRAIL is conferred by multiple agonistic and inhibitory receptors and proteins; these effects initiate a complex intracellular signaling mechanism involving an array of signaling adaptors and inhibitors (30, 49–53). Using the human MDM model, we first determined whether HIV-induced sensitization to TRAIL is through the regulation of TRAIL death receptors or decoy receptors. The two decoy receptors TRAIL-R3 and -R4 bind TRAIL, but are unable to induce apoptosis. TRAIL-R3 and -R4 were proposed as one manner in which normal cells and some cancer cell lines may resist TRAIL-induced apoptosis (16, 54, 55). Our results confirm the expression of TRAIL-R1, -R2, -R3, and -R4 mRNA and protein in human MDM. In accordance with the apoptosis assays and viral replication assays, we analyzed cell surface TRAIL-Rs expression by FACS 3 days after infection. HIV-1 infection and/or rhTRAIL treatment did not change the levels of TRAIL-R1 and -R2 or decoy TRAIL-R3 and -R4 at the time of HIV-1 infection and TRAIL treatment (Fig. 4 and Table I). HIV-1 has been reported to regulate TRAIL-Rs 10 days after infection in MDM (8). This discrepancy may be due to differences in the stage of infection in our cultured MDM. The regulation of TRAIL-Rs at the latter time point of productive HIV-1 infection in MDM and its role in the regulation of MDM survival merits further investigation. In the current report, we focused on the regulation of TRAIL-dependent intracellular signaling pathways upon HIV-1 infection and its effect on mediating MDM apoptosis.
The death response to TRAIL is cell-type specific and is characterized by two distinct apoptotic pathways. In the extrinsic pathway, extracellular signals lead to the increased expression and activation of caspase-8 and rapid cleavage of caspase-3 before the loss of mitochondrial transmembrane potential (48). In the intrinsic pathway, signals such as DNA damage lead to the release of cytochrome c and its down-stream activation of caspase-9, caspase-3, and other effector caspases (56). Bid has been reported as a plausible mechanistic link between the extrinsic and intrinsic pathways and apparently amplifies the apoptotic signal following death receptor activation (52, 56). In our studies, rhTRAIL treatment induced the release of cytochrome c and the activation of caspase-8, −9, and −3 (Fig. 5). Further, caspase-8, −9, or −3 inhibitors block HIV-1-sensitized rhTRAIL-induced apoptosis in MDM (Fig. 5D). These results provide evidence for the notion that TRAIL-mediated MDM apoptosis depends on the classical death receptor-initiated signaling cascade that involves both the extrinsic and intrinsic pathways.
In the present study, we demonstrate that the Akt-1-signaling pathway plays an essential role in regulating MDM escape from TRAIL-induced apoptosis. First, inhibition of the Akt-1 phosphorylation by a cell permeable PI3K inhibitor, LY294002, potentiates rhTRAIL-induced apoptosis (Fig. 7B). Second, dominant-negative Akt-1 infection was used to demonstrate the specific role of Akt in this process. Infection with a dominant-negative Akt-1 adenovirus induced significant MDM apoptosis and potentiated rhTRAIL-mediated apoptosis (Fig. 8B). Third, HIV-1 infection inhibited Akt phosphorylation (Fig. 6). Fourth, when macrophage overexpressed constitutively active Akt-1 adenovirus, rhTRAIL-induced apoptosis of HIV-infected MDM was partially blocked. The effects of partial blockade by constitutively active Akt-1 indicate Akt-1 may block only part of the signaling cascade of TRAIL-induced apoptosis, however, this partial effect may be due to the infection efficiency of MDM by the adenovirus, which generally approaches 70%. These results support TRAIL-mediated MDM apoptosis is dependent upon the inhibition of Akt-1, and that HIV-1 infection decreases Akt-1 phosphorylation, potentiating TRAIL-mediated apoptosis.
