

Abstract
Apoptotic protease activating factor-1 (Apaf-1) is a critical regulator of apoptosis and a crucial part of the apoptosome that is assembled in response to several cellular stresses like hypoxia. We have previously shown that hypoxia results in increased influx of nuclear Ca++ and increased expression of nuclear apoptotic proteins. The present study investigates that Apaf-1 is expressed during hypoxia in the cerebral cortex of newborn piglets and that administration of clonidine prevents the hypoxia induced increase expression of Apaf-1. Studies were conducted in 19 newborn piglets, 6 normoxic (Nx), 7 hypoxic (Hx FiO2 of 0.05-0.07 for 1 hr) and 6 clonidine-treated hypoxic (Hx-Clo) piglets. Tissue hypoxia was confirmed biochemically by determining the levels of high energy phosphates ATP and phosphocreatine (PCr). Neuronal nuclei, mitochondrial membranes and cytosolic fractions were isolated and separated by 12% SDS-PAGE and probed with specific antibodies to Apaf-1. The expression of Apaf-1 in neuronal nuclei was 48.86±5.27 in Nx, 108.43±6.37 in Hx and 78.53±7.00 in Hx-Clo. The Apaf-1 expression of in mitochondrial fraction was 72.73±11.76 in Nx, 132.27±16.15 in Hx and 85.17±5.64 in Hx-Clo. Similarly, the expression of Apaf-1 in cytosolic fraction was 86.79±6.97 in Nx, 193.95±15.41 in Hx and 111.07±7.91 in Hx-Clo. In summary, the results show that hypoxia results in increased expression of Apaf-1 proteins in neuronal nuclear, mitochondrial and cytosolic fractions. Administration of a high affinity Ca++-ATPase, prevented the hypoxia induced increased expression of Apaf-1 protein, suggesting that the hypoxia-induced increased expression of Apaf-1 proteins is nuclear Ca++-influx mediated. We conclude that cerebral hypoxia-induced increase in Apaf-1 protein will lead to increased activation of procaspase-9 to caspase-9 in the cytosolic compartment leading to a cascade of hypoxic neuronal death.
Keywords: Apaf-1, Apoptotic proteins, Hypoxia, Nuclear Ca++-influx, clonidine
INTRODUCTION
Apoptosis is an evolutionarily conserved, genetically controlled process of programmed cell death, used by multicellular organisms to eliminate cells in diverse physiological settings, such as development, homeostasis of tissues, and maintenance of integrity of the organism [1, 2]. The apoptotic cascade can be triggered through two major pathways. Extracellular signals such as members of the tumor necrosis factor (TNF) family can activate the receptor-mediated extrinsic pathway. Alternatively, stress signals such as DNA damage, hypoxia, and loss of survival signals may trigger the mitochondrial intrinsic pathway. In the latter, mitochondrial damage results in cytochrome c release and formation of the apoptosome, a multimeric protein complex containing apoptotic protease activating factor-1 (Apaf-1), cytochrome c, and pro-caspase-9. Once bound to the apoptosome, pro-caspase-9 is activated, and subsequently triggers a cascade of effector caspase activation and proteolysis, leading to apoptotic cell death. Anti-apoptotic Bcl-2 family members such as Bcl-2 prevent, cytochrome c release from mitochondria, thus blocking the downstream intrinsic pathway events of apoptosome formation and caspase activation. This function for Bcl-2 is supported by the findings that it is an integral membrane protein and is able to prevent cytochrome c release [3].
