

Abstract
Hereditary vitamin D resistant rickets (HVDRR) is caused by mutations in the vitamin D receptor (VDR). Here we describe a patient with HVDRR who also exhibited some hypotrichosis of the scalp but otherwise had normal hair and skin. A 102 bp insertion/duplication was found in the VDR gene that introduced a premature stop (Y401X). The patient's fibroblasts expressed the truncated VDR, but were resistant to 1,25(OH)2D3. The truncated VDR weakly bound [3H]-1,25(OH)2D3 but was able to heterodimerize with RXR, bind to DNA and interact with the corepressor hairless (HR). However, the truncated VDR failed to bind coactivators and was transactivation defective. Since the patient did not have alopecia or papular lesions of the skin generally found in patients with premature stop mutations this suggests that this distally truncated VDR can still regulate the hair cycle and epidermal differentiation possibly through its interactions with RXR and HR to suppress gene transactivation.
Keywords: Vitamin D, rickets, alopecia, mutation, vitamin D receptor, gene, calcitriol, hairless
INTRODUCTION
The steroid hormone 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] regulates a number of biological processes including calcium homeostasis, cellular differentiation, and immune function [1]. The activity of 1,25(OH)2D3 is mediated by the vitamin D receptor (VDR), a member of the steroid-thyroid-retinoid receptor superfamily of ligand-activated transcription factors [2-4]. The VDR contains an N-terminus DNA-binding domain (DBD) and a C-terminus ligand-binding domain (LBD). The VDR binds as a heterodimer with the retinoid X receptor (RXR) to specific vitamin D response elements (VDREs) in target genes via a two-zinc finger DBD. Crystallographic analysis of the VDR LBD has shown that it is composed of 13 α-helices (H1, H2, H2n-H12). Ligand binding induces a conformational change in the VDR that repositions the activation function 2 (AF-2) domain in helix H12 and allows for the recruitment and binding of coactivators. Coactivators such as SRC-1, a gp160 family member, and DRIP205, a member of the DRIP/TRAP/Mediator complex, modify chromatin and allow transcription via RNA polymerase II.
Heterogeneous mutations in the VDR gene cause the rare genetic disease hereditary vitamin D resistant rickets (HVDRR), also known as vitamin D dependent rickets type II (VDDR II) [3]. Mutations in the VDR result in partial or total hormone unresponsiveness. The clinical features of HVDRR include early onset rickets, hypocalcemia, and secondary hyperparathyroidism. Patients generally have significantly elevated serum levels of 1,25(OH)2D3 indicative of their resistance to the hormone. In addition, some patients exhibit total body alopecia together with dermal cysts [3]. To date, all HVDRR patients that have nonsense or frameshift mutations in the VDR gene that introduce premature stop signals have alopecia. Alopecia is also associated with HVDRR patients that have mutations that affect DNA binding or heterodimerization with RXR [5-15]. On the other hand, alopecia is not manifested in patients with HVDRR that have mutations that disrupt ligand binding or coactivator binding suggesting that ligand binding and coactivator interactions are not essential functions of the VDR for regulation of hair growth [8, 11, 12, 14-17]. VDR knockout mice, generated by disrupting exon 2 [18] or exon 3 [19] that constitute the DBD, also exhibit alopecia. Hair growth can be restored by specific expression of WT VDR in keratinocytes of VDR knockout mice [20, 21]. Importantly, keratinocyte-targeted expression of transactivation defective mutant VDRs with mutations that affect ligand binding or that prevent coactivator binding also restored hair growth [22]. The knockout mouse data further support our hypothesis based on analyses of HVDRR mutations that control of the hair cycle by the VDR is independent of ligand binding and gene transactivation [14]. The alopecia and skin lesions in some HVDRR patients have been shown to be a phenocopy of the disorder known as atrichia with papular lesions (APL) [23]. APL is caused by mutations in the Hairless gene. The hairless gene product, HR, like the VDR, is involved in regulating hair growth. HR has recently been shown to function as a corepressor with VDR [24], thyroid hormone receptor (TR) [25], and the retinoic acid receptor-related orphan receptor α (RORα) [26, 27]. It has been suggested that VDR and HR regulate a common pathway(s) involved in the hair cycle and epithelial cell differentiation [24].
In this report, we analyzed the molecular defect in a patient with HVDRR without alopecia. We identified a novel insertion/duplication mutation in the VDR gene that introduces a premature stop signal that truncates the VDR. The mutation disrupts ligand binding and coactivator interactions and results in loss of transactivation. This is the first report in which a premature stop mutation in the VDR did not cause total body alopecia or skin lesions in a patient with HVDRR.
MATERIALS AND METHODS
Patient consent and cultured fibroblasts
Informed consent was obtained from the patient and parents under a Stanford University IRB approved protocol. Dermal fibroblasts were cultured from a forearm skin biopsy of the patient as previously described [12].
[3H]1,25(OH)2D3 binding and Western blotting
Cultured fibroblasts from the patient were homogenized at ambient temperature for 10 min on a rotating mixer in M-PER extraction buffer (Pierce) containing 300 mM KCl, 5 mM dithiothreitol, and a complete protease inhibitor tablet (Roche). Cell debris was removed by centrifugation at 210,000 × g for 30 min at 4°C. The crude cell extracts were incubated with [3H]1,25(OH)2D3 with or without 250-fold excess of radioinert 1,25(OH)2D3 and hydroxylapatite was used to separate bound and free hormone as previously described [28]. For western blotting, samples were denatured in lithium-dodecylsulfate sample buffer for 10 min at 70°C and electrophoresed on 10% NuPAGE gels in MOPS-SDS running buffer (Invitrogen). Proteins were transferred to nitrocellulose and incubated with a rabbit anti-VDR polyclonal antibody N-20 (Santa Cruz Biotechnology, Santa Cruz, CA) as previously described [13]. Protein concentrations were determined by the Bradford method [29].
