

Abstract
We hypothesize that enzymatic switching during translesion synthesis (TLS) to relieve stalled replication forks occurs during transitions from preferential to disfavored use of damaged primer–templates, and that the polymerase or 3′-exonuclease used for each successive nucleotide incorporated is the one whose properties result in the highest efficiency and the highest fidelity of bypass. Testing this hypothesis requires quantitative determination of the relative lesion bypass ability of both TLS polymerases and major replicative polymerases. As a model of the latter, here we measure the efficiency and fidelity of cis–syn TT dimer and abasic site bypass using the structurally well-characterized T7 DNA polymerase. No bypass of either lesion occurred during a single round of synthesis, and the exonuclease activity of wild-type T7 DNA polymerase was critical in preventing TLS. When repetitive cycling of the exonuclease-deficient enzyme was allowed, limited bypass did occur but hundreds to thousands of cycles were required to achieve even a single bypass event. Analysis of TLS fidelity indicated that these rare bypass events involved rearrangements of the template and primer strands, insertions opposite the lesion, and combinations of these events, with the choice among these strongly depending on the sequence context of the lesion. Moreover, the presence of a lesion affected the fidelity of copying adjacent undamaged template bases, even when lesion bypass itself was correct. The results also indicate that a TT dimer presents a different type of block to the polymerase than an abasic site, even though both lesions are extremely potent blocks to processive synthesis. The approaches used here to quantify the efficiency and fidelity of TLS can be applied to other polymerase–lesion combinations, to provide guidance as to which of many possible polymerases is most likely to bypass various lesions in biological contexts.
Keywords: DNA damage, DNA synthesis fidelity, DNA polymerase, Replication errors, Translesion synthesis
Abbreviations: TLS, translesion synthesis; pol, polymerase; Dpo4, Sulfolobus solfataricus DNA polymerase 4; 8-oxoG, 8-oxo-7,8-dihydro-2′-deoxyguanosine; MF, mutation frequency
1. Introduction
Several DNA repair pathways exist to remove DNA damage before it can alter the genetic code of an organism [1]. However, no repair pathway is infallible and damage can occur at anytime during the cell cycle, including S phase. Thus, cells have evolved damage tolerance mechanisms to prevent replication blocking lesions from causing strand breaks that can lead to cell death. Since a large number of structurally distinct lesions can be generated by endogenous and exogenous environmental stresses, one mechanism to alleviate stalled replication forks is translesion synthesis (TLS) [2–7]. Depending on the nature of the lesion, TLS can be facile (e.g. bypass of uracil) or difficult, as is the case with lesions that eliminate base coding potential (e.g. an abasic site) and/or distort helix geometry (e.g. UV photoproducts). To deal with these diverse impediments, organisms encode multiple polymerases that have very different properties. These include the major replicative DNA polymerases (e.g. T7 DNA polymerase, pol δ, pol ɛ) that are highly accurate due to high geometric selectivity for correct incorporation and proofreading of errors by their intrinsic 3′ exonuclease activities [8]. Also included are the much less accurate Y-family DNA polymerases implicated in lesion bypass [2–5,9]. It is now clear that lesion-induced replication blockage triggers events that result in coordinated switching among these polymerases to allow TLS [9–11]. It is less clear exactly which polymerases are best suited to completely bypass different lesions. We have been investigating this subject based on the hypothesis that enzymatic switching during TLS occurs during transitions from preferential to disfavored use of damaged primer–templates, and that the polymerase or 3′ exonuclease used for each successive nucleotide incorporated is the one whose properties result in the highest efficiency and the highest fidelity of bypass.
To test this hypothesis, we developed assays to quantify the efficiency with which DNA polymerases bypass site-specific lesions during one cycle of processive synthesis, and to quantify base substitution and insertion/deletion error rates during TLS [12]. These assays provide efficiency parameters and TLS error rates for reactions performed with all four dNTPs in competition with each other during each stable incorporation event needed to completely bypass a lesion. We first applied these assays to human pol η and Sulfolobus solfataricus Dpo4, Y-family members that lack intrinsic 3′ exonuclease activity and copy undamaged DNA with low fidelity [3,6,13–15]. Both enzymes were found to bypass synthetic abasic sites and cis–syn thymine–thymine dimers with higher efficiencies than do the more accurate polymerases [12,16]. Intrinsically low fidelity and efficient lesion bypass by pol η and Dpo4 are consistent with structural studies revealing that Dpo4 does not contact the DNA minor groove and has a nascent base pair binding pocket comprised of smaller side chains that is more open and solvent exposed than those of higher fidelity polymerases [17].
When copying synthetic abasic sites, pol η and Dpo4 both preferentially incorporate dAMP [12]. This conforms to the well known “A rule” for misincorporation opposite non-instructional lesions (reviewed in [18–22]). Pol η and Dpo4 also preferentially incorporate dAMP opposite both bases of a TT dimer [16]. However, dAMP incorporation by Y-family polymerases during TT dimer bypass is likely to be instructed by the thymines (reviewed in [18]). Compelling evidence for this comes from Dpo4 structural studies revealing that incoming ddATP can correctly base pair with both the 3′ and the 5′ T [23]. Surprisingly, while ddATP is in the anti (Watson–Crick) conformation when paired with the 3′ T [23], it is in the syn (Hoogsteen) conformation when paired with the 5′ T. Thus Dpo4 solves the different challenges posed by the two covalently linked but structurally distinct bases by reconfiguring the incoming substrate as needed to permit efficient dimer bypass. While the anti–syn difference in base pairing permits correct synthesis at both damaged bases, it is also likely to be relevant to potentially mutagenic synthesis. This is suggested by the observation that the rate of stable misincorporation of dGMP during TT dimer bypass by both Dpo4 and human pol η is substantially higher at the 3′ T than at the 5′ T [16]. The higher fidelity at the 5′ T may result from less favorable mispairing between incorrect dGMP and the Hoogsteen face of thymine. Dpo4 and human pol η also generate single base deletion and insertion errors during complete bypass of both abasic sites and TT dimers [12,16]. Thus, substrate rearrangements during dimer bypass are not limited to anti–syn rotations of nucleotides, but include substrate misalignments that may involve the dNTP, each DNA strand, or both (see Section 4).
On the opposite end of the TLS spectrum from the Y-family enzymes are DNA polymerases in the A and B families which have lower ability to bypass template lesions that eliminate base coding potential and/or distort helix geometry. The purpose of the present study is to compare and contrast results with Y-family polymerases to the efficiency and fidelity of lesion bypass by a structurally well-characterized replicative DNA polymerase. Structural studies have shown that the nascent base pair binding pocket of these polymerases snugly accommodate a single correct undamaged base pair, likely contributing to their high nucleotide selectivity [8,24–29]. Among the most thoroughly studied of these enzymes is T7 DNA polymerase [25,30], a highly accurate A family enzyme that has an intrinsic proofreading exonuclease activity. Proofreading-proficient T7 DNA pol is unable to bypass abasic sites and UV photoproducts, but exonuclease-deficient T7 DNA pol can perform bypass, albeit inefficiently (reviewed in [18]). Kinetic studies of insertion of normal dNTPs and of a very large pyrene nucleotide [31,32] opposite abasic sites and UV photoproducts have led to the suggestion that, although photodimers are inherently instructional, they are accommodated in the nascent base pair binding pocket of T7 DNA pol in a manner that excludes base pairing with the 3′ base, thereby impeding phosphodiester bond formation and making this base behave like a non-instructional abasic site (reviewed in [18]).