Studies have shown that Bcl-2 and Apaf-1 do not bind in mammalian cells [4], supporting a membrane integrity model rather than a sequestration model for Bcl-2 function. These anti-apoptotic effects of Bcl-2 proteins are countered by the actions of the pro-apoptotic actions of Bax inhibition of apoptosome formation. Following cytochrome c release, the next step in the intrinsic pathway of apoptosis is the formation of the apoptosome. The modulation of apoptosome formation is a balance between pro-apoptotic and anti-apoptotic signals. It has been shown in in vitro experiments that apoptosis is regulated by intracellular potassium ion (K+) levels, with physiological intracellular K+ concentrations functioning to inhibit apoptosome formation by preventing Apaf-1 oligomerization. This inhibition is overcome by increasing cytoplasmic concentrations of cytochrome c and it is proposed that the mechanism for K+ dependent inhibition is through competition for cytochrome c binding sites on Apaf-1 [5]. This inhibition of apoptosome formation by physiologic concentrations of K+ may serve as a safety mechanism against initiation of the apoptotic cascade following the leakage of small amounts of cytochrome c from the mitochondria. Following cytochrome c release, the next step in the intrinsic pathway of apoptosis is apoptosome formation.
One critical regulator that plays a role in apoptosis under hypoxia is the apoptotic protease activating factor-1 (Apaf-1). Apaf-1 is a crucial part of the apoptosome that is assembled in response to several cellular stresses (i.e., hypoxia, DNA damage, oncogene activation, etc). Activation by these signals finally leads to caspase activation via the intrinsic mitochondrial pathway resulting in apoptotic cell death [6]. Apaf-1 knockout mice showed severe defects in the apoptotic response to hypoxic stimulation [7]. This finding demonstrates Apaf-1 as an essential component of the apoptotic response to hypoxia in vitro. Apaf-1 has been identified as the mammalian homolog of Caenorhabditis elegans ced-4, which is a core component of the cell death machine (together with ced-3 and ced-4) and plays a crucial role in the general apoptotic program of normal development and pathogenesis [8]. Understanding the pathways that lead to apoptosis and identifying strategies to regulate this pathway may have important clinical implications. In stress-induced apoptosis, mitochondria releases apoptogenic factors such as cytochrome c, [9]. Pro-apoptotic members of the Bcl-2 family such as Bax and Bid induce the release of apoptogenic factors, whereas anti-apoptotic members such as Bcl-2 or prevent their releases [10]. Cytochrome c binds to Apaf-1 and, when dATP is added to Bcl-XL the cytochrome c-bound Apaf-1, the oligomeric complex of Apaf-1 forms and leads to the recruitment of caspase-9 to form apoptosome and activates caspase-9 [11].
Previous studies on Apaf-1 and caspase-9 knockout mice suggest that Apaf-1 is involved in the control of cell numbers in the developing brain, retina, face, and limbs. Apaf-1 and caspase-9 are essential for caspase-3 activation and seem to play important roles in normal development [12]. Apaf-1-deficient mice exhibit reduced apoptosis in the brain and striking craniofacial abnormalities with hyperproliferation of neuronal cells. Apaf-1 and caspase-9 are also reported to control tumor development by functionally interacting with p53 [13]. Although some apoptosis regulated by Bcl-2 is activated independently of the apoptosome [14], these observations indicate that Apaf-1 plays a central role in the common events of mitochondria-dependent cell death pathways. Therefore, identification of such molecules as regulators of Apaf-1-mediated cell death will be important [15]. To our knowledge, the expression of Apaf-1 in hypoxia has not been investigated to date. The present study investigates that Apaf-1 is expressed during hypoxia in the cerebral cortex of newborn piglets and that administration of clonidine prevents the hypoxia induced increase expression of Apaf-1.
The present study specifically focuses on the mechanism of expression of apoptotic protease activating factor-1 (Apaf-1) protein during hypoxia and tests the hypothesis that hypoxia results in increased expression of apoptotic protease activating factor-1 in the neuronal nuclear, mitochondrial and cytosolic compartment and that the hypoxia-induced increased expression of apoptotic proteins is nuclear Ca++-influx-dependent.
