

Abstract
The neurodegenerative disease MPS III B (Sanfilippo syndrome type B) is caused by mutations in the gene encoding the lysosomal enzyme α-N-acetylglucosaminidase, with a resulting block in heparan sulfate degradation. A mouse model with disruption of the Naglu gene allows detailed study of brain pathology. In contrast to somatic cells, which accumulate primarily heparan sulfate, neurons accumulate a number of apparently unrelated metabolites, including subunit c of mitochondrial ATP synthase (SCMAS). SCMAS accumulated from 1 month of age, primarily in the medial entorhinal cortex and layer V of the somatosensory cortex. Its accumulation was not due to the absence of specific proteases. Light microscopy of brain sections of 6 months-old mice showed SCMAS to accumulate in the same areas as glycosaminoglycan and unesterified cholesterol, in the same cells as ubiquitin and GM3 ganglioside, and in the same organelles as Lamp 1 and Lamp 2. Cryo-immuno electron microscopy showed SCMAS to be present in Lamp positive vesicles bounded by a single membrane (lysosomes), in fingerprint-like layered arrays. GM3 ganglioside was found in the same lysosomes, but was not associated with the SCMAS arrays. GM3 ganglioside was also seen in lysosomes of microglia, suggesting phagocytosis of neuronal membranes. Samples used for cryo-EM and further processed by standard EM procedures (osmium tetroxide fixation and plastic embedding) showed the disappearance of the SCMAS fingerprint arrays and appearance in the same location of “zebra bodies”, well known but little understood inclusions in the brain of patients with mucopolysaccharidoses.
Introduction
Mucopolysaccharidosis III B (MPS III B, Sanfilippo syndrome type B) is a lysosomal storage disorder caused by mutation of the NAGLU gene; the resulting deficiency of the gene product, α-N-acetylglucosaminidase (EC 3.2.1.50) precludes a normal degradation of its substrate, heparan sulfate [1]. The disease is chronic and progressive; its manifestations are most pronounced in the central nervous system, with severe mental retardation and behavioral problems. The lifespan is usually less than two decades. The disease is very heterogeneous at the molecular level [2], with over a hundred mutations currently listed in the Human Gene Mutation Database at the Institute of Medicine in Cardiff, Wales (http://www.hgmd.org). There is no effective therapy, and management of this difficult disease is all that can be offered at the present time [1,3]. There are several animal models: the naturally occurring emu [4] and dog [5] models, and the mouse model generated by homologous recombination [6].
α-N-Acetylglucosaminidase is required for degradation of heparan sulfate, and deficiency of the enzyme causes accumulation of partially degraded heparan sulfate in lysosomes of most tissues examined (reviewed in [1]). However, the situation is more complex in the brain. On the one hand, soluble heparan sulfate is barely elevated in the brain of MPS III B mice [6], and only two-fold in the brain of human patients with MPS III B [7]. On the other hand, there is major accumulation of substances that do not need the participation of α-N-acetylglucosaminidase for their metabolism. The most studied are GM3 and GM2 gangliosides, which have been found significantly elevated in the brain of humans and animal models affected by MPS III B, other mucopolysaccharidoses and related lysosomal disorders (reviewed in [8]). Intense filipin staining (i.e., accumulation of unesterified cholesterol) has been recently reported in the brain of mouse models of several mucopolysaccharidoses, including MPS IIIB [9]. Ubiquitin accumulation has been found in some areas of the brain of mice with MPS VII [10] and MPS III A [11]. An accumulation of subunit c of mitochondrial ATP synthase (SCMAS) was shown by immunohistochemistry to accumulate in the brain of human patients affected by MPS I, II and III A [12].
SCMAS is a small hydrophobic protein that plays a major role in oxidative phosphorylation in mitochondria [13]. Pathologic accumulation of SCMAS is a hallmark of late infantile neuronal ceroid lipofuscinosis (NCL) as well as of the juvenile (Batten's disease) and other forms of NCL. The earliest report was that of accumulation in storage bodies, thought to be lysosomes, isolated from brain of an ovine model of NCL; the amino-terminal portion of SCMAS accounted for nearly half the protein in these bodies as determined by amino acid analysis [14]. Most subsequent studies used immunohistochemistry to identify abnormal accumulation of SCMAS; since mitochondrial SCMAS is not readily accessible to the antibody, presumably because it is buried in the inner mitochondrial membrane, immunoreactivity identifies a molecular species of SCMAS that is not in its usual environment. In late infantile NCL, the accumulation of immunoreactive SCMAS is caused by deficiency of tripeptidyl peptidase 1, a lysosomal protease which is required for its degradation [15-17]. The specific mechanism that causes accumulation of immunoreactive SCMAS in other NCL, such as Batten disease, is not known, since the primary deficiency is not that of a protease. Immunoreactive SCMAS has also been observed in the brain of mice with deficiency of the lysosomal protease cathepsin D [18] and of the lysosomal/endosomal chloride channel ClC7 [19]. We previously used accumulation of immunoreactive SCMAS as a convenient marker of neuropathology of the MPS III B mouse [20].