Structural studies of T7 DNA pol [33] show that the 5′ T of a cis–syn TT dimer can pair with incoming dATP, but when the 3′ T is the templating base, it is rotated out of the binding pocket, precluding normal base pairing with an incoming dNTP. In this case, the fingers subdomain is in an open conformation. This rationalizes why a TT dimer strongly blocks synthesis, and leaves open the issue of whether this non-instructional conformation of the 3′ T is on the pathway to either correct or potentially mutagenic synthesis that could result in a base-substitution or perhaps a single base deletion. One goal of the present study is to investigate specifically this issue and more generally the degree to which substrates are rearranged or not in order to accommodate the aberrant geometry created by two very different lesions and permit either correct or potentially mutagenic bypass.
We have previously hypothesized Y-family polymerases are strongly favored for lesion bypass because they display significantly higher efficiency of bypass compared to replicative polymerases. To investigate this hypothesis from the standpoint of a low-efficiency bypass polymerase, we present new data on the efficiency and fidelity of T7 DNA pol bypass of three different abasic sites and a cis–syn TT dimer and compare it to earlier results on the efficiency and fidelity of TLS by S. solfataricus Dpo4 and human pol η. This study is distinct from, yet complementary to, previous kinetic studies of insertion of individual dNTPs opposite lesions, because the parameters we report here are for complete lesion bypass requiring multiple incorporations when all four dNTPs are present in the TLS reactions.
2. Materials and methods
2.1. Materials
T4 polynucleotide kinase and restriction enzymes were purchased from New England Biolabs. Unlabeled dNTPs, [γ-32P] ATP and [α-32P] dCTP were purchased from Amersham Biosciences. Wild-type (exonuclease-proficient) and Δ6exo−(exonuclease-deficient) T7 DNA polymerase were expressed and purified as previously described [25]. Materials for fidelity assays were from previously described sources [12].
2.2. DNA substrates
Primer and template oligonucleotide sequences are shown in Fig. 1A. Undamaged and abasic site mimic (tetrahydrofuran residue, designated by x) oligonucleotides were purchased from Oligos, etc. (Wilsonville, OR). Template containing a cis–syn TT dimer, synthesized as described [34], was kindly provided by S. Iwai (Osaka University). Primer LBP-20 was 5′ end-labeled with [γ-32P] ATP and T4 polynucleotide kinase. Substrates were prepared by mixing primer and template strands in a 1:1.5 molar ratio in the presence of 20 mM Tris–Cl (pH 7.5) and 25 mM NaCl, heating to 75 °C, followed by cooling to room temperature over 3–4 h and storage at −20 °C.
Fig. 1.
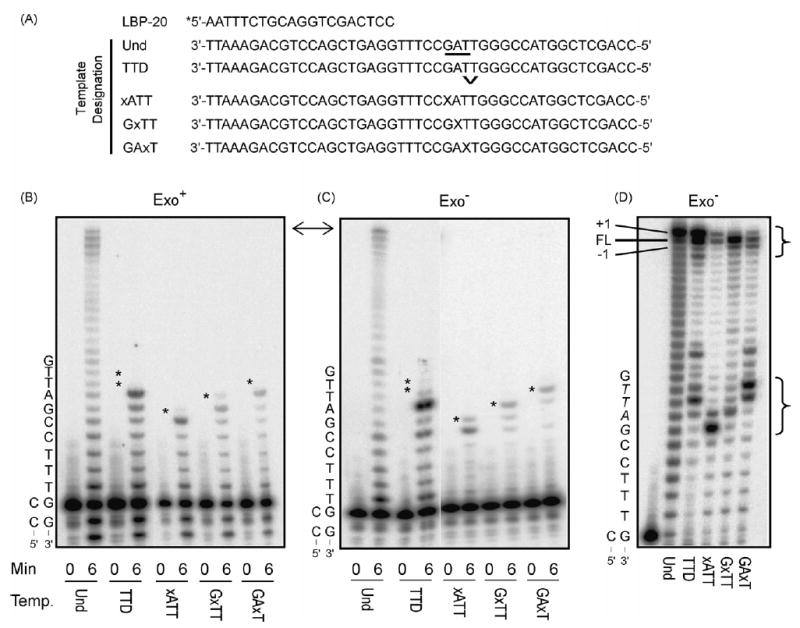
Effects of a TT dimer and abasic site on T7 DNA polymerase processive synthesis. (A) Schematic representation of substrates used. The 20-mer primer (LBP-20) was 5′ end-labeled with 32P (*) and annealed to the template 45-mers shown. The upside down caret (v) denotes the position of the cis–syn TT dimer. Tetrahydrofuran residue (X) mimics an abasic site. The underlined bases are the amber codon in the lacZα sequence. (B) Exonuclease-proficient (Exo+) T7 DNA polymerase reactions. Single hit reactions were performed using a 400-fold excess of substrate over enzyme. Reactions were performed for 0, 3, 6, 9 and 12 min and the products separated by 12% denaturing PAGE. The 0 and 6 min timepoints for each reaction are shown. A star (*) indicates the positions of the damaged bases for each substrate. A partial template sequence is given to the left of the gel. (C) Exonuclease-deficient (Exo−) T7 DNA polymerase reactions. The details are the same as described for panel B. (D) Exonuclease-deficient (Exo−) T7 DNA polymerase reactions using a two-fold excess of enzyme over substrate, incubated for 60 min. Full length (FL), −1, and +1 complete extension products are indicated on the left. Brackets on the right highlight the differences in both complete (upper bracket) and incomplete (lower bracket) bypass reactions for the different substrates. Reactions performed under these conditions using exonuclease-proficient enzyme display complete loss of signal, indicating degradation of the primer strand (data not shown).
2.3. Polymerase reactions
Reaction mixtures (30 μl) contained 40 mM Tris (pH 7.5), 5 mM MgCl2, 50 mM NaCl, 5 mM dithiothreitol, 25 μM each of the four dNTPs, and 4 pmol of primer:template. Reaction mixtures were pre-heated at 37 °C for 1 min and the reactions initiated by the addition of either wild-type (Exo+) or Δ6exo−(Exo−) T7 DNA polymerase in the amounts indicated in the figures and tables. Incubation was at 37 °C and aliquots removed at the times indicated in the figure legends were added to an equal volume of formamide loading buffer (95% deionized formamide, 25 mM EDTA, 0.1% bromophenol blue, 0.1% xylene cyanol) and stored on ice. Reaction products were separated by 12% denaturing polyacrylamide gel electrophoresis and product bands were quantified by phosphoimagery and the values were used to calculate the termination probability, insertion efficiency and extension efficiency at each template nucleotide, as described previously [12].