MATERIAL AND METHODS
Studies were performed in newborn piglets aged 2–5 days. The Institutional Animal Care and Use Committee of the Drexel University College of Medicine approved the animal protocol. Using 4% Isoflurane for induction and 0.8% Isoflurane for maintenance of anesthesia, a tracheostomy was performed and an endotracheal tube secured. Arterial and venous catheters were placed via the femoral vessels for blood sampling and administration of drugs. Lidocaine (1%) was used for local anesthesia at all incision sites. The animals were allowed to breathe spontaneously during instrumentation. Following surgery, halothane was withdrawn and the animals were placed on a volume ventilator with 72% nitrous oxide and 28% oxygen; tubocurarine (3 mg/kg) was administered, and fentanyl (30 μg/kg every 45 min) was used for analgesia. The ventilator settings and FiO2 were adjusted in order to maintain a PaCO2 of 35–45 mmHg and a PaO2 of 80–110 mmHg. Blood pressure, heart rate, core temperature, and end-tidal CO2 were monitored continuously. Core temperatures were maintained at 38.5–39°C using a servo-controlled warming blanket.
Piglets were assigned to one of the three groups: a normoxic control group, an untreated hypoxic group and a Clonidine-treated hypoxic group. Baseline measurements were obtained after 45 min in both control and experimental groups while maintaining normal arterial blood gas values. Following baseline measurements, the six piglets assigned to the control group were maintained under normoxic conditions for 60 min. The seven animals in the untreated hypoxic group were ventilated with an FiO2 of 0.06 for 1 h. The six piglets assigned to the Clonidine-treated hypoxic group received clonidine (12.5mg/kg) over 30 min before the induction of hypoxia. The control group of animals received normal saline. At the end of the study the cerebral cortex was harvested within 5 sec of decapitation in Tris–HCl buffer for isolation of neuronal nuclei, mitochondrial and cytosolic fractions; an additional section of brain was frozen in liquid nitrogen, and stored at −80°C for biochemical analysis.
Brain tissue concentrations of ATP and PCr
Tissue hypoxia was confirmed biochemically by determining the levels of high energy phosphates ATP and phosphocreatine (PCr). The concentrations of ATP and PCr in the cerebral cortex were determined by an enzyme-coupled assay using the method of Lamprecht et al [16]. Deproteinized cortical homogenate from 500 mg of frozen cortex was ground to a powder in 7% (v/v) perchloric acid (1 ml/100 mg brain) under liquid nitrogen, allowed to thaw on ice, then centrifuged at 4000 g for 5 min. Aliquots of supernatant were neutralized with KOH–K2CO3 and centrifuged at 2000 g for 5 min. ATP and PCr concentrations were determined in a 1-ml volume containing buffer (50 mM triethanolamine, 5 mM MgCl2, 1 mM EDTA, 2 mM glucose), 400 μl of the neutralized 2000 g supernatant, and 20 μl NADP. Readings were taken every 5 min after the addition of 10 μl hexokinase until a steady state was reached. The ATP concentration was calculated from the increase in absorbance at 340 nm during the 20 min after the addition of hexokinase. A 20-μl volume of ADP and 20 μl of creatine kinase were then added and readings taken at 5-min intervals until a second steady state was reached. PCr concentration was calculated from the increase in absorbance at 340 nm after the addition of creatine kinase.