Akt-1 promotes cell survival by regulating caspase-mediated apoptosis. Our observations are consistent with a variety of reports suggesting constitutively active Akt-1 is responsible for TRAIL resistance in tumor cells (57–59). Recent studies have identified molecular targets of Akt-1 in apoptotic pathways (see review in Ref. 60). For example, phosphorylation of Bcl2-antagonist of cell death in Ser136 by phospho-Akt-1 prevents the mitochondrial translocation of Bcl2-antagonist of cell death (61, 62); phospho-Akt-1 phosphorylates procaspase-9 on Ser196 (63); phosphorylation of the forkhead family of transcription factors by phospho-Akt-1 leads to decreased expression of proapoptotic genes such as Fas, TRAIL, or Bim (64, 65). Further, Akt-1 phosphorylation activates NF-κB, a transcription factor promoting the transcription of many essential survival genes (66). Akt-1 has previously been identified as a key modulator of TRAIL responsiveness (67, 68). Reports have shown caspases are able to cleave Akt-1 (69), and Akt-1 inhibition contributes to caspase activation (22). These findings imply a significant role for Akt in the regulation of a TRAIL-induced effect. The relationship of Akt-1 phosphorylation and caspase activation on MDM merits further investigation.
TRAIL-mediated apoptosis and the inhibition of viral replication on the HIV-1-infected macrophage seems intuitively linked. However, TRAIL-mediated inhibition of the Akt pathway may also play an important role in this process. It is interesting to note that LY294002, a cell permeable PI3K and Akt pathway inhibitor, has been reported to impact HIV replication in human lymphocytes and macrophages (70). Preliminary data from our laboratory showing a dose-dependent reduction of HIV replication using LY294002 also supports this notion (data not shown). The strong effect of TRAIL on the inhibition of HIV-1 replication (Fig. 3) and its effect on Akt phosphorylation (Fig. 6B) indicate TRAIL may also inhibit HIV-1 replication through inhibition of Akt pathways besides its direct effect on the enhancement of apoptosis of infected cells. This interesting possibility remains a target for further investigation.
In summary, the evidence presented in this study demonstrates that TRAIL preferentially provokes apoptosis in HIV-1-infected MDM, while having a reduced effect on uninfected MDM. TRAIL-induced apoptosis is reliant upon the inhibition of Akt-1 phosphorylation. These mechanisms illustrate TRAIL-mediated cell death in the HIV-1-infected macrophage. TRAIL also inhibits HIV-1 replication in these infected MDM. Whether TRAIL controls viral infection in vivo through this mechanism awaits further investigation. TRAIL and its associated inhibition of Akt may promote the elimination of HIV-1 reservoirs during disease, making this pathway a key therapeutic target during HIV-1 infection.
Acknowledgments
We kindly acknowledge Dr. Xiaoying Hou, Dr. Shinji Sato, James Buescher, Toluope Olade, Angelique Walstrom, Min Cui, Jacky Dar, and Eddie Arvisais, who provided technical support for this work. Drs. Howard E. Gendelman, Terry Hexum, Michael Mann, Mark Thomas, and Myron Toews provided valuable comments and suggestions about the manuscript. Dr. Charles Kuszynski, Linda Wilkie, and Victoria Smith performed the FACS analyses. Julie Ditter, Robin Taylor, Nellie Theresa Ingraham, Myhanh Che, and Emilie Scoggins provided outstanding administrative support.
Abbreviations used in this paper
- MP
mononuclear phagocyte
- ART
antiretroviral therapy
- MDM
monocyte-derived macrophage
- rh
recombinant human
- MOI
multiplicity of infection
- RT
reverse transcriptase
- VDAC
voltage-dependent anion channel
Footnotes
This work was supported in part by National Institutes of Health Research Grants R01 NS41858, P20 RR15635, and P01 NS043985 (each to J.Z.) and R01 MH092539 (to T.I.) and by research funds from the Department of Veterans Affairs (to J.S.D.).
Disclosures
All authors are employed by the University of Nebraska.