Gene amplification and DNA sequencing
Exons 2-9 of the VDR gene were amplified by PCR and directly sequenced at the Stanford protein and nucleic acid facility. For amplified fragment length polymorphism analysis, exon 9 was amplified from the patient and a normal control and PCR products separated on 1% agarose gels.
Real time RT-PCR
The patient's fibroblasts were treated with 1,25(OH)2D3 for 6 hr in medium containing 1% calf serum. RNA was isolated using RNAeasy spin columns (Qiagen). cDNA was prepared by reverse transcription using superscript III cDNA synthesis kit (Invitrogen). CYP24A1 and TBP genes were then amplified from the cDNA using SYBR-green qPCR kit (New England Biolabs) and semi-quantified using real time PCR.
Plasmid Construction
The Y401X mutation was recreated by site directed mutagenesis using the GeneEditor Mutagenesis kit (Promega) as previously described [14].
Gel Mobility Shift
Gel mobility shift assays were performed as previously described [15]. WT and mutant VDRs were expressed in COS-7 cells. Cell extracts were incubated with [32P]-labeled osteopontin VDRE in the presence and absence of 10 nM 1,25(OH)2D3 for 30 min at ambient temperature. For supershift assays, an α-VDR antibody (C-20, Santa Cruz Biotechnology) was added and incubation continued for 20 min. The samples were then resolved on non-denaturing gels and subjected to autoradiography.
GST-pull down assays
VDR proteins were synthesized using the quick-coupled in vitro transcription/translation system (Promega). For RXR binding, GST-RXR was incubated with in vitro synthesized [35S]-labeled VDR proteins. Bound proteins were subjected to SDS-PAGE and visualized by autoradiography as previously described [15]. For SRC-1 and DRIP205 binding, GST-SRC-1 or GST-DRIP205 was incubated with unlabeled in vitro synthesized VDR proteins. Bound proteins were subjected to western blotting and visualized using ECL plus (Amersham).
Co-immunoprecipitation
For co-immunoprecipitation, HR and VDRs were co-expressed in COS-1 cells. Co-immunoprecipitation was performed as previously described [24].
RESULTS
Case History
The patient is a young black Jamaican boy who was orphaned at birth so parental consanguinity is unknown. The child initially presented at 9 months of age with early onset severe rickets, secondary hyperparathyroidism, hypocalcemia and hypophosphatemia, elevated alkaline phosphatase, failure to thrive, and growth retardation. The patient exhibited hypotrichosis of the scalp with patchy hair and normal eyelashes and eyebrows (Fig. 1). His skin and teeth were normal. Joints were abnormal with widened, splayed wrists and a rachitic rosary. Height and weight, which had been 90th percentile at birth, had fallen to 5th percentile. Initial lab results were Ca 8.0, PO4 2.6, alkaline phosphatase 2990, PTH 1021 pg/mL, 25-hydroxyvitamin D 10.5 ng/mL and 1,25-dihydroxyvitamin D 138.3 pg/ml (Table 1). X-rays showed osteopenia and classical rickets. Urine calcium and phosphate were 0.04 and 0.55 mg/gm creatinine, respectively. Over the next 2 years the child failed to improve despite vitamin D, Rocaltrol© (calcitriol) and oral calcium and phosphate supplements. He was hypotonic and resisted bearing weight on his arms or legs. His milestones were all delayed. He stopped eating and eventually was fed via a gastrostomy tube. Repeat x-rays showed multiple old fractures in addition to classical findings of rickets. At 19 months of age it was noted that he had increased lanugo hair on his back, forehead and sideburns. Bowing of the legs was obvious.
Figure 1.
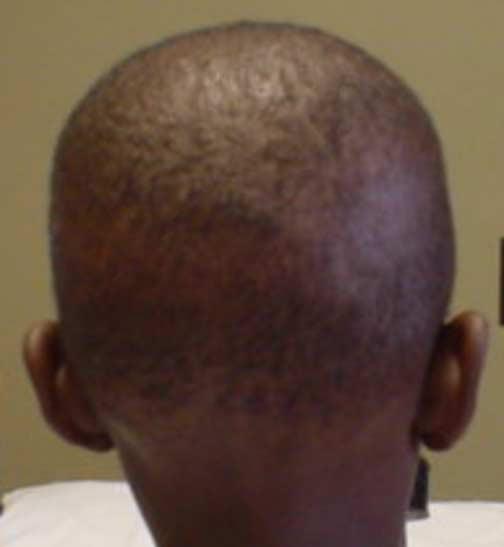
Photograph of patient's showing sparse and patchy hair on his scalp.
Table 1.
Patient's clinical findings over time.
| Analyte | NL. range | units | 9 mo | 23 mo | 37 mo | 43 mo | 46 mo |
|---|---|---|---|---|---|---|---|
| Calcium | 8.0-10.5 | mg/dL | 8.0 | 8.2 | 8.5 | 9.5 | 9.6 |
| Phosphorus | 3.2-6.3 | Mg/dL | 2.6 | 1.9 | 5.1 | 3.9 | |
| Alk p'tase | 100-390 | U/L | >2990 | 5714 | 1658 | 451 | 470 |
| PTH | 11-67 | pg/mL | 1021 | 144 | 51 | ||
| 1,25vitD | 15-90 | pg/mL | 138 | 185 |
IV Ca++ was started after 23 months. NL, normal; mo, months; Alk p'tase, Alkaline Phosphatase.