Isolation of cerebral cortical neuronal nuclei
Cerebral cortical nuclei were isolated by homogenizing 1-g of brain tissue in 15 volumes of a medium containing 0.32 M sucrose, 10 mM Tris–HCl and 1 mM MgCl2 (pH 6.8). The homogenate was filtered through a nylon bolting mesh (size 110 μm) and subsequently centrifuged at 850 g for 10 min. The nuclei were recovered through a discontinuous gradient with a final sucrose concentration of 2.1 M, which increases the yield of large neuronal nuclei. The nuclei were purified by centrifugation for 60 min at 70 000 g. The nuclear pellet was collected, re-homogenized and used as the nuclear preparation. The protein concentration was determined
Isolation of cerebral cortical mitochondria and cytosolic fraction
Cerebral tissue mitochondrial fraction was isolated by homogenizing one gram of cerebral cortical tissue by a Dounce-type glass homogenizer (seven strokes with pestle clearance 0.15 mm and seven strokes with pestle clearance 0.07 mm) in 30 ml of fresh isolation medium containing 0.32 M sucrose, 1 mM EDTA and 20 mM Tris–HCl buffer, pH 7.1. The homogenate was centrifuged for 3 min at 1500×g and the resulting supernatant was centrifuged at 15,000×g for 10 min to provide the crude mitochondrial pellet. To purify mitochondria, the crude mitochondrial pellet was suspended in 2.5 ml of isolation buffer and mixed with 12.5 ml of 12% ficoll solution and placed on the bottom of an ultracentrifuge centrifuge tube. Ten milliliters of 7% Ficoll solution was layered over it followed by 10 ml of isolation medium. The gradient was centrifuged for 30 min at 100,000×g. The mitochondrial pellet was washed and re-suspended in the isolation medium. The purity of mitochondrial preparation with respect to the possibility of contamination with cell nuclei was determined by immunoblotting using nuclear marker proteins lamin A and lamin C. There was an absence of lamin A and C in the mitochondrial fraction demonstrating that the mitochondrial preparation was free from nuclear contamination. The 15000 g supernatant was centrifuged at 100000 g for 660 min to obtain the cytosolic fraction. The mitochondrial and cytosolic preparations were diluted to a final concentration of 100 μg protein/100 μl.
Western Blot analysis
Proteins were separated on a 12% Separation of nuclear mitochondrial and cytosolic proteins and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel and transferred onto nitrocellulose paper. The nitrocellulose paper was blocked with 10% phosphate-buffered saline (PBS)–milk at 4°C with constant agitation for 6–8 h. Incubation with primary polyclonal anti-Apaf-1 antibody (Santa Cruz Biotech, Santa Cruz, CA, USA) in 3% PBS–milk was done overnight at 4°C. Subsequently the nitrocellulose was washed with distilled water (dH2O) and incubated with horseradish peroxidase conjugated secondary antibody (Rockland, Gilbertsville, PA, USA) in 3% milk for 1.5 h at room temperature with constant agitation. Following washings with dH2O and PBS–0.05% Tween 20 the specific complexes were detected using ECL reagents (Amersham Pharmacia Biotech, Buckinghamshire, UK) for 2–3 min. Protein bands were analyzed using imaging densitometry (GS-700 imaging densitometer, Bio-Rad, Hercules, CA, USA). The density of proteins was expressed as absorbance (OD×mm2).
Statistical analysis
Statistical analysis of biochemical measurements was performed using a one-way analysis of variance (ANOVA) and Tukey-test for comparison among groups. A P value <0.05 was considered significant. All values shown are mean ± standard deviation (S.D.).
RESULTS
Brain tissue concentrations of ATP significantly decreased in the untreated hypoxic group compared to the normoxic and the Clonidine-treated hypoxic groups (4.20± 0.36, Nx; 1.26 ± 0.16, Hx; 1.56± 0.66, Hx-Clo (p<0.05). Phosphocreatine levels were significantly lower in both hypoxic groups compared to the level for the control group (3.56 ± 0.18, Nx, 1.12 ± 0.17, Hx: 1.01 ± 0.14 Hx-Clo: p<0.05), confirming severe cerebral tissue hypoxia.