2.4. Lesion bypass fidelity assays
Reactions were performed essentially as described above with the following modifications: substrate primer did not contain a 5′-32P end label, 8 pmol of Exo− polymerase and 50 μCi of α-32P-dCTP was used and incubation was for 60 min. Newly synthesized, internally labeled 32-mer to 36-mer (−) strand DNA products of complete bypass that contain polymerization errors were purified, hybridized to gapped circular M13 DNA and used to infect Escherichia coli host cells, score plaque colors and quantify error rates, as described previously [12,16]. Error rates at the 3′ T of the cis–syn TT dimer and insertion specificity at abasic sites were determined from results of sequence analysis of independent M13 plaques. Error rates at the 5′ T of the cis–syn TT dimer were determined by plaque hybridization, as previously described [16].
3. Results
3.1. Efficiency of T7 DNA pol bypass of synthetic abasic sites and a cis–syn TT dimer
The ability of wild-type and exonuclease-deficient T7 DNA pol to perform TLS was examined using four different lesion-containing templates (Fig. 1A). One contains a cis–syn thymine–thymine dimer (TTD) and the other three contain a synthetic abasic site mimic (a tetrahydrofuran residue, designated x), each present in a different sequence context. For bypass efficiency measurements, TLS reactions were performed with a 400-fold excess of primer:template over enzyme, such that DNA products result from a single cycle of processive synthesis (see [12,35]). The DNA products were separated by gel electrophoresis (Fig. 1) and the band intensities were quantified and used to calculate the probability of termination of processive synthesis at each template nucleotide (Fig. 2) and efficiency values (Table 1) for insertion opposite the lesion, for extension from that primer terminus, and for lesion bypass relative to bypass of the equivalent undamaged base (see [12] for definitions and calculations).
Fig. 2.
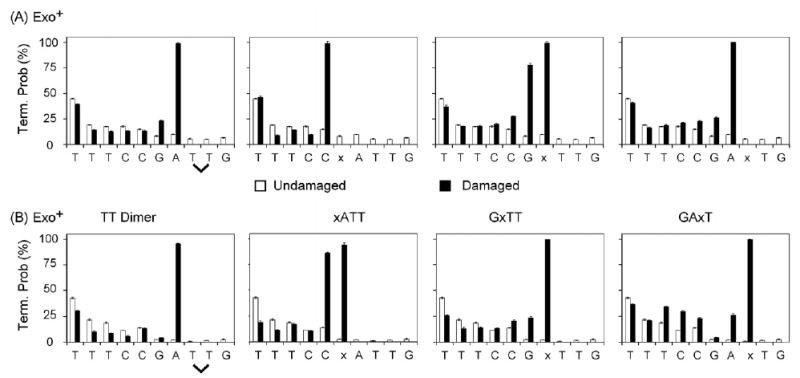
Termination probability of T7 DNA polymerase on TT dimer and abasic site template DNA. Quantitative analysis of all time points from reactions depicted in panels A and B in Fig. 1 was used to determine the termination probability at each template position for undamaged (white) and lesion containing DNA (black). Termination probability, a measure of how much of a given size primer is further extended to longer product, is calculated as the pixel intensity of a given band, divided by the total pixel intensity of all bands that size and larger. Values plotted are the average of eight data points (four time points each for two repeat experiments). Error bars represent the standard deviation.
Table 1.
Properties of T7 DNA polymerase while encountering a lesion
| TT dimer
|
AP site
|
||||
|---|---|---|---|---|---|
| 3′ T | 5′ T | xATT | GxTT | GAxT | |
| T7 Exo+ relative | |||||
| Insa | 1.6 | 2.0 | <0.2 | 24.7 | <0.2 |
| Ext | 2.0 | <0.2 | <0.2 | <0.2 | <0.2 |
| Byp | <0.2 | <0.2 | <0.2 | <0.2 | |
| T7 Exo− relative | |||||
| Ins | 2.7 | 2.5 | 15.4 | 78.2 | 75.2 |
| Ext | 2.5 | <0.2 | <0.2 | <0.2 | <0.2 |
| Byp | <0.2 | <0.2 | <0.2 | <0.2 | |
| Bypass events/cycle relativeb | 2.9 × l0−3 | 7.7 × 10−4 | 5.5 × 10−3 | 4.9 × 10−3 | |
| Pol η relativec | |||||
| Ins | 130 | 140 | 30 | 22 | ndd |
| Ext | 140 | 165 | 40 | 43 | nd |
| Byp | 300 | 13 | 10 | nd | |
| Dpo4c relative | |||||
| Ins | 31 | 80 | 38 | 55 | nd |
| Ext | 80 | 98 | 31 | 60 | nd |
| Byp | 24 | 13 | 30 | nd | |
Insertion (Ins), extension (Ext), and bypass (Byp) efficiencies (in percent) were calculated from single hit reactions, like those in Fig. 1, previously described [12,16]. The values shown are those of lesion containing templates compared to undamaged DNA (i.e. relative relative extension, and relative bypass). Values of <0.2 represent the limit of detection of the phosphor-imaging.
Values come from reactions containing a 10:1 substrate to enzyme ratio incubated for 15 min. The bypass percentages under these conditions are: TTD (8.7%), xATT (2.3%), GxTT (16.6%), GAxT (14.6%), with 97–99% primer utilization in all reactions. The total number of cycles estimated based on single hit reactions that give a cycling rate of 18–20 cycles/polymerase/min. This value was obtained by dividing number of DNA molecules that were acted on by the number of polymerase molecules present for each time point under single hit conditions, then dividing by the time of the reaction in minutes. The average of this value for 3, 6, 9, and 12 min time points for undamaged and TT templates was 18.8 and 21.4, respectively.
Data from human pol η [16] and S. solfataricus Dpo4 [12] are shown for comparison. These experiments contained a single base difference the substrates (3′-xATC instead of 3′-xATT; 3′GxTC instead of 3′GxTT).
No data.
For attempted bypass by wild-type T7 DNA pol, this exonuclease-proficient enzyme bound to and processed all four damaged primer:templates, as indicated by limited exonucleolytic degradation and by synthesis up to the lesion (Figs. 1B and 2A). For the abasic site in the GxTT substrate, insertion opposite the lesion was also observed (Figs. 1B and 2A). The exonuclease-proficient polymerase was unable to bypass any of the lesions during a single cycle of processive synthesis (Fig. 1B). Moreover, no bypass was observed even when the wild-type T7 DNA pol concentration was increased by as much as 800-fold, where the primer strand was substantially degraded (data not shown).