Intravenous (IV) calcium infusions were started at 2 years and 7 months of age and continued nightly. He received 42 mg of elemental calcium/kg/day or 0.95 gms/m2/day. On this treatment chemistries started to improve (Table 1) and the rickets and other signs and symptoms slowly started to regress. Within 6 months of starting IV calcium (37 months of age), chemistries showed substantial improvement. By 3 years and 4 months of age the excess hair on his face and back had receded. His scalp hair remained patchy. Gradually strength and motor skills improved, normal activity resumed and the child began to eat adequately so that the g-tube could be removed. By 3 years and 7 months the rachitic rosary had receded and the chest cage started to normalize. Splaying of the wrists was reduced and x-rays showed healing of the rickets. All of his chemistries had normalized except for a residual mild elevation in PTH (Table 1). Hypotonia improved and the child resumed normal activity. By 6 years and 6 months there was complete resolution of rickets. When IV calcium infusions were tried at every other night instead of nightly, his PTH levels doubled within 4 weeks and he was returned to nightly infusions. Over time growth velocity improved and there was slight catch-up growth seen. 1,25(OH)2D3 remained elevated, presumably because high doses of Rocaltrol© (3 μg/day) were continued. However, there was no hypercalcemia demonstrating continued resistance to calcitriol action.
Molecular Analyses
To analyze the molecular basis or HVDRR in this patient, a skin biopsy was obtained from the patient and used to generate cultured skin fibroblasts. We first determined whether the patient's fibroblasts could respond to treatment with 1,25-(OH)2D3 or whether they were resistant to 1,25-(OH)2D3. We treated normal fibroblasts and the patient's fibroblasts with increasing concentrations of 1,25-(OH)2D3 and measured the induction of the 24-hydroxylase gene (CYP24A1). As shown in Fig. 2, while normal fibroblasts exhibited a dose-dependent rise in 1,25-(OH)2D3 induction of CYP24A1 mRNA, the patient's cells failed to induce CYP24A1 gene expression when the cells were treated with up to 1 μM 1,25-(OH)2D3 clearly demonstrating total 1,25-(OH)2D3 resistance.
Figure 2.
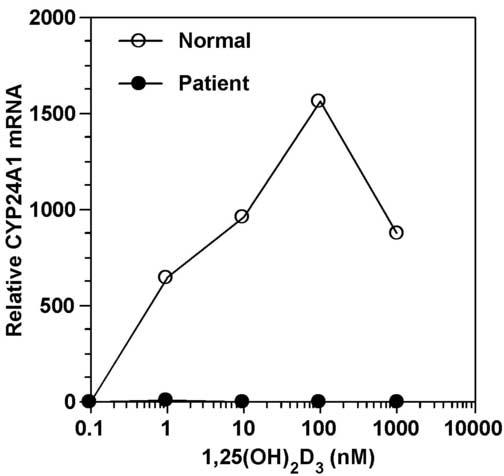
The patient's fibroblasts are resistant to 1,25(OH)2D3. Fibroblasts from the patient and a normal control were treated with 1,25(OH)2D3 for 6 hr. Induction of CYP24A1 gene expression by 1,25(OH)2D3 was analyzed by real time RT-PCR. Values were normalized to TBP expression.
The absence of a response to 1,25-(OH)2D3 in the patients fibroblasts suggested that there may be a defect in the VDR. To elucidate whether the molecular basis of the 1,25-(OH)2D3 resistance was due to a mutation in the VDR, we sequenced exons 2-9 of the VDR gene. A novel 102 bp insertion/duplication was found in exon 9 (Fig. 3A). The mutation introduced a premature stop signal at amino acid 401 (Y401X) in helix H11. The mutation deletes 27 amino acids from the C-terminus including all of helix H12 (Fig. 3B). Using the difference in length created by the 102 base pair insertion in exon 9 in the patient's DNA vs. the normal size of exon 9, we determined that the patient was homozygous for the insertion/duplication mutation (Fig. 3C).
Figure 3.
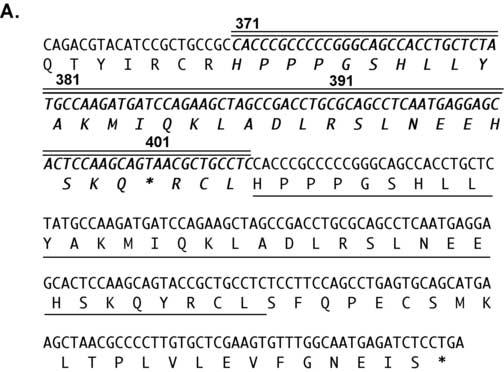
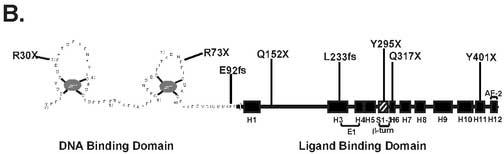
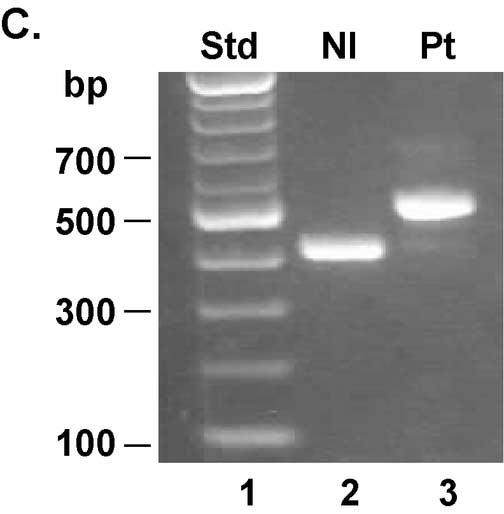
A unique 102 bp insertion/duplication was identified in exon 9 that introduced a premature stop codon truncating the VDR at amino acid 401. Panel A: Exon 9 of the VDR gene was amplified by PCR and sequenced directly. The 102 bp insertion/duplication is shown in boldface italics type with double lines above. The single line below the amino acid sequence is the region that is duplicated. The boldface asterisk indicates the location of the premature stop at amino acid 401. The un-bolded asterisk indicates the normal termination signal. Panel B: Schematic diagram of the VDR and location of premature stop signals due to nonsense or frameshift (fs) mutations. Panel C: Gel electrophoresis of PCR amplification products of exon 9 from a normal control (lane 2) and the patient (lane 3). Lane 1 shows size standard. The difference in the size of exon 9 in the patient is due to the 102 bp insertion.