The expression of Apaf-1 in neuronal nuclei was 48.86±5.27 in Nx; 108.43±6.37 in Hx; and 78.53±7.00 in Hx-Clo. The results show an increased expression of Apaf-1 protein in the hypoxic group. The density of Apaf-1 protein was lower in the clonidine pretreated hypoxic groups. The Apaf-1 expression of in mitochondrial fraction was 72.73±11.76 in Nx; 132.27±16.15in Hx; and 85.17±5.64 in Hx-Clo. The results show an increased expression of Apaf-1 in the hypoxic group Administration of Clonidine decreased the increase in Apaf-1 protein expression during hypoxia. Similarly, the expression of Apaf-1 in cytosolic fraction was 86.79±6.97 in Nx; 193.95±15.41 in Hx; and 111.07±7.91 in Hx-Clo. The results show an increased expression of Apaf-1 protein in the hypoxic group and the administration of clonidine prevented the hypoxia-induced increased expression of Apaf-1 protein (figure 1). The density data of the apoptotic protease activating factor-1 (Apaf-1) presented as mean±standard deviation in the nuclear, mitochondrial and cytosolic fraction is shown in Figure 2a, 2b and 2c respectively.
Figure 1.
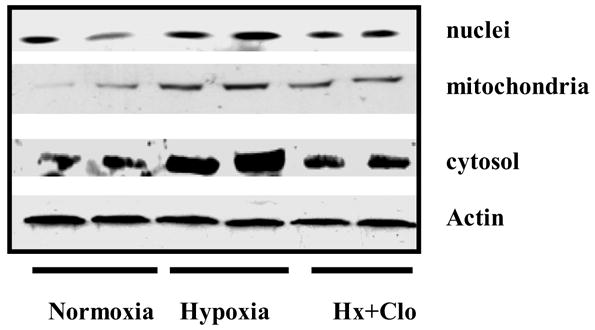
Immunoblots of apoptotic protease activating factor-1 (Apaf-1) in neuronal nuclear membranes, mitochondrial membranes and cytosolic fraction of the cerebral cortex of normoxic, hypoxic and clonidine-treated hypoxic newborn piglets.
Figure 2.
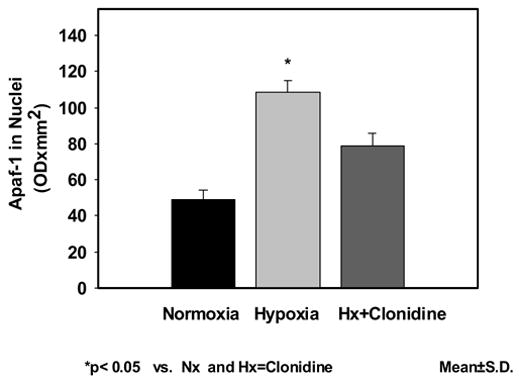
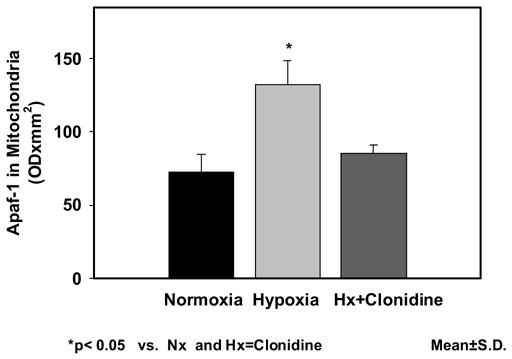
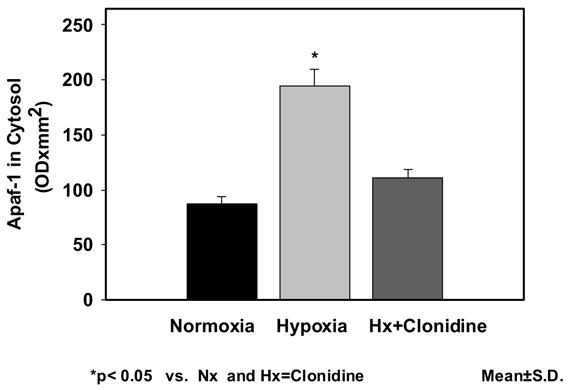
Figure 2a Expression of apoptotic protease activating factor-1 (Apaf-1) neuronal nuclear membranes of the cerebral cortex of normoxic, hypoxic and clonidine-treated hypoxic newborn piglets. Data is presented as mean±standard deviation. *p<0.05.