In the present study, we have examined the pathological accumulation of SCMAS at the level of both light and electron microscopy, in order to determine its intracellular location and its relationship to accumulation of other metabolites found in neurons of the MPS III B mouse brain.
Materials and Methods
Antibodies and other reagents
Rabbit polyclonal antibody against the amino-terminal peptide of mature SCMAS, DIDTAAKFIGA [21], was custom-made by Biosource International (Camarillo, CA); a gift from Dr. E. Kominami was used in preliminary experiments. Rabbit antiserum raised against the entire SCMAS molecule [22] was a gift from Dr. D.N. Palmer. Rat monoclonal antibodies against mouse Lamp-1 (clone 1D4B) or Lamp-2 (clone ABL93) were purchased from the Developmental Studies Hybridoma Bank (Iowa City, IA) or Santa Cruz Biotechnology Inc. (Santa Cruz, CA) ; mouse monoclonal antibody against GM3 ganglioside (clone GMR6) was from Seikagaku America Inc. (Rockville, MD); mouse monoclonal antibody against ubiquitin (clone Ubi-1) was from Novus Biologicals (Littleton, CO); mouse monoclonal antibody against protein disulfide isomerase (PDI, clone ID3) was from Stressgen Bioreagents Co. (Victoria, BC, Canada); rabbit polyclonal antibody against mitochondrial preprotein translocase of the outer membrane (TOM20) was from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Secondary antibodies conjugated with Alexa dye and Prolong-Gold antifade reagent were from Molecular Probes/Invitrogen (Carlsbad, CA). Secondary antibodies conjugated with biotin, Fab donkey anti-mouse IgG and bovine serum albumin were from Jackson ImmunoResearch Lab (West Grove, PA). Secondary antibodies conjugated to gold particles were from Ted Pella, Inc. (Redding, CA) or Jackson ImmunoResearch Laboratories (West Grove, PA). Vecstatin Elite ABC kit, diaminobenzidine (DAB), Vectabond, Vectashield and hematoxylin were from Vector laboratories (Burlingame, CA). Filipin was from Sigma/Aldrich (St. Louis, MO). Substrates for assay of tripeptidyl peptidase1, cathepsin H, and dipeptidyl peptidase II were obtained from Sigma (St. Louis, MO); those for assay of cathepsin L +B and cathepsin D were obtained from Peptide Institute, Inc. (Louisville, KY). Polyvinylpyrrolidone (PVP-10) was purchased from Sigma. Other reagents were of analytical grade.
Mice
The mouse model of MPS III B (Naglu −/−) had been generated by targeted disruption of the mouse Naglu gene [6], and the mutant gene was placed on a C57BL/6 background by repeated backcrossing. Wild type control mice (Naglu +/+) were derived from the same colony. For collection of brain tissue, the mice were deeply anesthetized with pentobarbital (100 mg/kg) and perfused from the left ventricle with phosphate-buffered saline. For light microscopy, perfusion was continued with Protocol Formalin (3.7% phosphate-buffered formaldehyde, Fisher Scientific, Pittsburgh, PA). For electron microscopy, the perfusion solution contained 4% formaldehyde or 4% formaldehyde plus 0.1% glutaraldehyde in phosphate-buffered saline. The animal studies were approved by the UCLA Animal Research Committee.
Enzyme assays
Brains from 9-11 month-old mice (not perfused) were stored frozen, then homogenized in 0.9% NaCl/ 0.2% Triton 100. Peptidase/ protease assays used MCA substrates. The fluorescent product, 7-amino-4-methylcoumarin, was measured in an HTP 7000 Plus Bioassay Reader (Perkins Elmer) at 370 nm excitation and 460 nm emission. Tripeptidyl peptidase I (TPPI) was assayed with Ala-Ala-Phe-MCA ([23]; dipeptidylpetidase II, with Lys-Ala-MCA [24]; cathepsin B+L , with Z-Phe-Arg-MCA [25,26]; cathepsin H with Arg-MCA [27]; cathepsin D with MOCAc-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys(Dnp)γ-NH2 [28].
Preparation of samples for light microscopy and confocal microscopy
After perfusion (see above), brains were post-fixed overnight in formalin at 4°C. Sections, 40 μm thick, were cut sagittally on vibratome VT1000S (Leica Microsystems, Bannockburn, IL). Glycosaminoglycan was detected by the Muller-Mowry colloidal iron stain [29] and unesterified cholesterol by binding of the fluorescent reagent, filipin [30].
For immunohistochemistry, slices were permeabilized with ice-cold methanol or acetone, washed in 1% H2O2, and incubated sequentially with Fab donkey anti-mouse IgG, primary antibody and biotin-labeled F(ab′)2 donkey secondary antibody. The signal was detected by the Vecstatin ABC Elite kit, visualized with diaminobenzidine and counterstained with hematoxylin.
For double immunostaining, slices were permeabilized and incubated sequentially with Fab donkey anti-mouse IgG, primary antibody and secondary antibody conjugated to Alexa 488. Then they were incubated in another primary antibody and with secondary antibody conjugated to Alexa 647. For double immunostaining of GM3 ganglioside and SCMAS, the procedure was modified by permeabilizing the slices with 0.02% saponin [9]. Images were acquired by laser scanning confocal microscopy (Pascal, Carl Zeiss Microimaging, Thornwood, NY), and processed by Axovision software (Zeiss) and Adobe Photoshop.