References
- 1.Gartner S, Popovic M. Macrophage tropism of HIV-1. AIDS Res Hum Retroviruses. 1990;6:1017–104. doi: 10.1089/aid.1990.6.1017. [DOI] [PubMed] [Google Scholar]
- 2.Gendelman HE, Orenstein JM, Baca LM, Weiser B, Burger H, Kalter DC, Meltzer MS. The macrophage in the persistence and pathogenesis of HIV-1 infection. AIDS. 1989;3:475–495. doi: 10.1097/00002030-198908000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Gabuzda DH, Ho DD, Monte MSDL, Rota TR, Sobel RA. Immunohistochemical identification of HTLV-III antigen in brains of patients with AIDS. Ann Neurol. 1986;20:289–295. doi: 10.1002/ana.410200304. [DOI] [PubMed] [Google Scholar]
- 4.Gartner S, Markovits P, Markovits DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III LAV infection. Science. 1986;233:214. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 5.Koenig S, Gendelman HE, Orenstein JM, Canto MCD, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 6.Shieh JTC, Albright AV, Sharron M, Gartner S, Strizki J, Doms RW, Gonzalez-Scarano F. Chemokine receptor utilization by human immundeficiency virus type 1 isolates that replicate in microglia. J Virol. 1998;72:4243–4249. doi: 10.1128/jvi.72.5.4243-4249.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gendelman HE, Morahan P. The macrophage in viral infections. In: Lewis C, McGee J, editors. The Natural Immune System Series: The Macrophage. Oxford University Press; London: 1992. pp. 156–232. [Google Scholar]
- 8.Lum JJ, Pilon AA, Sanchez-Dardon J, Phenix BN, Kim JE, Mihowich J, Jamison K, Hawley-Foss N, Lynch DH, Badley AD. induction of cell death in human immunodeficiency virus-infected macrophages and resting memory CD4 T cells by TRAIL/Apo2L. J Virol. 2001;75:11128–11136. doi: 10.1128/JVI.75.22.11128-11136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Y, Erdmann N, Zhao J, Zheng J. The signaling and apoptotic effects of TNF-related apoptosis-inducing ligand in HIV-1 associated dementia. Neurotox Res. 2005;8:135–148. doi: 10.1007/BF03033825. [DOI] [PubMed] [Google Scholar]
- 10.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–12690. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 11.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 12.Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–114. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 13.Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, Timour MS, Gerhart MJ, Schooley KA, Smith CA, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–5397. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seol DW, Li J, Seol MH, Park SY, Talanian RV, Billiar TR. Signaling events triggered by tumor necrosis factor-related apoptosis- inducing ligand (TRAIL): caspase-8 is required for TRAIL-induced apoptosis. Cancer Res. 2001;61:1138–1143. [PubMed] [Google Scholar]
- 15.Pan G, Ni J, Yu GL, Wei YF, Dixit VM. TRUNDD a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett. 1998;424:41–45. doi: 10.1016/s0014-5793(98)00135-5. [DOI] [PubMed] [Google Scholar]
- 16.Degli-Esposti MA, Smolak PJ, Walczak H, Waugh J, Huang CP, DuBose RF, Goodwin RG, Smith CA. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J Exp Med. 1997;186:1165–1170. doi: 10.1084/jem.186.7.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–14367. doi: 10.1074/jbc.273.23.14363. [DOI] [PubMed] [Google Scholar]
- 18.Ryan LA, Williams C, Ghorpade A, Gendelman HE, Zheng J. Macrophage activation and neuronal injury: a potential role for TRAIL in HIV-1 associated dementia. Soc Neurosci Abstr. 2001;27:235.4 . (Abstr.) [Google Scholar]
- 19.Ryan LA, Peng H, Erichsen DA, Huang Y, Persidsky Y, Zhou Y, Gendelman HE, Zheng J. TNF-related apoptosis-inducing ligand mediates human neuronal apoptosis: links to HIV-1 associated dementia. J Neuroimmunol. 2004;148:127–139. doi: 10.1016/j.jneuroim.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Ehrlich S, Infante-Duarte C, Seeger B, Zipp F. Regulation of soluble and surface-bound TRAIL in human T cells: B cells, and monocytes. Cytokine. 2003;24:244–253. doi: 10.1016/s1043-4666(03)00094-2. [DOI] [PubMed] [Google Scholar]
- 21.Herbeuval JP, Boasso A, Grivel JC, Hardy AW, Anderson SA, Dolan MJ, Chougnet C, Lifson JD, Shearer GM. TNF-related apoptosis-inducing ligand (TRAIL) in HIV-1-infected patients and its in vitro production by antigen-presenting cells. Blood. 2005;105:2458–2464. doi: 10.1182/blood-2004-08-3058. [DOI] [PubMed] [Google Scholar]
- 22.Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages: role of Mcl-1, independent of nuclear factor (NF)- κB, Bad, or caspase activation. J Exp Med. 2001;194:113–126. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–654. [PMC free article] [PubMed] [Google Scholar]