We next determined whether the truncated VDR was expressed in the patient's fibroblasts. As shown in the western blot in Fig. 4, the normal VDR was detected in cell extracts from normal fibroblasts (lane 1) while a truncated VDR was present in the patient's fibroblasts (lane 2) consistent with the size predicted by the Y401X mutation. In contrast, a truncated form of the VDR was not detected in fibroblasts from a patient with HVDRR due to a premature stop signal at amino acid 295 (Y295X) (lane 3). We have previously demonstrated that the Y295X mutant VDR is undetectable in patient's fibroblasts most likely due to nonsense-mediated decay of the messenger RNA [28].
Figure 4.
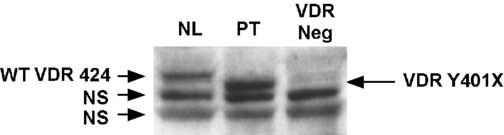
The patient's fibroblasts express a truncated VDR. Cell extracts from the patient's fibroblasts were subjected to western blotting using a VDR polyclonal antibody to the N-terminus of the VDR. The antibody detected the WT VDR protein (424 amino acids) in normal fibroblasts and a truncated protein (400 amino acids) in the patient's fibroblasts. No VDR protein was detected in fibroblasts from an HVDRR patient that does not express the mutant protein due to nonsense-mediated decay. NS, non-specific bands.
Since the mutation deleted a significant part of helix H11 and all of helix H12 in the VDR LBD, we next analyzed whether the truncated VDR was capable of binding 1,25(OH)2D3. Using [3H]1,25-(OH)2D3 binding assays, no specific binding was observed at low concentrations of [3H]1,25-(OH)2D3 and only a trace of specific binding was observed at 1 nM [3H]1,25-(OH)2D3 (data not shown). We did not examine the binding at higher concentrations of [3H]1,25-(OH)2D3.
To demonstrate that the mutation we described in exon 9 actually causes the 1,25-(OH)2D3 resistance, we recreated the Y401X mutation in the VDR cDNA. In transactivation assays in COS-7 cells (Fig. 5), the WT VDR exhibits a dose-response to 1,25-(OH)2D3 in activating the CYP24A1 promoter reporter while the Y401X mutant VDR was totally defective in transactivation.
Figure 5.
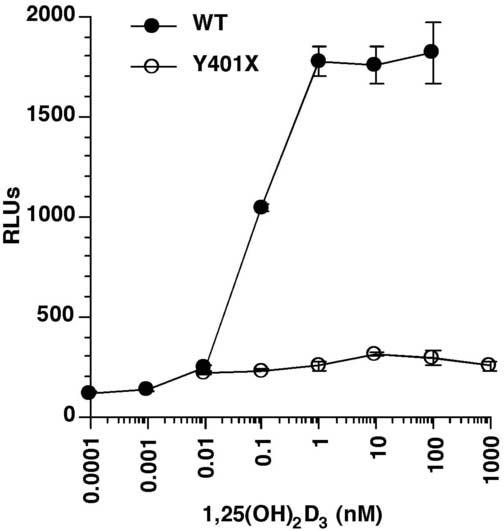
The truncated VDR is transactivation incompetent. The Y401X mutant and WT VDRs were transfected into COS-7 cells along with the CYP24A1 promoter luciferase reporter. The cells were treated with graded concentrations of 1,25(OH)2D3 for 24 hr and luciferase activity determined. The mutant VDR showed no stimulation of the CYP24A1 promoter as compared to the WT VDR that exhibited a dose-dependent stimulation of the promoter activity.
Based on the lesion that was discovered one might predict that the truncated VDR would retain the ability to form a heterodimer with RXRα and to bind to DNA. As shown in GST-pull down assays in Fig. 6A, the WT VDR bound RXRα in the absence of 1,25-(OH)2D3. Addition of graded concentrations of 1,25-(OH)2D3 resulted in an increase in WT VDR binding to RXRα. On the other hand, the truncated VDR bound RXRα in the absence of 1,25-(OH)2D3 similar to the WT VDR but failed to exhibit an increase in binding to RXRα in the presence of graded concentrations of 1,25-(OH)2D3. The presence of several forms of VDR seen in Fig. 6A is most likely due to usage of different methionine start sites in the in vitro translation reaction. As shown in gel mobility shift assays in Fig. 6B, the truncated VDR bound to the osteopontin VDRE in the absence of 1,25-(OH)2D3 similar to the WT VDR demonstrating that the mutant VDR retained the ability to interact with DNA. However, addition of 1,25-(OH)2D3 only slightly increased the binding of the mutant VDR to the osteopontin VDRE whereas the WT VDR exhibited a substantial increase in binding in the presence of 1,25-(OH)2D3. Addition of anti-VDR antibodies that recognized the C-terminal region of the VDR caused a supershift of the WT VDR but no shift in the mutant VDR. Only faint bands were observed in the pSG5 vector control.
Figure 6.
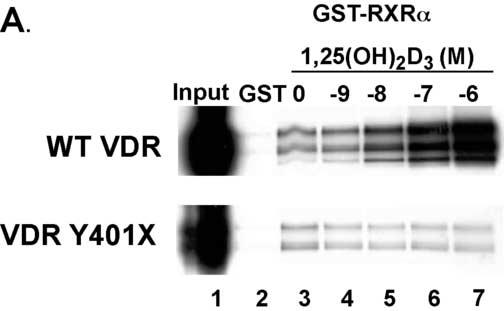
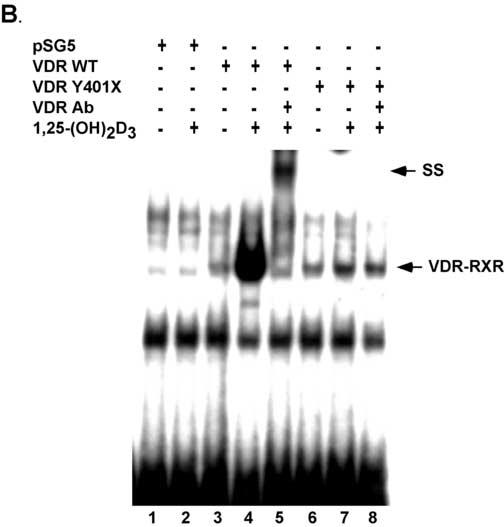
The truncated VDR binds RXRα and to VDREs. Panel A. VDRs labeled with [35S]-methionine by in vitro coupled transcription-translation were incubated with GST-RXRα in the presence of vehicle or graded concentrations of calcitriol. The samples were then subjected to GST-pull down assays and SDS-PAGE. Bands were visualized by autoradiography. The autoradiograph shows that the truncated VDR binds weakly to GST-RXRα and does not exhibit a 1,25(OH)2D3 dependent increase in binding as exhibited by the WT VDR. Panel B. Empty vector (pSG5) and WT and Y401X VDRs were expressed in COS-7 cells. Cell extracts were incubated with [32P]-labeled osteopontin VDRE with and without 10 nM 1,25(OH)2D3. For supershift assays VDR antibody was added. SS, supershift.