Figure 2b: Expression of apoptotic protease activating factor-1 (Apaf-1) in mitochondrial membranes of the cerebral cortex of normoxic, hypoxic and clonidine-treated hypoxic newborn piglets. Data is presented as mean±standard deviation. *p<0.05.
Figure 2c: Expression of apoptotic protease activating factor-1 (Apaf-1) in the cytosolic fraction of the cerebral cortex of normoxic, hypoxic and clonidine-treated hypoxic newborn piglets. Data is presented as mean±standard deviation. *p<0.05.
DISCUSSION
Apaf-1, a key regulator of apoptosis, has a major role in programmed cell death [17]. In the presence of cytochrome c and dATP, Apaf-1 oligomerizes to form a very large protein complex, apoptosome [18]. Hypoxia is an important factor in the pathogenesis of several major diseases such as stroke and cancer [19]. Several lines of evidence indicate that, under certain conditions, hypoxia induces cell death through the Apaf-1-mediated mitochondrial pathway. Hypoxia results in translocation of Bax from the cytosol to the mitochondria and activation of caspases. In addition, the cells overexpressing the anti-apoptotic Bcl-2 family proteins have been shown to prevent hypoxia-induced apoptosis by inhibiting the release of cytochrome c [20]. Thus, apoptotic signals during hypoxia seem to occur through the release of cytochrome c and the Apaf-1-mediated activation of caspase-9 [21].
Taken together, the data presented here specifically focus on investigating the effect of hypoxia on expression of Apaf-1 protein in the neuronal nuclei, mitochondria and the cytosolic fractions of the cerebral cortex of newborn piglets and tests the hypothesis that hypoxia results in increased expression of Apaf-1 protein in all the three fractions and the hypoxia-induced increased expression of Apaf-1 protein is nuclear Ca++-influx dependent. Therefore, administration of a high affinity Ca++-ATPase inhibitor, Clonidine, that blocks hypoxia-induced increase in neuronal nuclear Ca++-influx, will prevent the increased expression of Apaf-1 protein during hypoxia in all the three sub-cellular fractions of the cerebral cortex of newborn piglets. The data shows that hypoxia results in increase in apoptotic protease activating factor-1 (Apaf-1) protein in all the three cell fractions in the cerebral cortex of newborn piglets. In addition, the hypoxia-induced increase in apoptotic protease activating factor-1 (Apaf-1) protein was prevented by administration of high affinity Ca++-ATPase inhibitor, clonidine that prevents the hypoxia-induced increase in neuronal nuclear Ca++. These results indicate that the hypoxia-induced increased expression of pro-apoptotic proteins is nuclear Ca++-influx –dependent-induced.
In our previous studies, we have shown that cerebral hypoxia results in increased expression of Bax protein in neuronal nuclei of the cerebral cortex of newborn piglets [22]. We have demonstrated that pretreatment with clonidine prevents the hypoxia-induced increase in nuclear Ca++-influx and Ca++-ATPase activity in the cortical neuronal nuclei of newborn piglets. We have also recently shown that hypoxia results in increased expression of proapoptotic proteins Bax and Bad in neuronal nuclear, mitochondrial as well as cytosolic fractions. The expression of anti-apoptotic proteins Bcl-xl and Bcl-2 did not increase in any of these fractions. The hypoxia-induced increase in proapoptotic proteins was prevented by administration of high affinity Ca++-ATPase inhibitor clonidine that prevents the hypoxia-induced increase in neuronal nuclear Ca++. These results suggest that the hypoxia-induced increased expression of pro-apoptotic proteins is nuclear Ca++-influx–dependent. We have also shown that hypoxia results in increased fragmentation of nuclear DNA and the increase in DNA fragmentation was a function of cerebral hypoxia [23].