- 24.Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- 25.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 26.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng J, Thylin M, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng Y, Gelbard H, et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J Neuroimmunol. 1999;98:185–200. doi: 10.1016/s0165-5728(99)00049-1. [DOI] [PubMed] [Google Scholar]
- 28.Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, Rosenzweig A, Kahn CR, Abel ED, Walsh K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem. 2002;277:37670–37677. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 29.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–774. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 31.Sedger LM, Shows DM, Blanton RA, Peschon JJ, Goodwin RG, Cosman D, Wiley SR. IFN-γ mediates a novel antiviral activity through dynamic modulation of TRAIL and TRAIL receptor expression. J Immunol. 1999;163:920–926. [PubMed] [Google Scholar]
- 32.Clarke P, Tyler KL. Reovirus-induced apoptosis: a mini review. Apoptosis. 2003;8:141–144. doi: 10.1023/a:1022966508671. [DOI] [PubMed] [Google Scholar]
- 33.Kotelkin A, Prikhod’ko EA, Cohen JI, Collins PL, Bukreyev A. Respiratory syncytial virus infection sensitizes cells to apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J Virol. 2003;77:9156–9172. doi: 10.1128/JVI.77.17.9156-9172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McArthur JC, Sacktor N, Selnes O. Human immunodeficiency virus-associated dementia. Semin Neurol. 1999;19:129–124. doi: 10.1055/s-2008-1040831. [DOI] [PubMed] [Google Scholar]
- 35.Carpenter CC, Cooper DA, Fischl MA, Gatell JM, Gazzard BG, Hammer SM, Hirsch MS, Jacobsen DM, Katzenstein DA, Montaner JS, et al. Antiretroviral therapy in adults: updated recommendations of the International AIDS Society-USA Panel. J Am Med Assoc. 2000;283:381–390. doi: 10.1001/jama.283.3.381. [DOI] [PubMed] [Google Scholar]
- 36.Krebs FC, Ross H, McAllister J, Wigdahl B. HIV-1-associated central nervous system dysfunction. Adv Pharmacol. 2000;49:315–314. doi: 10.1016/s1054-3589(00)49031-9. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, Li X, Pang X, Ding L, Wood O, Clouse K, Hewlett I, Dayton AI. Identification of a potential HIV-induced source of bystander-mediated apoptosis in T cells: upregulation of trail in primary human macrophages by HIV-1 tat. J Biomed Sci. 2001;8:290–296. doi: 10.1007/BF02256603. [DOI] [PubMed] [Google Scholar]
- 38.Mongkolsapaya J, Grimes JM, Chen N, Xu XN, Stuart DI, Jones EY, Screaton GR. Structure of the TRAIL-DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat Struct Biol. 1999;6:1048–1053. doi: 10.1038/14935. [DOI] [PubMed] [Google Scholar]
- 39.Dorr J, Bechmann I, Waiczies S, Aktas O, Walczak H, Krammer PH, Nitsch R, Zipp F. Lack of tumor necrosis factor-related apoptosis-inducing ligand but presence of its receptors in the human brain. J Neurosci. 2002;22:RC209. doi: 10.1523/JNEUROSCI.22-04-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miura Y, Misawa N, Kawano Y, Okada H, Inagaki Y, Yamamoto N, Ito M, Yagita H, Okumura K, Mizusawa H, Koyanagi Y. Tumor necrosis factor-related apoptosis-inducing ligand induces neuronal death in a murine model of HIV central nervous system infection. Proc Natl Acad Sci USA. 2003;100:2777–2782. doi: 10.1073/pnas.2628048100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price R, Brew B, Sidtis J, Rosenblum M, Scheck A, Cleary P. The brain and AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science. 1988;239:586–592. doi: 10.1126/science.3277272. [DOI] [PubMed] [Google Scholar]
- 42.Persidsky Y, Buttini M, Limoges J, Bock P, Gendelman HE. An analysis of HIV-1-associated inflammatory products in brain tissue of humans and SCID mice with HIV-1 encephalitis. J Neurovirol. 1997;3:401–416. doi: 10.3109/13550289709031186. [DOI] [PubMed] [Google Scholar]
- 43.Weiss JM, Nath A, Major EO, Berman JW. HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and up- regulates CCR5 expression on human monocytes. J Immunol. 1999;163:2953–2959. [PubMed] [Google Scholar]
- 44.Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26:1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Pietro R, Zauli G. Emerging non-apoptotic functions of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)/Apo2L. J Cell Physiol. 2004;201:331–340. doi: 10.1002/jcp.20099. [DOI] [PubMed] [Google Scholar]