We next tested whether the truncated VDR could interact with coactivators since coactivators bind at the AF-2 domain at the C-terminus. Since the AF-2 domain was deleted one might predict that the coactivators would not bind. Using GST-pull down assays (Fig. 7A) we show that the WT VDR bound the coactivators SRC-1 and DRIP205 in a ligand-dependent manner. As expected the truncated VDR failed to bind either coactivator. We then tested another regulatory protein, the corepressor HR, for binding to the truncated VDR. Since HR has been shown to interact with a laboratory created truncated VDR (VDRΔ403-427) [24] one might predict that HR would bind to the Y401X VDR. To examine the HR interaction with VDR we co-expressed c-myc-tagged rat HR with WT VDR or Y401X VDR and immunoprecipitated the VDR using anti-VDR antibodies. HR was then detected in the immunoprecipitates on western blots using anti-c-myc antibodies. As shown in Fig. 7B, using in vivo co-immunoprecipitation assays we demonstrate that the truncated VDR interacts with HR similar to the WT VDR.
Figure 7.
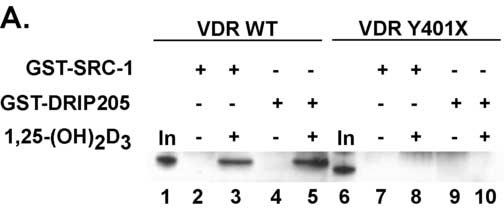
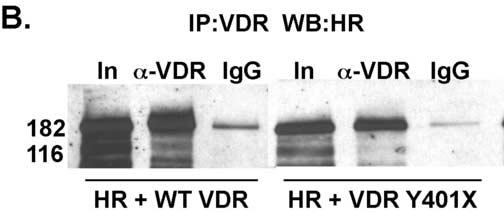
The mutant VDR fails to bind coactivators but interacts with the corepressor hairless. Panel A. VDRs were synthesized by in vitro coupled transcription-translation and incubated with GST-SRC-1 or GST-DRIP205 in the presence of vehicle or 100 nM 1,25(OH)2D3. The samples were then subjected to GST-pull down assays. Bands were detected by western blot. No interactions were observed between the truncated VDR and the coactivators while the WT VDR exhibited a ligand-dependent interaction with both coactivators. Panel B. Mutant and WT VDRs were co-expressed with myc-tagged rat HR in COS-7 cells. Cell extracts were prepared and proteins immunoprecipitated with VDR or non-specific IgG antibodies. HR was detected by western blot using α-c-myc antibodies. HR was immunoprecipitated by both the WT VDR and Y401X mutant VDR demonstrating that the mutant VDR interacts with HR in vivo. A small amount of binding was observed with the control IgG. In, input; α-VDR, immunoprecipitating antibody; IgG, non-specific control immunoprecipitating antibody.
DISCUSSION
The child described here, a young boy born in Jamaica, exhibited the classical clinical pattern of HVDRR. He initially presented with early onset rickets, secondary hyperparathyroidism, and hypocalcemia. He also exhibited elevated serum 1,25-(OH)2D3 levels. He exhibits hypotrichosis or sparse and patchy hair on his scalp and has normal eyelashes and eyebrows and therefore does not have total alopecia. We identified a novel 102 bp insertion/duplication in exon 9 of the VDR gene. The 102 bp sequence is a duplication of the sequence that encodes amino acids 371 to 404. Also in this 102 bp insertion there is a C to A conversion in the codon for amino acid Y401. This conversion creates a premature stop signal (TAC to TAA). The resulting Y401X mutation truncates 27 amino acids from the VDR and is the molecular cause of 1,25-(OH)2D3 resistance in this patient with HVDRR.
The patient's fibroblasts expressed the truncated VDR but the cells were resistant to 1,25-(OH)2D3 treatment as evidenced by failure of even high concentrations (1 μM) of 1,25-(OH)2D3 to induce CYP24A1. Previous studies using a laboratory created 25 amino acid deletion of the VDR (Δ403-427) demonstrated that the VDRΔ403-427 bound 1,25-(OH)2D3 weakly and failed to activate gene transcription [30]. However, the VDRΔ403-427 was able to form a complex with RXR and bind to a VDRE. Similarly, the truncated VDR Y401X mutant described here had defective 1,25-(OH)2D3 binding and was inactive in transactivation assays but was able to heterodimerize with RXR and bind to DNA although not in a ligand-dependent manner.
The VDR LBD is composed of 12 α-helices and 3 β-sheets forming a hydrophobic core that is occupied by the ligand [31]. The ligand-binding pocket is formed by helixes H1, H3, H4, H5, H7, H8, H9, H10 and H11 (Fig. 3B) [31]. When the receptor is bound to ligand, helix H12 is repositioned forming a binding site for coactivators. The premature termination signal in helix H11 at amino acid Y401X deletes part of helix H11 including Y401 that is a contact point for the ligand thereby greatly attenuating ligand binding [31]. The mutation also deletes all of helix H12 resulting in the elimination of the coactivator-binding site. As expected, the truncated VDR failed to interact with the coactivators SRC-1 and DRIP205.