Preparation of samples for cryo-electron microscopy
After perfusion and removal from the skull, brain was sliced sagittally into 0.5 mm-thick pieces and further fixed overnight at 4°C. Slices were then processed for cryo electron microscopy by the method of Tokuyasu [31-33] with modifications [34]. Briefly, slices were incubated overnight in 1.85 M sucrose/ 20% PVP-10/ 50 mM HEPES pH 7.4. The area of interest (layer V, somatosensory cortex) was determined and small pieces of tissue (1 × 2 × 0.5 mm) were cut out from that area. Each piece of tissue was mounted on an aluminum pin and quick frozen in liquid nitrogen. Ultra-thin sections were prepared using a Leica Microsystems (Austria) UC6 microtome with an F6 cryo-attachment and a Diatome (Switzerland) cryoimmuno (35°) diamond knife. Conditions were as follows: temperature for sample/knife/chamber, −120°C; cutting speed, 1 mm/sec; thickness, 0.75 nm. Sections were collected on nickel grids with plastic/carbon film and stained with antibodies. The secondary antibodies were conjugated to 5, 10 or 12 nm gold particles. Samples were analyzed using a JEM 1200-EX transmission microscope (JEOL, Japan) equipped with a BioScan 600W digital camera (Gatan, Inc., Pleasanton, CA).
Results
Age dependence and distribution of immunoreactive SCMAS studied by light microcopy
Immunoreactive SCMAS was barely visible in brain of 1month-old MPS III B (Naglu −/−) mice, but increased with time, as shown in Fig 1. When brain of 6 months-old mutant mice was examined, immunoreactive SCMAS was found primarily in the pyramidal neurons of layers II/III of the medial entorhinal cortex and layer V of the somatosensory cortex (Fig. 2A and 2C). It could also be found elsewhere in the brain, especially in mice older than 6 months, but the staining in these other regions was generally less intense. Immunoreactive SCMAS was not seen in brain of wild type mice (Fig. 2B and 2D) and heterozygous mice (not shown) under the conditions used for preparing the samples, indicating that the SCMAS present in the mitochondrial inner membrane did not react with the antibody . However, some caution is required in choosing the fixative, because Bouin's solution, a very acidic fixative, caused mitochondrial SCMAS to react with the antibody (probably by extracting the epitope out of the mitochondrial membrane) and obliterated the difference between normal and mutant brain (not shown).
Fig. 1.
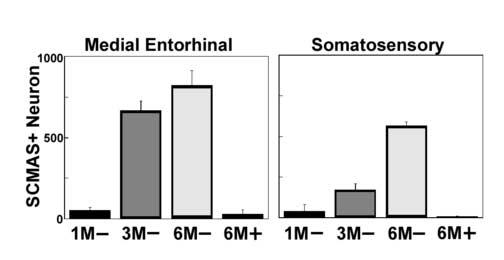
Age-dependent increase of neurons staining positively for SCMAS in the medial entorhinal cortex (left panel) and the somatosensory cortex (right panel). The barrel field of the somatosensory cortex (S1BF) was used for counting stained cells after confirmation by the mouse brain atlas [47]. The age in months and the genotype (− for MPS III B, + for wild type control) of the mice are indicated on the horizontal axis. The data represent the mean and standard deviation for the number of SCMAS-positive neurons in a brain hemisphere from each of three mice.
Fig. 2.
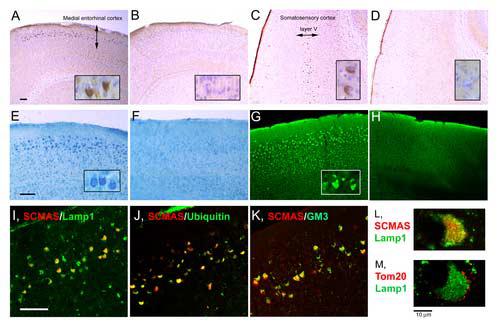
Storage products revealed by staining and immunostaining in selected areas of the brain of 6 months-old mice. All panels show sections from the medial entorhinal cortex, except for Panels C and D which show sections from the somatosensory cortex. Panels A - D: SCMAS immunostaining and hematoxylin counterstain; A and C from MPS III B mouse, B and D, from wild type control. Panels E and F: glycosaminoglycan stained with colloidal iron in sections from MPS III B (E) and wild type control (F) mice. Panels G and H: unesterified cholesterol fluorescence upon reaction with filipin in sections from MPS III B (G) and wild type control (H) mice. Panels I -L: double immunofluorescence of SCMAS with Lamp 1, ubiquitin and GM3, as indicated on the figure; Panel M: double immunofluorescence of Tom 20 with Lamp1. The scale bars represent 100 μm for panels A-K, while the insets show cells at 10 times higher magnification for panels A-D and 4 times higher magnification for panels E and G. The scale bars for panels L and M represent 10 μm.