Recent evidence indicates that oxygen deprivation induces cell death through apoptosis, and not necrosis. Apoptotic signaling during oxygen deprivation occurs through the release of cytochrome c and Apaf-1 mediated caspase-9 activation. How oxygen deprivation couples to activation of pro-apoptotic Bcl-2 family members remains unknown. Furthermore, it has been described that Bcl-2 prevents the mitochondrial release of other proapoptotic factors, including AIF and endonuclease G, which might be involved in caspase-independent cell death [24].
Although caspase-independent forms of apoptosis have been described, activation of caspases plays a central role in most apoptotic pathways [25]. Upon formation of the death-inducing signaling complex in the extrinsic death receptor pathway or the apoptosome in the intrinsic mitochondrial pathway, initiator caspases are autoproteolytically processed resulting in the activation of downstream caspases and cleavage of numerous death substrates [26]. It is widely believed that Bcl-2 family proteins regulate mitochondrial membrane pores that release cytochrome c and other apoptogenic factors. However, Bcl-2 proteins can also induce or suppress caspase-independent nonapoptotic cell death [27]. Moreover, recent reports have shown that Bcl-2 can regulate activation of initiator caspases independently of the classical apoptosome, arguing that they operate by mechanisms other than by regulating caspase-activating proteins such as Apaf-1 [28]. Although most studies have focused on the anti-apoptotic role of mitochondrial localized Bcl-2, recent evidence has suggested that Bcl-2 can also act as a pro-apoptotic. Recent studies have indicated that one putative subcellular localization where the pro-apoptotic action of Bcl-2 can be exerted is at the nuclear compartment, specifically the perinuclear area. These studies have far reaching implications because they reveal a dual role for Bcl-2 as a cell protector or cell killer, dependent on subcellular localization. These studies thus open the possibility for therapeutic intervention by directing the Bcl-2 subcellular distribution to dictate cell fate [29].
In summary, the results of the present study show that hypoxia results in increased expression of Apaf-1 proteins in neuronal nuclear, mitochondrial and cytosolic fractions. Administration of a high affinity Ca++-ATPase prevented the hypoxia induced increased expression of Apaf-1 proteins indicating that the hypoxia-induced increased expression of Apaf-1 proteins is nuclear Ca++-influx mediated. We conclude that cerebral hypoxia-induced increase in Apaf-1 protein will lead to increased activation of procaspase-9 to caspase-9 in the cytosolic compartment leading to a cascade of hypoxic neuronal death.
Acknowledgments
This study was supported by grants from the National Institutes of Health, NIH-HD-38079 and NIH-HD20337. The authors express their thanks to Ms. Joanna Kubin and Ms. Anli Zhu for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raff M. Cell suicide for beginners. Nature. 1998;396:119–122. doi: 10.1038/24055. [DOI] [PubMed] [Google Scholar]
- 2.Ameisen JC. On the origin, evolution, and nature of programmed cell death: A timeline of four billion years. Cell Death Differ. 2002;9:367–393. doi: 10.1038/sj.cdd.4400950. [DOI] [PubMed] [Google Scholar]
- 3.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 4.Moriishi K, Huang DC, Cory S, Adams JM. Bcl-2 family members do not inhibit apoptosis by binding the caspase activator Apaf-1. Proc Natl Acad Sci USA. 1999;96:9683– 9688. doi: 10.1073/pnas.96.17.9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cain K, Langlais C, Sun XM, Brown DG, Cohen GM. Physiological concentrations of K+ inhibit cytochrome c -dependent formation of the apoptosome. J Biol Chem. 2001;276:41985–41990. doi: 10.1074/jbc.M107419200. [DOI] [PubMed] [Google Scholar]