Parallel experiments with exonuclease-deficient T7 DNA pol resulted in primer extension up to and including variable amounts of insertion opposite the lesions (Figs. 1C and 2B). Even so, for these reactions in which the polymerase only encountered the lesion once, no bypass products were observed (Fig. 1C and Table 1). However, when the polymerase concentration was increased to allow many cycles of polymerization (up to two-fold enzyme excess), exonuclease-deficient T7 DNA pol was able to substantially bypass all four lesions (i.e. Fig. 1D). To investigate how strong a block these lesions are to T7 DNA polymerase, we first determined how often a given polymerase cycles during synthesis. Based on the amount of primer extended and how much polymerase was present in the single hit reactions, we calculated that each polymerase molecule cycled ~18–20 times per minute (see Table 1, note b). Using this information, in reactions that contained a 10:1 ratio of DNA to T7 DNA pol that were incubated for 15 min (the earliest time and lowest polymerase amount combination in which bypass was observed) we estimated the total number of polymerase cycles to be 7.2 × 1013 (0.4 pmol, 15 min, 20 cycles/min). We then compared this to the total number of bypassed DNA molecules (4 pmol starting substrate times the bypass percents listed in note b of Table 1; between 5.5 × 1010 and 4.0 × 1011 depending on substrate). The resulting “bypass per cycle” values were very low for each damaged template (Table 1). They indicate that at least ~350 cycles of polymerization (inverse of bypass per cycle value) were required to achieve a single dimer bypass event. The values varied from ~200 to over 1300 cycles for the three different AP sites templates (Table 1). During this multi-cycle bypass, the four different lesion templates also generated several distinct distributions of DNA chain lengths for the products of both blocked (lower bracket in Fig. 1D) and complete (upper bracket in Fig. 1D) lesion bypass. The latter include full-length products, chains that are one nucleotide longer than the template (resulting from both end addition [36,37] and +1 insertions), and chains that are shorter than full length (see Section 4).
3.2. T7 DNA pol incorporation specificity during lesion bypass
To determine error rates for lesion bypass by exonuclease-deficient T7 DNA pol, DNA products representing the chain lengths bracketed at the top of Fig. 1D were recovered and hybridized to a derivative of gapped M13 DNA that contains the lacZ α-complementation gene. This DNA was then introduced into E. coli cells to score plaque colors (see Section 2 and [12,38]). The products of correct DNA synthesis yield faint blue plaques due to the presence of a slightly leaky amber codon in the lacZ α-complementation gene (underlined in Fig. 1A). Errors made during TLS were detected as darker blue plaques (which typically contain base substitutions) and colorless plaques (typically insertions or deletions). The changes in α-complementation gene coding sequence responsible for the altered plaque colors were determined by sequence analysis of DNA from independent lacZ mutants.
The lacZ mutant frequencies (MF) resulting from copying the undamaged DNA (Table 2) were similar to those observed in previous studies of T7 DNA pol using this assay [35]. For the DNA products of TT dimer bypass, the colorless mutant frequency was similar to that for copying the undamaged template, while the dark blue frequency was ~2-fold higher than that for the undamaged DNA template. Bypass of all three synthetic abasic sites resulted in much higher mutant frequencies for dark blue and/or colorless plaques, whose proportions strongly depended on sequence context (Table 2). For example, bypass in the xATT sequence context resulted in mostly dark blue plaques, whereas bypass of an abasic site present in the adjacent template position (the GxTT sequence context) resulted in mostly colorless plaques.
Table 2.
Fidelity of TT dimer bypass and insertion specificity of abasic site bypass by exonuclease-deficient T7 DNA polymerase
| Plaque counts (%)b |
|||
|---|---|---|---|
| Templatea (3′ → 5′) | Total | Dark blue | Colorless |
| GATT | 15610 | 11 (0.1%) | 304 (1.9%) |
| GATT | 17596 | 32 (0.2%) | 383 (2.2%) |
| xATT | 10119 | 6420 (63%) | 107 (1.1%) |
| GxTT | 1072 | 55 (5.1%) | 520 (49%) |
| GAxT | 4618 | 8 (0.2%)c | 1428 (31%) |
TT, cis–syn thymine–thymine dimer; x, tetrahydrofuran residue (abasic site mimic); bases in italics designate the amber stop codon in the lacZα complementation gene.
Plaque numbers and the calculated mutation frequencies for dark blue and colorless plaques are shown. A value of ~60% is the maximum detectable mutation frequency of this assay due to the relative expression of the (+) and (−) strands during transfection [51].
Note that for the GAxT substrate, dAMP insertion opposite the AP site is a ‘correct’ event and therefore not detectable by color screening.
For all of these reactions, the sequence changes responsible for the plaque colors were determined by sequencing DNA from independent dark blue and colorless plaques (Table 3). From this information, the rates (Table 4) at which single base substitutions, single base deletions and insertions, and more complex errors were generated during TLS were calculated (see Ref. [12] and table legends). Because the 5′ T of the TT dimer is not in the amber codon, single base substitutions at the 5′ T are not detectable by color screening. Therefore, we performed plaque hybridization [16] to screen for T to C changes at this position and used the information to calculate the error rate (Table 5). Overall, the results reveal that bypass resulted in a variety of different errors whose rates depended on the lesion and its sequence context. The implications of these fidelity data, and the bypass efficiency parameters mentioned above, are discussed in detail below.
Table 3.
Observed errors during TT dimer and AP site bypass by exonuclease-deficient T7 DNA polymerase
| Dark blue plaques | Colorless plaques |
|---|---|
| GATT | |
| 10/11: Single base substitution in ambera codon (4 G → C, 3 T → C, 1 G → T, 1 A → G, 1 T → A) | 1/16: Single base deletion in amber codon (CGATT → CATT) |
| 1/11: Single base substitution outside amber codon (C → A) | 13/16: Single base deletion outside amber codon (6 CTGAG → CTAG, 2 GGCCA → GGCA, 2 TGGGC → TGGC, 1 TGGCT → TGCT, 1 GGCTC → GGTC, 1 AGGTT → AGTT) |
| 2/16: Single base substitution outside amber codon (C → A, T → G) | |
| GATT | |
| 25/28: Single base substitution in amber codon (T → C) | 15/16: Single base deletion outside amber codon (1 CTGAG → CTAG, 14 TTGGGC → TTGGC) |
| 2/28: Single base substitution outside amber codon (T → G, C → G) | 1/16: Single base substitution outside amber codon (T → G) |
| 1/28: Separate single base insertion (TGGG → TGGGG) and deletion (TGAGG → TGGG)b flanking amber codon | |
| xATT | |
| 15/15: dAMP insertion opposite AP site | 5/16: dAMP insertion opposite AP site with single base insertion outside amber codon (2 TGGC → TGGGC, GGCT → GGCCT, GTTTC → GTTTTC, TCGA → TCGGA) |
| 4/16: dAMP insertion opposite AP site with single base deletion outside amber codon (AGGGT → AGGT, CGGAC → CGAC, TTGGG → TTGG, CATGG → CAGG) | |
| 4/16: Deletion of AP site (CCxATT → CCATT) | |
| 2/16: dAMP insertion opposite AP site coupled with potential single base deletion in amber codon (TCCxATTG → TCCTATG) | |
| 1/16: Four base deletion including AP site (TTCCxATT → TATT) | |
| GxTT | |
| 9/14: dAMP insertion opposite AP site | 16/16: Deletion of AP site (CGxTT → CGTT) |
| 5/14: Deletion of AP site coupled with single base insertion (3 TGGG → TGGGG, 2 CCAT → CCAAT) | |
| GAxT | |
| 2/8: dTTP insertion opposite AP site | 13/15: dAMP insertion opposite AP site with single base deletion outside amber codon (GAxTGGGC → GATTGGC) |
| 2/8: Coupled single base deletion (TGGGC → TGGC) and insertion in amber codon (CGATT → CGGATT, CGATT → CGAATT) | 1/15: Single base deletion of either the AP site or the adjacent T (GAxTGG → GATGG) |
| 1/8: Three base deletion of AP site and flanking bases (CGAxTG → CGG) | 1/15: Two distinct single base deletions (GAxTGG → GATGG, TTTCCG → TTTCG) |
| 1/8: Coupled single base deletion (TTGGGC → TTGGC) and insertion outside amber codon (GCCA → GCCCA) | |
| 1/8: Single base substitution outside amber codon (G → C) | |
| 1/8: Three base deletion outside amber codon (GGCCAT → GAT) | |
The amber codon (shown in italics in the left-most column) is the target sequence in which each lesion is located. All sequence information is of the template strand in the 3′ → 5′ orientation (see Fig. 1 for full sequence).