Normal protease/peptidase activities in brain of mutant mice
The activities of tripeptidyl peptidase I, dipeptidyl peptidase II, cathepsins D, B plus L, and H were as high in brain homogenates of the MPS III B mice as of wild type mice (Table 1), showing that there was no global deficiency of these enzymes. Thus unlike the SCMAS accumulation in the late infantile form of neuronal ceroid lipofuscinosis (TPP I deficiency) and in deficiency of cathepsin D, SCMAS accumulation in MPS III B was not caused by global deficiency of the proteases tested.
Table 1.
Activity of peptidases/proteases in brain homogenates
| Enzyme | MPS III B (μmol/min/mg) | Wild type (μmol/min/mg) |
|---|---|---|
| Tripeptidyl peptidase I | (1.6 ± 0.16) × 10−5 | (1.4 ± 0.39) × 10−5 |
| Dipeptidyl peptidase II | (1.1 ± 0.23) × 10−4 | (1.0 ± 0.49) × 10−4 |
| Cathepsin D | (7.5 ± 1.5) × 10−2 | (4.4 ± 0.73) × 10−2 |
| Cathepsin (B+L) | (1.4 ± 0.36) × 10−2 | (0.9 ± 0.20) × 10−2 |
| Cathepsin H | (1.9 ± 0.06) × 10−3 | (1.9 ± 0.23) × 10−3 |
Activity, mean ± SD, was obtained by assay of brain of 4 mice each of MPS III B (Naglu −/−) and wild type (Naglu +/+) genotype.
Other abnormalities found in the same areas as immunoreactive SCMAS
Vacuolation of numerous neurons was seen throughout the brain of the MPS III B mice [6]. However, a number of additional histologic abnormalities were found in the same areas as immunoreactive SCMAS. These areas stained strongly with colloidal iron, indicating storage of glycosaminoglycan (Fig. 2E) and with filipin, indicating the presence of unesterified cholesterol (Fig. 2G); the corresponding areas in wild type mouse brain were not stained (Fig 2F and 2H). Excess ubiquitin accumulation was localized not only to the same areas, but to the same neurons as immunoreactive SCMAS (Fig. 2J), as was GM3 ganglioside (Fig. 2K). In each case, the staining of MPS III B brain sections was compared to that of wild type controls (not shown).
Intracellular localization of immunoreactive SCMAS seen by confocal microscopy
Double staining was performed to determine the intracellular site of the immunoreactive SCMAS. It occupied a large area of the soma, where it was co-localized with the lysosomal/endosomal marker, Lamp 1 (Fig. 2I, 2L). Similar results were seen with the lysosomal marker, Lamp 2 (not shown). On the other hand, the mitochondrial outer membrane protein TOM 20 was found at the periphery of the cell body and around Lamp 1 (Fig. 2M), a location very different from that of immunoreactive SCMAS. The immunoreactive SCMAS did not co-localize with protein disulfide isomerase (PDI), a marker of the endoplasmic reticulum (not shown). The data obtained by confocal microscopy therefore indicate that immunoreactive SCMAS represents a lysosomal/endosomal pool.
Observation of SCMAS at the electron microscopic level
We turned to examination of SCMAS at the level of electron microscopy in order to obtain more detailed information on its localization. In the ultrathin sections used for cryo-electron microscopy, all SCMAS is available to the antibody, whether in mitochondrial membranes or in other organelles. However, mitochondrial SCMAS was stained equally in neurons of MPS III B and wild type mice (not shown). In addition to the mitochondrial SCMAS, the MPS III B mouse brain contained immunoreactive SCMAS in large vesicles bounded by a single membrane (Fig. 3A), but in no other organelle. The staining was seen in or at the edges of fingerprint-like arrays within the vesicles. At higher magnification, the arrays were found to have a layered structure with a distance of about 9 nm between the layers (Fig. 3B). The vesicles could be labeled with a mixture of antibodies against Lamp1 and Lamp2 (Fig. 3C). Because antibodies against Lamp1/2 and against SCMAS interfered with each other, it was not possible to use them simultaneously for double labeling; however, the unique appearance of the arrays served to identify SCMAS within the vesicles (Fig. 3C and 3D). The reactivity with Lamp1/2 antibodies as well as the single membrane identifies the vesicles as lysosomes. Similar SCMAS-positive fingerprint-like arrays were seen in lysosomes of 3 months-old mouse brain, but in lesser abundance and more loosely packed (not shown). In the wild-type (Naglu +/+) brain, lysosomes were smaller and few in number, but occasionally contained SCMAS in fingerprint-like arrays similar to those seen in the MPS III B mouse brain.
Fig. 3.

Cryoimmuno EM showing SCMAS in lysosomes of a pyramidal neuron in layer V of somatosensory cortex. Panel A: gold-labeled antibody identifies SCMAS (asterisks); Panel B: higher magnification shows SCMAS in layered finger-print-like array; Panel C: gold-labeled antibodies (arrowheads) identify Lamp at the surface of a single membrane of a vacuole containing a multilayered array (asterisk); Panel D: higher magnification of the array shows the characteristic fingerprint-like structure of SCMAS. Scale bars are 0.2 μm; gold particles, 10 nm.