- 46.Walczak H, Krammer PH. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res. 2000;256:58–66. doi: 10.1006/excr.2000.4840. [DOI] [PubMed] [Google Scholar]
- 47.Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 2003;14:337–348. doi: 10.1016/s1359-6101(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 48.LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10:66–64. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 49.Zhang XD, Zhang XY, Gray CP, Nguyen T, Hersey P. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of human melanoma is regulated by smac/DIABLO release from mitochondria. Cancer Res. 2001;61:7339–7348. [PubMed] [Google Scholar]
- 50.Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001;61:1314–1319. [PubMed] [Google Scholar]
- 51.Ricci-Vitiani L, Pedini F, Mollinari C, Condorelli G, Bonci D, Bez A, Colombo A, Parati E, Peschle C, De Maria R. Absence of caspase 8 and high expression of PED protect primitive neural cells from cell death. J Exp Med. 2004;200:1257–1266. doi: 10.1084/jem.20040921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 53.Choi C, Kutsch O, Park J, Zhou T, Seol DW, Benveniste EN. Tumor necrosis factor-related apoptosis-inducing ligand induces caspase-dependent interleukin-8 expression and apoptosis in human astroglioma cells. Mol Cell Biol. 2002;22:724–736. doi: 10.1128/MCB.22.3.724-736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grotzer MA, Eggert A, Zuzak TJ, Janss AJ, Marwaha S, Wiewrodt BR, Ikegaki N, Brodeur GM, Phillips PC. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression. Oncogene. 2000;19:4604–4610. doi: 10.1038/sj.onc.1203816. [DOI] [PubMed] [Google Scholar]
- 55.Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-κB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–820. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 56.Fulda S, Meyer E, Friesen C, Susin SA, Kroemer G, Debatin KM. Cell type specific involvement of death receptor and mitochondrial pathways in drug-induced apoptosis. Oncogene. 2001;20:1063–1075. doi: 10.1038/sj.onc.1204141. [DOI] [PubMed] [Google Scholar]
- 57.Kandasamy K, Srivastava RK. Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in non-small cell lung cancer cells. Cancer Res. 2002;62:4929–4937. [PubMed] [Google Scholar]
- 58.Chen X, Thakkar H, Tyan F, Gim S, Robinson H, Lee C, Pandey SK, Nwokorie C, Onwudiwe N, Srivastava RK. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene. 2001;20:6073–6083. doi: 10.1038/sj.onc.1204736. [DOI] [PubMed] [Google Scholar]
- 59.Larribere L, Khaled M, Tartare-Deckert S, Busca R, Luciano F, Bille K, Valony G, Eychene A, Auberger P, Ortonne JP, et al. PI3K mediates protection against TRAIL-induced apoptosis in primary human melanocytes. Cell Death Differ. 2004;11:1084–1091. doi: 10.1038/sj.cdd.4401475. [DOI] [PubMed] [Google Scholar]
- 60.Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT–a major therapeutic target. Biochim Biophys Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 61.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 62.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 63.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–134. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 64.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 65.Burgering BM, Medema RH. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J Leukocyte Biol. 2003;73:689–701. doi: 10.1189/jlb.1202629. [DOI] [PubMed] [Google Scholar]
- 66.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 67.Cenni V, Maraldi NM, Ruggeri A, Secchiero P, Del Coco R, De Pol A, Cocco L, Marmiroli S. Sensitization of multidrug resistant human osteosarcoma cells to Apo2 Ligand/TRAIL-induced apoptosis by inhibition of the Akt/PKB kinase. Int J Oncol. 2004;25:1599–1608. [PubMed] [Google Scholar]
- 68.Puduvalli VK, Sampath D, Bruner JM, Nangia J, Xu R, Kyritsis AP. TRAIL-induced apoptosis in gliomas is enhanced by Akt-inhibition and is independent of JNK activation. Apoptosis. 2005;10:233–243. doi: 10.1007/s10495-005-6078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee YJ, Froelich CJ, Fujita N, Tsuruo T, Kim JH. Reconstitution of caspase-3 confers low glucose-enhanced tumor necrosis factor-related apoptosis-inducing ligand cytotoxicity and Akt cleavage. Clin Cancer Res. 2004;10:1894–1900. doi: 10.1158/1078-0432.ccr-03-0136. [DOI] [PubMed] [Google Scholar]
- 70.Francois F, Klotman ME. Phosphatidylinositol 3-kinase regulates human immunodeficiency virus type 1 replication following viral entry in primary CD4+ T lymphocytes and macrophages. J Virol. 2003;77:2539–2549. doi: 10.1128/JVI.77.4.2539-2549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]