Changes occurring in the primer:template duplex region are shown in italics in the main table body and are assumed to be caused by errors during chemical synthesis of the oligonucleotides.
Table 4.
Error frequency for classes of errors during AP site bypass
| Error frequency (%)a |
||||
|---|---|---|---|---|
| AP site
|
GATTGGGCb |
|||
| A insertion | Deletion | ΔG (TGGC) | ∇G (TGGGGC) | |
| T7exo− DNA Pol | ||||
| GATT | ≤0.2c | ≤0.2c | 0.4 | ≤0.2 |
| xATT | >99 | 0.6 | 0.1 | ≤0.1 |
| GxTT | 5.5 | 85 | ≤5.1 | 1.8 |
| GAxT | 45 | 3.5 | 45 | ≤3.4 |
| Pol ηd | ||||
| xATC | 67 | 4.1 | nd | nd |
| GxTC | 51 | 27 | nd | nd |
| Dpo4d | ||||
| xATC | 69 | 22 | nd | nd |
| GxTC | 75 | 14 | nd | nd |
Error rates, expressed as a percentage of all events, are calculated using the formula: ((N/T) × (MF))/0.6, where N is the observed number of selected events, T the total number of sequenced samples, MF is the mutation frequency of plaque color sequenced, and 0.6 is to correct for the average minus strand expression percentage during transfection [51]. Rates are totals from sequencing of dark blue and colorless plaques.
Deletion (Δ) and addition (∇) of G at the 3G run downstream of the lesion.
Error rates at each position in the amber codon were the same.
Table 5.
Rates for T → C errors during dimer bypass
| Error rate (×10−4)
|
||||||
|---|---|---|---|---|---|---|
| T7exo− |
Pol ηa |
Dpo4a |
||||
| 3′ Tb | 5′ Tc | 3′ T | 5′ T | 3′ T | 5′ T | |
| Undamaged | 5 | 1 (5↓) | 400 | 140 (3↓) | 100 | 36 (3↓) |
| TT dimer | 37 | 4 (9↓) | 390 | 32 (12↓) | 1000 | 15 (67↓) |
4. Discussion
Unlike the specialized Y-family polymerases that are strongly implicated in bypassing lesions that distort DNA helix geometry, the major biological function of T7 DNA polymerase is to replicate undamaged DNA. It does so accurately [39] due to its intrinsic 3′ exonucleolytic proofreading activity and a nascent base pair binding pocket that strictly selects correct nucleotides based on geometry [30]. Exonucleolytic proofreading and nucleotide selectivity are both relevant to the present results on the efficiency and fidelity of translesion DNA synthesis by T7 DNA polymerase. We first consider the role of the exonuclease, then discuss the ability of the polymerase to insert opposite the lesion, to extend the resulting termini, and to achieve complete lesion bypass, albeit very inefficiently, while generating a remarkably diverse set of bypass products.
4.1. Exonucleolytic suppression of TLS
The observations of aborted lesion bypass by wild-type T7 DNA pol (Figs. 1 and 2 and Table 1) have three interesting implications. First, wild-type T7 DNA pol was unable to bypass the four lesions studied here under any condition tested. In contrast, exonuclease-deficient T7 did bypass all four lesions when multiple polymerization cycles were permitted (Fig. 1D), and in so doing generated a variety of products (Tables 2–5).
This demonstrates that the various matched, mismatched and/or misaligned bypass intermediates generated as T7 DNA pol attempts to bypass a TT dimer or abasic site are degraded by its intrinsic 3′ exonuclease activity rather than extended by polymerization. In other words, the exonuclease activity of T7 DNA pol effectively suppresses TLS of these particular lesions, whether accurate or potentially mutagenic (see below).
Second, the extent of exonucleolytic degradation of the various possible bypass intermediates varies with sequence context. For example, wild-type T7 DNA pol failed to generate a product representing insertion opposite abasic sites in two sequence contexts (xATT and GAxT), whereas the exonuclease-deficient enzyme did insert opposite these lesions. The simplest interpretation is that the polymerase inserts opposite these lesions, but this is followed by exonucleolytic removal rather than continued polymerization from the damaged primer termini. However, the efficiency of this idling is sequence dependent because results with the substrate containing the abasic site in the GxTT context show that both wild-type and exonuclease-deficient T7 DNA pol generate a DNA product band indicating stable insertion of a nucleotide opposite the abasic site (Figs. 1B and 2A, and 25% relative insertion efficiency opposite the lesion by wild-type T7 DNA pol, Table 1). It may be that in this sequence context, wild-type T7 DNA pol inserts dAMP (see below), but its removal by the exonuclease is partially suppressed by an ability of dAMP to pair with either of the T residues in the downstream template.
Third, the influence of the exonuclease activity strongly varies with the lesion. For example, the relative probability of observing insertion opposite the abasic site in the GAxT sequence context is more than 370-fold higher for exonuclease-deficient T7 DNA pol in comparison to wild-type T7 DNA pol (Table 1). In contrast, the relative probability of observing insertion opposite the 3′ T and the 5′ T of the TT dimer is similar for exonuclease-deficient and wild-type T7 DNA pol (Table 1). Moreover, the exonucleolytic suppression of T7 DNA pol bypass of a TT dimer and abasic sites observed here contrasts sharply with earlier results indicating that the exonuclease has very little if any effect on the efficiency or the fidelity of T7 DNA pol bypass of template 8-oxo-guanine [35]. Thus, differences in the structures and coding potential (or lack thereof) of abasic sites, TT dimers and 8-oxo-G result in differences in the extent of exonucleolytic degradation of various bypass intermediates. The 3′ exonuclease activities of other polymerases also affect their ability to conduct translesion synthesis [40–44]. The remainder of the discussion here focuses on the TLS properties of T7 DNA pol in the absence of its 3′ exonuclease activity, with the two main incorporation events required for bypass considered sequentially.