The greater intensity of immunostaining at the edges of the arrays could be explained either by better accessibility of the antibody at the edges or by loss of the epitope in deeper portions of the array. Use of antiserum raised against full-length SCMAS [22] yielded similar results (not shown) as the antibody against the amino terminus. It is therefore likely that the edges of arrays were stained preferentially because they provided better accessibility to the antibody.
GM3 ganglioside at the EM level
GM3 ganglioside was present in the same vacuoles as SCMAS but was not associated with the SCMAS arrays, as shown by double staining for SCMAS and GM3 (Fig. 4A). Thus SCMAS and GM3 are two independent storage products in neuronal lysosomes. GM3 ganglioside was also seen in the large lysosomes of microglia (Fig. 4B). The presence of GM3 ganglioside in microglia had been observed previously in the MPS III B mice at the level of light microscopy [35].
Fig. 4.
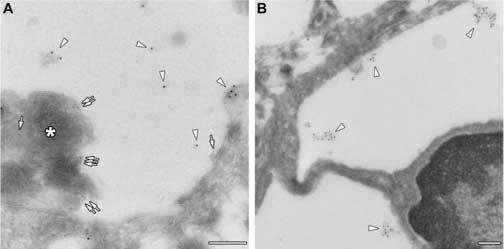
Cryo-immuno electron microscopic identification of GM3 ganglioside. Panel A: GM3 ganglioside is identified by 10 nm gold particles (arrowheads) whereas SCMAS is identified by 5 nm gold particles (arrows) and by the characteristic fingerprint-like array (asterisk) in lysosome of a pyramidal neuron. Panel B: GM3 is identified by 10 nm gold particles (arrowheads) in lysosome of a microglial cell. Scale bars are 0.2 μm.
SCMAS arrays and zebra bodies
Since the regular fingerprint-like arrays of SCMAS shown above had not been reported previously in electron micrographs of MPS III B brain, mouse or human, we suspected that the very mild processing used for cryo-immuno EM may have been responsible for preserving these structures. Therefore, a sample already studied by cryo-EM and found rich in SCMAS-containing vacuoles was fixed and processed as for standard EM (osmium tetroxide fixation and plastic embedding). The SCMAS arrays disappeared whereas structures known as zebra bodies appeared in the same place (Fig. 5). The zebra bodies also contained arrays, but these were coarse, irregular and distantly spaced. Yet occasional zebra bodies had arrays that appeared finer and more closely spaced, almost suggesting an intermediate stage between the fingerprint arrays and the zebra stripes.
Fig. 5.
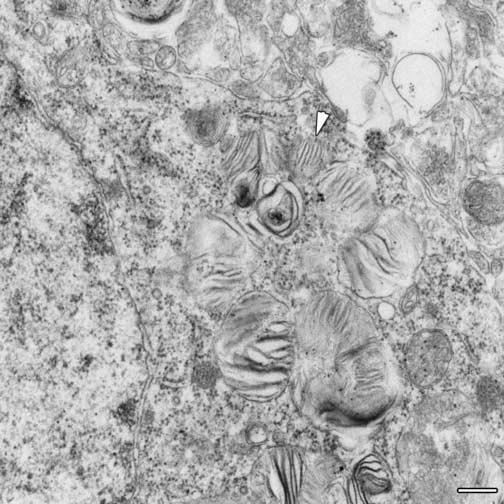
Zebra bodies in neuron after processing for standard electron microscopy. No fingerprint-like arrays are visible, but the arrowhead indicates a zebra body with relatively well organized striations. Scale bar is 0.2 μm.
Discussion
The results above show that SCMAS, normally a mitochondrial protein, accumulates in an age-dependent manner in lysosomes of neurons in certain parts of the brain of MPS III B mice- primarily in layer II/III of the medial entorhinal cortex and layer V of the somatosensory cortex. At the level of light microscopy, this lysosomal SCMAS can be distinguished by its immunoreactivity from mitochondrial SCMAS, which is not immunoreactive, probably because the epitope is embedded in the membrane. At the level of electron microscopy, in which all SCMAS is immunoreactive, lysosomal SCMAS can be recognized by its distinctive appearance as regular fingerprint-like arrays in large vacuoles that could be identified as lysosomes by their single membrane and reactivity with Lamp1/2 antibodies. Such arrays were seen only when fixation and processing was carried out under the very mild conditions used for cryo-immuno EM; under standard EM processing, the finger-print like SCMAS arrays disappeared and zebra bodies appeared in the same place. Zebra bodies have long been known to occur in brain of patients with mucopolysaccharidoses [36], including MPS III (e.g., [37]). While our data do not exclude the presence of other lysosomal components within zebra bodies - e.g., GM3 ganglioside [9] and other proteins, we suggest that the characteristic stripes of the zebra bodies in the Naglu −/− mouse brain may be due to aggregation of SCMAS induced by standard EM procedures.