Of the HVDRR patients that have been analyzed to date, alopecia has been associated with mutations that affect DNA binding [5-7, 32-34], RXR heterodimerization [11, 13, 35] and mutations that create premature stop signals [8, 28, 35-41]. All of the previously described premature stop mutations occurred more N-terminal to the Y401X stop mutation described here (Fig. 3B) and disrupt DNA and/or RXR binding. These findings suggest that RXR heterodimerization and DNA binding are essential functions of the VDR that are required to prevent alopecia. On the other hand, alopecia is not present in HVDRR patients with mutations in the VDR that affect ligand binding [8, 12, 16, 17] or coactivator interactions [14]. These findings suggest that ligand binding and coactivator interactions as well as gene transactivation are dispensable functions of the VDR in the prevention of alopecia.
Several cases of HVDRR caused by premature stop signals have been described [8, 28, 35-41]. In some cases the mutant mRNA was detected while in other cases the mutant mRNA was not detected possibly due to a phenomenon known as nonsense-mediated RNA decay [28]. However, all of the previously reported cases with premature stop signals exhibited total body alopecia. It is interesting to note that the patient in this study does not have alopecia. He does have patchy hair distribution on the scalp and he also has normal eyebrows and eyelashes. The truncated VDR, that we have shown is expressed in the patient's fibroblasts, apparently retains enough function to allow for some hair growth and prevent alopecia. We have no explanation for the transient increase in lanugo hair on the boy's back and face that eventually regressed.
Since the truncated VDR lacks transactivation, what is the activity that allows hair growth? The Y401X mutation severely reduces ligand binding and eliminates coactivator interactions but retains some ability to heterodimerize with RXR, interact with the co-repressor HR and bind to DNA. These activities may prevent alopecia. Although the mutant receptor fails to activate gene transcription, it is apparently able to function as a repressor of gene transcription due to its ability to interact with HR. This case strengthens the hypothesis that one function of the unliganded VDR is to regulate a gene or set of genes involved in hair cycling by suppressing gene transactivation by interaction with the co-repressor HR [14, 22, 24]. Mutations in VDR and HR that disrupt the ability to suppress gene transactivation cause dysregulation of these important hair cycling genes that when inappropriately expressed disrupt the normal hair cycling process.
Recent evidence has indicated a possible mechanism for alopecia involving the Wnt signaling pathway. Thompson's group has shown that the Wnt inhibitor Wise/Ectodin/Sosdc1 is up-regulated in the skin of HR knockout mice [42]. Since the Wnt-signaling pathway is important in hair cycling this suggests that dysregulation of Wise/Ectodin/Sosdc1 during the hair cycle may inhibit Wnt signaling and provide a mechanism to prevent normal hair cycle development.
Like some HVDRR patients, VDR knockout mice also have alopecia. Chen et al [20] and Kong et al [21] have shown that specific expression of WT VDR in keratinocytes of VDR knockout mice can restore hair growth. Skorija et al [22] have recently shown that keratinocyte-targeted expression of either a mutant VDR (L233S) that does not bind ligand or a mutant VDR (L417S) that is defective in binding coactivators can restore hair growth in VDR knockout mice. However, the mice with the L417S VDR transgene exhibited significant progressive hair loss over time. The authors speculated that the VDR complex may require interactions with other factors for maintaining long-term hair follicle homeostasis [22]. Whether the mechanism for maintaining hair growth in mice is similar in humans remains to be determined. Based on the findings in transgenic mice one might expect that the patient with the deletion of helix H12 (including residue L417) would exhibit a case of more severe hair loss. However, the patient described here does not have alopecia. We have also previously described an E420K mutation in helix H12 in the VDR that eliminated coactivator interactions and transactivation in an HVDRR patient that did not have alopecia [14]. Whether the E420K mutation will have the same affect as the L417S mutation on causing the child to develop alopecia overtime is unknown. Unlike the mouse model, we are unaware of any cases of HVDRR that developed alopecia later in life after having normal hair as a child. It will be interesting to see if these two patients lose their hair as they get older.
In conclusion, we have identified a novel 102 bp insertion/duplication in the VDR gene associated with a premature stop mutation at codon 401 that truncates the VDR as the cause of 1,25-(OH)2D3 resistance in a patient with HVDRR without alopecia. Our findings support the hypothesis that VDR regulates hair growth by ligand and coactivator independent mechanisms through its interactions with RXR and HR to suppress gene transactivation.