4.2. Insertion efficiency opposite lesions during attempted bypass
The results for insertion opposite the lesions by exonuclease-deficient T7 DNA pol have several implications. First, the probability of insertion opposite abasic sites (relative to insertion opposite an undamaged base in the equivalent location) is substantially higher than the probability of insertion opposite either T of the dimer (Table 1). Moreover, the probability of insertion opposite the two Ts of the dimer are similar to each other (2.5% and 2.7%). Both observations are interesting in light of the hypothesis mentioned in the Introduction that T7 DNA pol inserts dAMP opposite the 3′ T of a TT dimer without instruction from the template base, whereas it inserts dAMP opposite the 5′ T of a TT dimer using its base coding potential [18,31,33]. While these insertion efficiency data and the fidelity data (discussed below) do not exclude this general idea, they nonetheless clearly reveal that in a complete reaction containing all four dNTPs and involving a single cycle of processive synthesis, both template thymines in the dimer are more problematic for this critical insertion step in a bypass reaction than are synthetic abasic sites, the prototypical non-instructional lesion.
Second, the relative efficiency of insertion opposite an abasic site flanked by one or two 5′ template Ts (78% and 75% for GxTT and GAxT, respectively, Table 1) is five-fold higher than the relative efficiency of insertion opposite an abasic site flanked by a 5′ template A (15%, xATT). Given that insertion of dAMP opposite an abasic site is strongly preferred (see below and [18]), the 15% value likely reflects direct dAMP insertion without any template instruction. However, the five-fold higher insertion efficiencies for abasic sites in the other two sequence contexts may partly reflect misalignment of the abasic site to permit the next template base to instruct incorporation of dAMP. This misalignment idea is supported by the fact that bypass of both GxTT and GAxT generates a high proportion of DNA chains one nucleotide shorter than full length (top of Fig. 1D) and generates a high rate of deletion errors as detected in the plaque assay (Table 4). The sequence dependence of insertion efficiency may be relevant to the probability of mutagenic bypass of abasic site in vivo, perhaps even more so when non-optimal polymerases may be forced to participate in TLS in the absence of a dedicated TLS polymerase.
Third, a comparison of the relative insertion efficiencies of exonuclease-deficient T7 DNA pol and naturally exonuclease-deficient Y-family polymerases reveals several interesting features. We were surprised that the exonuclease-deficient T7 DNA pol relative insertion efficiency opposite an abasic site in the xAT sequence context (15%) was only about two-fold lower than the values for human pol η and Dpo4 (30% and 38%, respectively, Table 1). Moreover, in the GxT sequence context, the relative insertion efficiency opposite an abasic site is even higher for T7 DNA pol than for the Y-family pols (Table 1). This is remarkable given very substantial differences in the structures of the active sites of T7 DNA pol [30] and Dpo4 [17,45], the former being more selective for correct geometry compared to Y-family pols. Also of interest is the observation that the relative insertion efficiency of pol η opposite abasic sites in the xAT and GxT sequence contexts are similar to each other (30% and 22%), whereas they differ by five-fold for exonuclease-deficient T7 DNA pol (Table 1). This implies that these two enzymes differ with respect to how they partly solve the challenge of bypassing this lesion, i.e. success by T7 DNA pol is increased if a misalignment mechanism is available by virtue of the sequence context, whereas pol η is less sensitive in this regard. Finally, in contrast to somewhat similar insertion probabilities opposite abasic sites, T7 DNA pol is 10- to 50-fold less efficient than the Y-family polymerases at inserting a nucleotide opposite either template base in the dimer (Table 1). Such large differences can be rationalized on structural grounds, as extensively discussed previously [23,35,45].
4.3. Efficiency of extending damaged template–primer termini
The results in Table 1 reveal low relative extension efficiencies during a single cycle of processive synthesis to bypass abasic sites and the TT dimer. This indicates that T7 DNA pol generally has great difficulty extending damaged template–primers resulting from insertion opposite lesions. This extension barrier is consistent with partitioning of the wild-type enzyme into the exonuclease mode, thereby suppressing bypass. It differs strongly from results with pol η and Dpo4, both of which are more efficient for this key incorporation step needed to achieve complete bypass (Table 1).
4.4. T7 DNA pol performs very inefficient TLS
During a single cycle of processive synthesis by either wild-type or exonuclease-deficient T7 DNA polymerase, we did not detect complete bypass of abasic sites or the TT dimer. However, the situation is lesion-specific because T7 DNA pol has substantially higher relative bypass efficiency when performing a single cycle of processive synthesis to bypass an 8-oxoG in the template [35]. In the present study, bypass was only observed when exonuclease-deficient T7 DNA pol was permitted to attempt multiple cycles of polymerization. Values for the number of bypass events per cycle (Table 1) indicated that greater than 99% of bypass attempts were failures. This is in contrast to Y-family polymerases that can bypass either AP sites or a TTD under single hit conditions [12,16], indicative of a substantially lower number of cycles that result in successful bypass. The fact that between 180 and 1300 attempted cycles by this replicative polymerase (Table 1) were needed to achieve only one complete bypass event implies that an enzyme with such behavior would be highly unlikely to contribute to lesion bypass in vivo under normal conditions. However, under conditions where a dedicated TLS polymerase is unavailable (i.e. genetically or perhaps under conditions of heavy lesion load), forced bypass by replicative polymerases may become a critical process in cell survival. In that respect, these results, while showing a very low efficiency of bypass, are relevant to damaged-induced mutagenesis in vivo. Currently, only very limited quantitative data exists for most polymerase/lesion combinations for either efficiency or fidelity of bypass. Therefore, assigning a role in either error-free or mutagenic bypass of a given lesion to any single polymerase is difficult. On that basis, although many DNA polymerases have been shown to be capable of some degree of TLS, we believe that biologically relevant interpretations regarding their possible roles in bypassing various lesions would benefit by quantifying relative bypass efficiency per attempted polymerization cycle. Given the differences reported here for a single polymerase with two different lesions, it is possible that other replicative polymerases will display vastly different bypass properties than previously reported when investigated in a more rigorous, quantitative manner such as described here.
4.5. Fidelity of lesion bypass
The requirement for multiple polymerization cycles to achieve even one bypass event may reflect the fact that T7 DNA pol has high geometric selectivity and must make many attempts to find a few catalytically competent solutions to the barriers imposed by lesions. These solutions may be lesion specific and they may involve rare mismatched and/or misaligned substrates during one or more of the several incorporations needed to complete bypass. To examine the possibilities, we determined the fidelity of the TLS reactions, and compared the results to the bypass fidelity of the Y-family polymerases that are much more efficient in catalyzing TLS.