By comparing EM data from 3 and 6 months-old mice, we can construct a hypothetical scheme for the evolution of the SCMAS. At first, SCMAS is present in small, layered structures attached to the lysosomal membrane. In time, new layers grow around these “seeds”, creating the fingerprint-like structures, which keep growing and eventually fill most of the lysosomal space. During the maturation process, the fingerprint-like arrays become denser in the center so that the layered structure is disturbed and finally disappears. Lysosomes with dense, mature fingerprint arrays were also occasionally detected in wild type mouse brain, though in very low abundance.
How would a mitochondrial protein accumulate in lysosomes? Lysosomes are known to engulf mitochondria by autophagy [38], a process that occurs normally and that may be essential for neuronal survival [39]. Mice made genetically deficient in autophagy suffer from neurodegeneration [39,40]. Once inside lysosomes, mitochondrial proteins are destined for degradation. A lysosomal defect may prevent their degradation, so that SCMAS accumulates in the absence of TPP1 and of cathepsin D, two proteolytic enzymes that are required for the degradation of SCMAS [15] [18]. However, these enzymes are not deficient in MPS III B. Rather, it is likely that some unfavorable condition in lysosomes slows down the degradation of SCMAS, allowing this very hydrophobic protein to pack into closely stacked arrays that may be even harder to digest.
Since heparan sulfate is the primary storage product caused by α-N-acetyl-glucosaminidase deficiency and is observed in large amounts in somatic organs, its limited storage in the MPS III B brain (previously determined by direct analysis [41] and by colloidal iron staining in this study) may seem puzzling. However, lack of heparan sulfate accumulation may be explained by the existence of endo-heparanases, which provide an alternate degradative pathway [42]. The massive accumulation of a heparan sulfate-derived disaccharide (hexosamine-N-sulfate [α1,4]hexuronic acid) in brain and other organs of the MPS III A mouse [43] strongly supports the activity of such an alternate pathway in brain when the normal pathway is blocked. The storage of glycosaminoglycan detected by colloidal iron in the medial entorhinal cortex and in layer V of the somatosensory cortex suggests that this amount of accumulated heparan sulfate was so great that it could not be removed by the presumed alternate pathway.
The neurons that accumulated GAG and SCMAS were also the ones that accumulated ubiquitin, GM3 ganglioside and unesterified cholesterol. These three metabolites are associated with the endosomal/ lysosomal system. Accumulation of unesterified cholesterol is characteristic of fibroblasts from patients with Niemann-Pick disease type C and has been postulated to inhibit traffic and membrane recycling by inhibiting rab 4 [44]. Unesterified cholesterol and glycosphingolipids have an affinity for each other, and it has been suggested that accumulation of one may lead to accumulation of the other [45]. Ubiquitin, originally thought to be involved only in the degradation of proteins by proteasomes, is now known to be involved in the normal endocytosis and trafficking of endocytic vesicles [46]. On the other hand, SCMAS accumulation should depend on the level of autophagy and the rate of degradation of this protein within lysosomes. We don't know whether the accumulation of SCMAS is the cause or effect of the accumulation of the other metabolites, or occurs independently. Regardless of the mechanism, lysosomal accumulation of SCMAS is a marker for neuropathology in MPS III B and, as shown previously, can be used for gauging the results of therapy [20].
Acknowledgments
We thank Dr. E. Kominami (Juntendo University, Tokyo, Japan) and Dr. D.N. Palmer (Lincoln University, Canterbury, New Zealand) for antibodies raised against the SCMAS amino terminus and the entire molecule, respectively. This work was supported in part by grants from the National Institutes of Health (NS 22376), the Children's Medical Research Foundation and the Sanfilippo Children's Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3421–3452. [Google Scholar]
- 2.Yogalingam G, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type IIIA and IIIB: Diagnostic, clinical, and biological implications. Hum Mutat. 2001;18:264–281. doi: 10.1002/humu.1189. [DOI] [PubMed] [Google Scholar]
- 3.Cleary MA, Wraith JE. Management of mucopolysaccharidosis type III. Arch Dis Child. 1993;69:403–406. doi: 10.1136/adc.69.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aronovich EL, Johnston JM, Wang P, Giger U, Whitley CB. Molecular basis of mucopolysaccharidosis type IIIB in emu (Dromaius novaehollandiae): an avian model of Sanfilippo syndrome type B. Genomics. 2001;74:299–305. doi: 10.1006/geno.2001.6552. [DOI] [PubMed] [Google Scholar]