Footnotes
Supported by NIH Grant DK42482 (to D.F.)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Feldman D, Glorieux FH, Pike JW, Vitamin D, editors. Elsevier Academic Press; San Diego: 2005. [Google Scholar]
- 2.Haussler MR, Whitfield GK, Haussler CA, Hsieh J, Thompson PD, Selznick SH, Dominguez CE, Jurutka PW. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J. Bone Min. Res. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 3.Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25- dihydroxyvitamin D-resistant rickets. Endocr Rev. 1999;20:156–188. doi: 10.1210/edrv.20.2.0359. [DOI] [PubMed] [Google Scholar]
- 4.Rachez C, Freedman LP. Mechanisms of gene regulation by vitamin D3 receptor: a network of coactivator interactions. Gene. 2000;246:9–21. doi: 10.1016/s0378-1119(00)00052-4. [DOI] [PubMed] [Google Scholar]
- 5.Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D, O'Malley BW. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science. 1988;242:1702–1705. doi: 10.1126/science.2849209. [DOI] [PubMed] [Google Scholar]
- 6.Sone T, Marx SJ, Liberman UA, Pike JW. A unique point mutation in the human vitamin D receptor chromosomal gene confers hereditary resistance to 1,25-dihydroxyvitamin D3. Mol. Endocrinol. 1990;4:623–631. doi: 10.1210/mend-4-4-623. [DOI] [PubMed] [Google Scholar]
- 7.Saijo T, Ito M, Takeda E, Huq AH, Naito E, Yokota I, Sone T, Pike JW, Kuroda Y. A unique mutation in the vitamin D receptor gene in three Japanese patients with vitamin D-dependent rickets type II: utility of single-strand conformation polymorphism analysis for heterozygous carrier detection. Am. J. Hum. Genet. 1991;49:668–673. [PMC free article] [PubMed] [Google Scholar]
- 8.Kristjansson K, Rut AR, Hewison M, O'Riordan JL, Hughes MR. Two mutations in the hormone binding domain of the vitamin D receptor cause tissue resistance to 1,25 dihydroxyvitamin D3. J. Clin. Invest. 1993;92:12–16. doi: 10.1172/JCI116539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malloy PJ, Weisman Y, Feldman D. Hereditary 1 alpha,25-dihydroxyvitamin D-resistant rickets resulting from a mutation in the vitamin D receptor deoxyribonucleic acid-binding domain. J. Clin. Endocrinol. Metab. 1994;78:313–316. doi: 10.1210/jcem.78.2.8106618. [DOI] [PubMed] [Google Scholar]
- 10.Lin NU-T, Malloy PJ, Sakati N, Al-Ashwal A, Feldman D. A novel mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor gene causes hereditary 1,25-dihydroxyvitamin D resistant rickets. J. Clin. Endocrinol. Metab. 1996;81:2564–2569. doi: 10.1210/jcem.81.7.8675579. [DOI] [PubMed] [Google Scholar]
- 11.Whitfield GK, Selznick SH, Haussler CA, Hsieh JC, Galligan MA, Jurutka PW, Thompson PD, Lee SM, Zerwekh JE, Haussler MR. Vitamin D receptors from patients with resistance to 1,25-dihydroxyvitamin D3: point mutations confer reduced transactivation in response to ligand and impaired interaction with the retinoid X receptor heterodimeric partner. Mol Endocrinol. 1996;10:1617–1631. doi: 10.1210/mend.10.12.8961271. [DOI] [PubMed] [Google Scholar]
- 12.Malloy PJ, Eccleshall TR, Gross C, Van Maldergem L, Bouillon R, Feldman D. Hereditary vitamin D resistant rickets caused by a novel mutation in the vitamin D receptor that results in decreased affinity for hormone and cellular hyporesponsiveness. J Clin Invest. 1997;99:297–304. doi: 10.1172/JCI119158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malloy PJ, Zhu W, Zhao XY, Pehling GB, Feldman D. A novel inborn error in the ligand-binding domain of the vitamin D receptor causes hereditary vitamin D-resistant rickets. Mol Genet Metab. 2001;73:138–148. doi: 10.1006/mgme.2001.3181. [DOI] [PubMed] [Google Scholar]
- 14.Malloy PJ, Xu R, Peng L, Clark PA, Feldman D. A novel mutation in helix 12 of the vitamin D receptor impairs coactivator interaction and causes hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Mol Endocrinol. 2002;16:2538–2546. doi: 10.1210/me.2002-0152. [DOI] [PubMed] [Google Scholar]
- 15.Malloy PJ, Xu R, Peng L, Peleg S, Al-Ashwal A, Feldman D. Hereditary 1,25-dihydroxyvitamin D resistant rickets due to a mutation causing multiple defects in vitamin D receptor function. Endocrinology. 2004;145:5106–5114. doi: 10.1210/en.2004-0080. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen TM, Adiceam P, Kottler ML, Guillozo H, Rizk-Rabin M, Brouillard F, Lagier P, Palix C, Garnier JM, Garabedian M. Tryptophan missense mutation in the ligand-binding domain of the vitamin D receptor causes severe resistance to 1,25-dihydroxyvitamin D. J Bone Miner Res. 2002;17:1728–1737. doi: 10.1359/jbmr.2002.17.9.1728. [DOI] [PubMed] [Google Scholar]
- 17.Malloy PJ, Xu R, Cattani A, Reyes L, Feldman D. A unique insertion/substitution in helix H1 of the vitamin D receptor ligand binding domain in a patient with hereditary 1,25-dihydroxyvitamin D-resistant rickets. J Bone Miner Res. 2004;19:1018–1024. doi: 10.1359/jbmr.2004.19.6.1018. [DOI] [PubMed] [Google Scholar]
- 18.Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, Kawakami T, Arioka K, Sato H, Uchiyama Y, Masushige S, Fukamizu A, Matsumoto T, Kato S. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genet. 1997;16:391–396. doi: 10.1038/ng0897-391. [DOI] [PubMed] [Google Scholar]
- 19.Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–9835. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen CH, Sakai Y, Demay MB. Targeting expression of the human vitamin D receptor to the keratinocytes of vitamin D receptor null mice prevents alopecia. Endocrinology. 2001;142:5386–5389. doi: 10.1210/endo.142.12.8650. [DOI] [PubMed] [Google Scholar]