4.6. Incorporation specificity during lesion bypass
Collective results for bypass of the abasic sites indicate four distinct catalytically competent solutions for bypass of this canonical non-coding lesion. One is direct misincorporation of dAMP opposite the abasic site followed by extension. This can account for the vast majority (>99%) of the complete bypass events in the xATT sequence and about half (45%) of the events in the GAxT sequence, where dAMP incorporation is phenotypically silent due to retention of the TAG codon but can be detected during other error events (Table 4). And while less than 5% of the total events in the GxTT sequence were dAMP incorporations, they did comprise a majority of the events leading to dark blue plaques (Table 3). Such preferential incorporation of dAMP is consistent with earlier kinetic studies and conforms to the classical “A rule” for nucleotide incorporation opposite non-instructional lesions (reviewed in [18–22]. The second solution is deletion of the abasic site.
Although rarely observed in the 3′-xATT sequence, this deletion represents over 80% of the bypass events in the 3′-GxTT sequence and also occurs, albeit at a lower frequency, in the 3′-GAxT sequence (Table 4). This deletion may be explained either by (i) insertion of dAMP followed by primer relocation to pair that dAMP with an adjacent template T or (ii) by misalignment that allows the abasic site to simply be skipped [22]. We favor the former hypothesis because it would allow formation of a correct primer terminus that could lessen the strong barrier to extension of the damaged template–primer (see [46] and references therein). The third and fourth outcomes of complete bypass are deletion and insertion of a base within a template GGG run downstream of the abasic site. Deletion of a G from the run was observed in both dark blue and colorless plaques from the 3′-GAxT substrate. All of the G deletions are accompanied by incorporation of dAMP opposite the abasic site. In the 3′-GxTT sequence, dark blue plaques that had a deleted AP site also showed an insertion of a G in this run. Both of these events are dependent on the abasic sites, because the frequencies are from 9- to over 100-fold higher than observed among the products of copying undamaged DNA. Interestingly, bypass of the TT dimer also resulted in a large number (14 of the 16 sequenced colorless plaques) of single base deletions of a template G in the GGG run downstream of the dimer (Table 3), and these were accompanied by correct incorporation of dAMP opposite both thymines in the dimer. The results suggest that as bypass proceeds, the presence of an abasic site-dAMP “pair” or two T-A pairs in a dimer in the upstream duplex bound by T7 DNA pol increases the probability of subsequent strand misalignment. Since complete bypass requires multiple cycles of polymerization, the misalignments could arise within the confines of the polymerase bound to the template:primer, during enzyme dissociation or reassociation, or when the polymerase is not bound. Curiously, only deletions that result from an unpaired G in the template strand are seen with the 3′-GAxT sequence, whereas only additions resulting from an extra C in the primer strand are seen with the 3′-GxTT sequence. We currently have no explanation for these asymmetries. These events are another manifestation of the general concepts of lesion-dependent “untargeted mutagenesis” and “action-at-a-distance”, ideas that have been proposed to explain polymerization errors in several other systems [47,48], including substitutions at undamaged bases flanking abasic sites and 8-oxoG lesions [49,50]. Overall, the data on bypass of the same lesion in three different sequence contexts clearly illustrate the remarkably strong influence of sequence context on mutagenic specificity during TLS. The results also differ markedly from similar measurements using Y-family DNA polymerases (Table 4), where the relative levels of the different types of errors is less affected by the sequence context (Table 4 and [12,16]).
The majority of the products generated during complete bypass of the TT dimer represented the incorporation of dAMP opposite both the 3′ and 5′ thymines in the dimer. It is possible that largely “accurate” bypass of the 3′ T could reflect preferential insertion of dAMP in a non-instructional manner, as has been proposed based on kinetic and structural data [18]. Our data are consistent with this possibility, but do not rule out the possibility that some mis-instructional insertion occurs. The observation that the error rate for incorporating dGMP opposite the 3′ T of the dimer is substantially higher than at the corresponding undamaged T (Table 5) is similar to the case with Dpo4 [16], and structural evidence [23] strongly indicates that Dpo4 does perform template dependent bypass at both thymines of a TT dimer. Thus, our data is also consistent with the occasional misinsertion of dGMP opposite the 3′ T during TT dimer bypass due to rare, albeit incorrect, use of the base coding potential during one of the many cycles of polymerization needed to achieve bypass. T7 DNA pol fidelity for misincorporating dGMP opposite the 5′ T of the dimer is ninefold higher than for copying the 3′ T, and not substantially different from that for copying the corresponding undamaged 5′ T (Table 5). Similar asymmetries in error rates at the two thymines were also observed for bypass of a cis–syn dimer by human pol η and by Dpo4 [16]. Explanations for these asymmetries in error rates need not be the same for A- and Y-family polymerases, and include the just mentioned possibility that the 3′ T is non-instructional, whereas the 5′ T retains coding potential, or that the bypass of the 3′ T involves Watson–Crick base pairing, whereas the 5′ T involves Hoogsteen base pairing [23].
Acknowledgments
We thank Drs. Xuejun Zhong and Mercedes Arana for constructive comments on the manuscript. This research was funded by the Intramural Research Program of the NIEHS/NIH.