- 6.Ferraro E, Corvaro M, Cecconi F. Physiological and pathological roles of Apaf1 and the apoptosome. J Cell Mol Med. 2003;7:21–34. doi: 10.1111/j.1582-4934.2003.tb00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- 8.Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 9.Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- 10.Tsujimoto Y. Cell death regulation by the Bcl-2 protein family in the mitochondria. J Cell Physiol. 2003;195:158–167. doi: 10.1002/jcp.10254. [DOI] [PubMed] [Google Scholar]
- 11.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP-mediated Oligomerization of Apaf-1 Is a Prerequisite for Procaspase-9 Activation. J Biol Chem. 1999;274:17941–7945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- 12.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 Homolog) Regulates Programmed Cell Death in Mammalian Development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 13.Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW. Apaf-1 and Caspase-9 in p53-Dependent Apoptosis and Tumor Inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- 14.Marsden VS, O’Connor L, O’Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DC, Cecconi F, Kuida K, Tomaselli KJ, Roy S, Nicholson DW, Vaux DL, Bouillet P, Adams JM, Strasser A. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 2002;419:634–637. doi: 10.1038/nature01101. [DOI] [PubMed] [Google Scholar]
- 15.Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol. 2000;8:476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- 16.Lamprecht W, Stein P, Heinz F, Weisser H. Creatine phosphate. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. Vol. 4. Academic Press; New York: 1974. pp. 1777–1781. [Google Scholar]
- 17.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 18.Zou H, Li Y, Liu X, Wang X. An APAF-1·Cytochrome c Multimeric Complex Is a Functional Apoptosome That Activates Procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 19.Saikumar P, Dong Z, Patel Y, Hall K, Hopfer U, Weinberg JM, Venkatachalam MA. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene. 1998;17:3401–3415. doi: 10.1038/sj.onc.1202590. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu S, Eguchi Y, Kosaka H, Kamiike W, Matsuda H, Tsujimoto Y. Prevention of hypoxia-induced cell death by Bcl-2 and Bcl-xL. Nature. 1995;374:811–813. doi: 10.1038/374811a0. [DOI] [PubMed] [Google Scholar]
- 21.McClintock DS, Santore MT, Lee VY, Brunelle J, Budinger GR, Zong WX, Thompson CB, Hay N, Chandel NS. Bcl-2 Family Members and Functional Electron Transport Chain Regulate Oxygen Deprivation-Induced Cell Death. Mol Cell Biol. 2002;22:94–104. doi: 10.1128/MCB.22.1.94-104.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravishankar S, Ashraf QM, Fritz KI, Mishra O, Delivoria-Papadopoulos M. Expression of Bax and Bcl-2 proteins during hypoxia in cerebral cortical neuronal nuclei of newborn piglets: effect of administration of magnesium sulfate. Brain Res. 2001;901:23–29. doi: 10.1016/s0006-8993(01)02109-6. [DOI] [PubMed] [Google Scholar]
- 23.Akhter W, Ashraf QM, Zanelli S, Mishra O, Delivoria-Papadopoulos M. Effects of graded hypoxia on cerebral cortical genomic DNA fragmentation in newborn piglets. Biol Neonate. 2001;79:187–193. doi: 10.1159/000047089. [DOI] [PubMed] [Google Scholar]
- 24.Haraguchi M, Torii S, Matsuzawa S, Xie Z, Kitada S, Krajewski S, Yoshida H, Mak T, Reed J. Apoptotic Protease Activating Factor 1 (Apaf-1)–independent Cell Death Suppression by Bcl-2. J Exp Med. 2000;191:1709–1720. doi: 10.1084/jem.191.10.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter M. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254:439–459. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- 26.Fischer U, Janicke R, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kane D, Ord T, Anton R, Bredesen D. Expression of bcl-2 inhibits necrotic neural cell death. J Neurosci Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- 28.Marsden V, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-Only Proteins and More. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- 29.Portier B, Taglialatela G. Bcl-2 Localized at the Nuclear Compartment Induces Apoptosis after Transient Overexpression. J Biol Chem. 2006;281:40493–40502. doi: 10.1074/jbc.M606181200. [DOI] [PubMed] [Google Scholar]