- 5.Ellinwood NM, Wang P, Skeen T, Sharp NJ, Cesta M, Decker S, Edwards NJ, Bublot I, Thompson JN, Bush W, Hardam E, Haskins ME, Giger U. A model of mucopolysaccharidosis IIIB (Sanfilippo syndrome type IIIB): N-acetyl-alpha-D-glucosaminidase deficiency in Schipperke dogs. J Inherit Metab Dis. 2003;26:489–504. doi: 10.1023/a:1025177411938. [DOI] [PubMed] [Google Scholar]
- 6.Li HH, Yu WH, Rozengurt N, Zhao HZ, Lyons KM, Anagnostaras S, Fanselow MS, Suzuki K, Vanier MT, Neufeld EF. Mouse model of Sanfilippo syndrome type B produced by targeted disruption of the gene encoding alpha-N-acetylglucosaminidase. Proc. Natl. Acad. Sci. USA. 1999;96:14505–14510. doi: 10.1073/pnas.96.25.14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hadfield MG, Ghatak NR, Nakoneczna I, Lippman HR, Myer EC, Constantopoulos G, Bradley RM. Pathologic findings in mucopolysaccharidosis type IIIB (Sanfilippo's sydnrome B) Arch Neurol. 1980;37:645–650. doi: 10.1001/archneur.1980.00500590069012. [DOI] [PubMed] [Google Scholar]
- 8.Walkley SU. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin Cell Dev Biol. 2004;15:433–444. doi: 10.1016/j.semcdb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 9.McGlynn R, Dobrenis K, Walkley SU. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J Comp Neurol. 2004;480:415–426. doi: 10.1002/cne.20355. [DOI] [PubMed] [Google Scholar]
- 10.Heuer GG, Passini MA, Jiang K, Parente MK, Lee VM, Trojanowski JQ, Wolfe JH. Selective neurodegeneration in murine mucopolysaccharidosis VII is progressive and reversible. Ann Neurol. 2002;52:762–770. doi: 10.1002/ana.10373. [DOI] [PubMed] [Google Scholar]
- 11.Savas PS, Hemsley KM, Hopwood JJ. Intracerebral injection of sulfamidase delays neuropathology in murine MPS-IIIA. Mol Genet Metab. 2004;82:273–285. doi: 10.1016/j.ymgme.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Elleder M, Sokolová J, Hrebícek M. Follow-up study of subunit c of mitochondrial ATP synthase (SCMAS) in Batten disease and in unrelated lysosomal disorders. Acta Neuropathol (Berl) 1997;93:379–390. doi: 10.1007/s004010050629. [DOI] [PubMed] [Google Scholar]
- 13.Boyer PD. A research journey with ATP synthase. J Biol Chem. 2002;277:39045–39061. doi: 10.1074/jbc.X200001200. [DOI] [PubMed] [Google Scholar]
- 14.Palmer DN, Martinus RD, Cooper SM, Midwinter GG, Reid JC, Jolly RD. Ovine ceroid lipofuscinosis. The major lipopigment protein and the lipid-binding subunit of mitochondrial ATP synthase have the same NH2-terminal sequence. J Biol Chem. 1989;264:5736–5740. [PubMed] [Google Scholar]
- 15.Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- 16.Ezaki J, Tanida I, Kanehagi N, Kominami E. A lysosomal proteinase, the late infantile neuronal ceroid lipofuscinosis gene (CLN2) product, is essential for degradation of a hydrophobic protein, the subunit c of ATP synthase. J Neurochem. 1999;72:2573–2582. doi: 10.1046/j.1471-4159.1999.0722573.x. [DOI] [PubMed] [Google Scholar]
- 17.Warburton MJ, Bernardini F. Tripeptidyl-peptidase I deficiency in classical late-infantile neuronal ceroid lipofuscinosis brain tissue. Evidence for defective peptidase rather than proteinase activity. J Inherit Metab Dis. 2000;23:145–154. doi: 10.1023/a:1005665732189. [DOI] [PubMed] [Google Scholar]
- 18.Koike M, Nakanishi H, Saftig P, Ezaki J, Isahara K, Ohsawa Y, Schulz-Schaeffer W, Watanabe T, Waguri S, Kametaka S, Shibata M, Yamamoto K, Kominami E, Peters C, von Figura K, Uchiyama Y. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–6906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poet M, Steinfeld R, Schweizer M, Kornak U, Jentsch TJ. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005;24:1079–1091. doi: 10.1038/sj.emboj.7600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Y, Ryazantsev S, Ohmi K, Zhao HZ, Rozengurt N, Kohn DB, Neufeld EF. Retrovirally transduced bone marrow has a therapeutic effect on brain in the mouse model of mucopolysaccharidosis IIIB. Mol Genet Metab. 2004;82:286–295. doi: 10.1016/j.ymgme.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Higuti T, Kawamura Y, Kuroiwa K, Miyazaki S, Tsujita H. Molecular cloning and sequence of two cDNAs for human subunit c of H(+)-ATP synthase in mitochondria. Biochim Biophys Acta. 1993;1173:87–90. doi: 10.1016/0167-4781(93)90249-d. [DOI] [PubMed] [Google Scholar]
- 22.Palmer DN, Bayliss SL, Westlake VJ. Batten disease and the ATP synthase subunit c turnover pathway: raising antibodies to subunit c. Am J Med Genet. 1995;57:260–265. doi: 10.1002/ajmg.1320570230. [DOI] [PubMed] [Google Scholar]