- 21.Kong J, Li XJ, Gavin D, Jiang Y, Li YC. Targeted expression of human vitamin d receptor in the skin promotes the initiation of the postnatal hair follicle cycle and rescues the alopecia in vitamin D receptor null mice. J Invest Dermatol. 2002;118:631–638. doi: 10.1046/j.1523-1747.2002.01727.x. [DOI] [PubMed] [Google Scholar]
- 22.Skorija K, Cox M, Sisk JM, Dowd DR, MacDonald PN, Thompson CC, Demay MB. Ligand-independent actions of the vitamin D receptor maintain hair follicle homeostasis. Mol Endocrinol. 2005;19:855–862. doi: 10.1210/me.2004-0415. [DOI] [PubMed] [Google Scholar]
- 23.Miller J, Djabali K, Chen T, Liu Y, Ioffreda M, Lyle S, Christiano AM, Holick M, Cotsarelis G. Atrichia caused by mutations in the vitamin D receptor gene is a phenocopy of generalized atrichia caused by mutations in the hairless gene. J Invest Dermatol. 2001;117:612–617. doi: 10.1046/j.0022-202x.2001.01438.x. [DOI] [PubMed] [Google Scholar]
- 24.Hsieh JC, Sisk JM, Jurutka PW, Haussler CA, Slater SA, Haussler MR, Thompson CC. Physical and functional interaction between the vitamin D receptor and hairless corepressor, two proteins required for hair cycling. J Biol Chem. 2003;278:38665–38674. doi: 10.1074/jbc.M304886200. [DOI] [PubMed] [Google Scholar]
- 25.Potter GB, Beaudoin GM, 3rd, DeRenzo CL, Zarach JM, Chen SH, Thompson CC. The hairless gene mutated in congenital hair loss disorders encodes a novel nuclear receptor corepressor. Genes Dev. 2001;15:2687–2701. doi: 10.1101/gad.916701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moraitis AN, Giguere V, Thompson CC. Novel mechanism of nuclear receptor corepressor interaction dictated by activation function 2 helix determinants. Mol Cell Biol. 2002;22:6831–6841. doi: 10.1128/MCB.22.19.6831-6841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moraitis AN, Giguere V. The co-repressor hairless protects RORalpha orphan nuclear receptor from proteasome-mediated degradation. J Biol Chem. 2003;278:52511–52518. doi: 10.1074/jbc.M308152200. [DOI] [PubMed] [Google Scholar]
- 28.Malloy PJ, Hochberg Z, Tiosano D, Pike JW, Hughes MR, Feldman D. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. J. Clin. Invest. 1990;86:2071–2079. doi: 10.1172/JCI114944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 30.Nakajima S, Hsieh JC, MacDonald PN, Galligan MA, Haussler CA, Whitfield GK, Haussler MR. The C-terminal region of the vitamin D receptor is essential to form a complex with a receptor auxiliary factor required for high affinity binding to the vitamin D-responsive element. Mol. Endocrinol. 1994;8:159–172. doi: 10.1210/mend.8.2.8170472. [DOI] [PubMed] [Google Scholar]
- 31.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 32.Yagi H, Ozono K, Miyake H, Nagashima K, Kuroume T, Pike JW. A new point mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor in a kindred with hereditary 1,25-dihydroxyvitamin D-resistant rickets. J. Clin. Endocrinol. Metab. 1993;76:509–512. doi: 10.1210/jcem.76.2.8381803. [DOI] [PubMed] [Google Scholar]
- 33.Rut AR, Hewison M, Kristjansson K, Luisi B, Hughes MR, O'Riordan JL. Two mutations causing vitamin D resistant rickets: modelling on the basis of steroid hormone receptor DNA-binding domain crystal structures. Clin. Endocrinol. 1994;41:581–590. doi: 10.1111/j.1365-2265.1994.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 34.Lin NU, Malloy PJ, Sakati N, al-Ashwal A, Feldman D. A novel mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. J Clin Endocrinol Metab. 1996;81:2564–2569. doi: 10.1210/jcem.81.7.8675579. [DOI] [PubMed] [Google Scholar]
- 35.Cockerill FJ, Hawa NS, Yousaf N, Hewison M, O'Riordan JL, Farrow SM. Mutations in the vitamin D receptor gene in three kindreds associated with hereditary vitamin D resistant rickets. J Clin Endocrinol Metab. 1997;82:3156–3160. doi: 10.1210/jcem.82.9.4243. [DOI] [PubMed] [Google Scholar]
- 36.Ritchie HH, Hughes MR, Thompson ET, Malloy PJ, Hochberg Z, Feldman D, Pike JW, O'Malley BW. An ochre mutation in the vitamin D receptor gene causes hereditary 1,25-dihydroxyvitamin D3-resistant rickets in three families. Proc. Natl. Acad. Sci. USA. 1989;86:9783–9787. doi: 10.1073/pnas.86.24.9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiese RJ, Goto H, Prahl JM, Marx SJ, Thomas M, al-Aqeel A, DeLuca HF. Vitamin D-dependency rickets type II: truncated vitamin D receptor in three kindreds. Mol. Cell. Endocrinol. 1993;90:197–201. doi: 10.1016/0303-7207(93)90152-a. [DOI] [PubMed] [Google Scholar]
- 38.Hawa NS, Cockerill FJ, Vadher S, Hewison M, Rut AR, Pike JW, O'Riordan JL, Farrow SM. Identification of a novel mutation in hereditary vitamin D resistant rickets causing exon skipping. Clin. Endocrinol. 1996;45:85–92. [PubMed] [Google Scholar]
- 39.Mechica JB, Leite MO, Mendonca BB, Frazzatto ES, Borelli A, Latronico AC. A novel nonsense mutation in the first zinc finger of the vitamin D receptor causing hereditary 1,25-dihydroxyvitamin D3-resistant rickets. J Clin Endocrinol Metab. 1997;82:3892–3894. doi: 10.1210/jcem.82.11.4384. [DOI] [PubMed] [Google Scholar]
- 40.Zhu W, Malloy PJ, Delvin E, Chabot G, Feldman D. Hereditary 1,25-dihydroxyvitamin D-resistant rickets due to an opal mutation causing premature termination of the vitamin D receptor. J Bone Miner Res. 1998;13:259–264. doi: 10.1359/jbmr.1998.13.2.259. [DOI] [PubMed] [Google Scholar]
- 41.Malloy PJ, Zhu W, Bouillon R, Feldman D. A novel nonsense mutation in the ligand binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. Mol Genet Metab. 2002;77:314–318. doi: 10.1016/s1096-7192(02)00173-7. [DOI] [PubMed] [Google Scholar]
- 42.Beaudoin GM, 3rd, Sisk JM, Coulombe PA, Thompson CC. Hairless triggers reactivation of hair growth by promoting Wnt signaling. Proc Natl Acad Sci U S A. 2005;102:14653–14658. doi: 10.1073/pnas.0507609102. [DOI] [PMC free article] [PubMed] [Google Scholar]