- 23.Vines D, Warburton MJ. Purification and characterisation of a tripeptidyl aminopeptidase I from rat spleen. Biochim Biophys Acta. 1998;1384:233–242. doi: 10.1016/s0167-4838(98)00012-0. [DOI] [PubMed] [Google Scholar]
- 24.Huang K, Takagaki M, Kani K, Ohkubo I. Dipeptidyl peptidase II from porcine seminal plasma: purification, characterization, and its homology to granzymes, cytotoxic cell proteinases (CCP 1-4) Biochim Biophys Acta. 1996;1290:149–156. doi: 10.1016/0304-4165(96)00013-x. [DOI] [PubMed] [Google Scholar]
- 25.Barrett AJ, Kirschke H. Cathepsin B, Cathepsin H, and cathepsin L. Methods Enzymol. 1981;80(Pt C):535–561. doi: 10.1016/s0076-6879(81)80043-2. [DOI] [PubMed] [Google Scholar]
- 26.Yoshizaki N, Moriyama A, Yonezawa S. Purification and properties of embryonic cysteine proteinase which participates in yolk-lysis of Xenopus laevis. Comp Biochem Physiol B Biochem Mol Biol. 1998;119:571–576. doi: 10.1016/s0305-0491(98)00030-3. [DOI] [PubMed] [Google Scholar]
- 27.Aranishi F, Hara K, Osatomi K, Ishihara T. Substrate specificity of carp Cyprinus carpio cathepsin H with methylcoumarylamide substrates. Comp Biochem Physiol B Biochem Mol Biol. 1997;116:203–208. doi: 10.1016/s0305-0491(96)00249-0. [DOI] [PubMed] [Google Scholar]
- 28.Yasuda Y, Kageyama T, Akamine A, Shibata M, Kominami E, Uchiyama Y, Yamamoto K. Characterization of new fluorogenic substrates for the rapid and sensitive assay of cathepsin E and cathepsin D. J Biochem (Tokyo) 1999;125:1137–1143. doi: 10.1093/oxfordjournals.jbchem.a022396. [DOI] [PubMed] [Google Scholar]
- 29.Sheehan DC, B.B. H. Theory and Practice of Histotechnology. C.V. Mosby; St. Louis: 1980. [Google Scholar]
- 30.Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 31.Tokuyasu KT. A technique for ultracryotomy of cell suspensions and tissues. J Cell Biol. 1973;57:551–565. doi: 10.1083/jcb.57.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tokuyasu KT. Application of cryoultramicrotomy to immunocytochemistry. J Microsc. 1986;143(Pt 2):139–149. doi: 10.1111/j.1365-2818.1986.tb02772.x. [DOI] [PubMed] [Google Scholar]
- 33.Tokuyasu KT. Use of poly(vinylpyrrolidone) and poly(vinyl alcohol) for cryoultramicrotomy. Histochem J. 1989;21:163–171. doi: 10.1007/BF01007491. [DOI] [PubMed] [Google Scholar]
- 34.Liou W, Geuze HJ, Slot JW. Improving structural integrity of cryosections for immunogold labeling. Histochem Cell Biol. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- 35.Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. U S A. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aleu FP, Terry RD, Zellweger H. Electron Microscopy of Two Cerebral Biopsies in Gargoylism. J Neuropathol Exp Neurol. 1965;24:304–317. doi: 10.1097/00005072-196504000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Ghatak NR, Fleming DF, Hinman A. Neuropathology of Sanfilippo syndrome. Ann. Neurol. 1977;2:161–166. [Google Scholar]
- 38.Knecht E, Martinez-Ramón A, Grisolia S. Autophagy of mitochondria in rat liver assessed by immunogold procedures. J Histoch Cytochem. 1988;36:1433–1440. doi: 10.1177/36.11.3171166. [DOI] [PubMed] [Google Scholar]
- 39.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 40.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 41.Li HH, Yu WH, Rozengurt N, Anagnostaras S, Fanselow M, Gomez-Pinilla F, Suzuki K, Vanier M, Neufeld EF. A mouse model for Sanfilippo syndrome type B, a heritable neurological disorder. J. Neurochem. 1999;73:S23. [Google Scholar]
- 42.Kindler A, Klein U, von Figura K. Characterization of glycosaminoglycans stored in mucopolysaccharidosis III A: evidence for a generally occuring degradation of heparan sulfate by endoglycosidases. Hoppe Seylers Z Physiol Chem. 1977;358:1431–1438. doi: 10.1515/bchm2.1977.358.2.1431. [DOI] [PubMed] [Google Scholar]
- 43.King B, Savas P, Fuller M, Hopwood J, Hemsley K. Validation of a heparan sulfate-derived disaccharide as a marker of accumulation in murine mucopolysaccharidosis type IIIA. Mol Genet Metab. 2006;87:107–112. doi: 10.1016/j.ymgme.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 44.Choudhury A, Sharma DK, Marks DL, Pagano RE. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol Biol Cell. 2004;15:4500–4511. doi: 10.1091/mbc.E04-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marks DL, Pagano RE. Endocytosis and sorting of glycosphingolipids in sphingolipid storage disease. Trends Cell Biol. 2002;12:605–613. doi: 10.1016/s0962-8924(02)02399-1. [DOI] [PubMed] [Google Scholar]
- 46.Aguilar RC, Wendland B. Ubiquitin: not just for proteasomes anymore. Curr Opin Cell Biol. 2003;15:184–190. doi: 10.1016/s0955-0674(03)00010-3. [DOI] [PubMed] [Google Scholar]
- 47.Paxinos G, Franklin KBJ. The Mouse Brain In Stereotactic Coordinates. Academic Press; San Diego: 2001. [Google Scholar]