References
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, DC: 2005. [Google Scholar]
- 2.Friedberg EC. Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol. 2005;6:943–953. doi: 10.1038/nrm1781. [DOI] [PubMed] [Google Scholar]
- 3.Bebenek K, Kunkel TA. Functions of DNA polymerases in DNA repair and replication. In: Yang W, editor. Advances in Protein Chemistry. Elsevier Academic Press; Boston: 2004. [DOI] [PubMed] [Google Scholar]
- 4.Rattray AJ, Strathern JN. Error-prone DNA polymerases: when making a mistake is the only way to get ahead. Annu Rev Genet. 2003;37:31–66. doi: 10.1146/annurev.genet.37.042203.132748. [DOI] [PubMed] [Google Scholar]
- 5.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 6.Boudsocq F, Ling H, Yang W, Woodgate R. Structure-based interpretation of missense mutations in Y-family DNA polymerases and their implications for polymerase function and lesion bypass. DNA Repair (Amst) 2002;1:343–358. doi: 10.1016/s1568-7864(02)00019-8. [DOI] [PubMed] [Google Scholar]
- 7.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 8.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69 doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 9.Friedberg EC, Lehmann AR, Fuchs RP. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 10.McCulloch SD, Kokoska RJ, Kunkel TA. Efficiency, fidelity and enzymatic switching during translesion DNA synthesis. Cell Cycle. 2004;3:580–583. [PubMed] [Google Scholar]
- 11.Plosky BS, Woodgate R. Switching from high-fidelity replicases to low-fidelity lesion bypass polymerases. Curr Opin Genet Dev. 2004;14:113–119. doi: 10.1016/j.gde.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Kokoska RJ, McCulloch SD, Kunkel TA. The efficiency and specificity of apurinic/apyrimidinic site bypass by human DNA polymerase η and Sulfolobus solfataricus Dpo4. J Biol Chem. 2003;278:50537–50545. doi: 10.1074/jbc.M308515200. [DOI] [PubMed] [Google Scholar]
- 13.Kokoska RJ, Bebenek K, Boudsocq F, Woodgate R, Kunkel TA. Low fidelity DNA synthesis by a Y-family DNA polymerase due to misalignment in the active site. J Biol Chem. 2002;277(22):19633–19638. doi: 10.1074/jbc.M202021200. [DOI] [PubMed] [Google Scholar]
- 14.Matsuda T, Bebenek K, Masutani C, Hanaoka F, Kunkel TA. Low fidelity DNA synthesis by human DNA polymerase-η. Nature. 2000;404:1011–1013. doi: 10.1038/35010014. [DOI] [PubMed] [Google Scholar]
- 15.Matsuda T, Bebenek K, Masutani C, Rogozin IB, Hanaoka F, Kunkel TA. Error rate and specificity of human and murine DNA polymerase η. J Mol Biol. 2001;312:335–346. doi: 10.1006/jmbi.2001.4937. [DOI] [PubMed] [Google Scholar]
- 16.McCulloch SD, Kokoska RJ, Masutani C, Iwai S, Hanaoka F, Kunkel TA. Preferential cis–syn thymine dimer bypass by DNA polymerase η occurs with biased fidelity. Nature. 2004;428:97–100. doi: 10.1038/nature02352. [DOI] [PubMed] [Google Scholar]
- 17.Ling H, Boudsocq F, Woodgate R, Yang W. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell. 2001;107:91–102. doi: 10.1016/s0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 18.Taylor JS. New structural and mechanistic insight into the A-rule and the instructional and non-instructional behavior of DNA photoproducts and other lesions. Mutat Res. 2002;510:55–70. doi: 10.1016/s0027-5107(02)00252-x. [DOI] [PubMed] [Google Scholar]
- 19.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 20.Strauss BS. The “A” rule revisited: polymerases as determinants of mutational specificity. DNA Repair (Amst) 2002;1:125–135. doi: 10.1016/s1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- 21.Strauss BS. The ‘A rule’ of mutagen specificity: a consequence of DNA polymerase bypass of non-instructional lesions? Bioessays. 1991;13:79–84. doi: 10.1002/bies.950130206. [DOI] [PubMed] [Google Scholar]
- 22.Goodman MF, Creighton S, Bloom LB, Petruska J. Biochemical basis of DNA replication fidelity. Crit Rev Biochem Mol Biol. 1993;28:83–126. doi: 10.3109/10409239309086792. [DOI] [PubMed] [Google Scholar]
- 23.Ling H, Boudsocq F, Plosky BS, Woodgate R, Yang W. Replication of a cis–syn thymine dimer at atomic resolution. Nature. 2003;424:1083–1087. doi: 10.1038/nature01919. [DOI] [PubMed] [Google Scholar]
- 24.Goodman MF. Hydrogen bonding revisited: geometric selection as a principal determinant of DNA replication fidelity. Proc Natl Acad Sci USA. 1997;94:10493–10495. doi: 10.1073/pnas.94.20.10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doublié S, Ellenberger T. The mechanism of action of T7 DNA polymerase. Curr Opin Struct Biol. 1998;8:704–712. doi: 10.1016/s0959-440x(98)80089-4. [DOI] [PubMed] [Google Scholar]
- 26.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 27.Kool ET. Active site tightness and substrate fit in DNA replication. Annu Rev Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 28.Beard WA, Wilson SH. Structural insights into the origins of DNA polymerase fidelity. Structure. 2003;11:489–496. doi: 10.1016/s0969-2126(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 29.Echols H, Goodman MF. Fidelity mechanisms in DNA replication. Annu Rev Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 30.Doublié S, Tabor S, Long AM, Richardson CC, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Wang M, Kool ET, Taylor JS. Pyrene nucleotide as a mechanistic probe: evidence for a transient abasic site-like intermediate in the bypass of dipyrimidine photoproducts by T7 DNA polymerase. Biochemistry. 2000;39:14603–14610. doi: 10.1021/bi001446v. [DOI] [PubMed] [Google Scholar]
- 32.Matray TJ, Kool ET. A specific partner for abasic damage in DNA. Nature. 1999;399:704–708. doi: 10.1038/21453. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Dutta S, Doublie S, Bdour HM, Taylor JS, Ellenberger T. Nucleotide insertion opposite a cis–syn thymine dimer by a replicative DNA polymerase from bacteriophage T7. Nat Struct Mol Biol. 2004;11:784–790. doi: 10.1038/nsmb792. [DOI] [PubMed] [Google Scholar]
- 34.Murata T, Iwai S, Ohtsuka E. Synthesis and characterization of a substrate for T4 endonuclease V containing a phosphorodithioate linkage at the thymine dimer site. Nucl Acids Res. 1990;18:7279–7286. doi: 10.1093/nar/18.24.7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brieba LG, Eichman BF, Kokoska RJ, Doubliâe S, Kunkel TA, Ellenberger T. Structural basis for the dual coding potential of 8-oxoguanosine by a high-fidelity DNA polymerase. EMBO J. 2004;23(17):3452–3461. doi: 10.1038/sj.emboj.7600354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark JM, Joyce CM, Beardsley GP. Novel blunt-end addition reactions catalyzed by DNA polymerase I of Escherichia coli. J Mol Biol. 1987;198:123–127. doi: 10.1016/0022-2836(87)90462-1. [DOI] [PubMed] [Google Scholar]
- 37.Clark JM. Novel non-templated nucleotide addition reactions catalyzed by prokaryotic and eucaryotic DNA polymerases. Nucl Acids Res. 1988;16:9677–9686. doi: 10.1093/nar/16.20.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCulloch SD, Kunkel TA. Measuring the fidelity of translesion DNA synthesis. Methods Enzymol. 2006;408 doi: 10.1016/S0076-6879(06)08021-9. [DOI] [PubMed] [Google Scholar]
- 39.Kunkel TA, Patel SS, Johnson KA. Error-prone replication of repeated DNA sequences by T7 DNA polymerase in the absence of its processivity subunit. Proc Natl Acad Sci USA. 1994;91:6830–6834. doi: 10.1073/pnas.91.15.6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith CA, Baeten J, Taylor JS. The ability of a variety of polymerases to synthesize past site-specific cis–syn, trans–syn-II, (6-4), and Dewar photoproducts of thymidylyl-(3′ → 5′)-thymidine. J Biol Chem. 1998;273:21933–21940. doi: 10.1074/jbc.273.34.21933. [DOI] [PubMed] [Google Scholar]
- 41.Le Gac NT, Delagoutte E, Germain M, Villani G. Inactivation of the 3′–5′ exonuclease of the replicative T4 DNA polymerase allows translesion DNA synthesis at an abasic site. J Mol Biol. 2004;336:1023–1034. doi: 10.1016/j.jmb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Eckert KA, Opresko PL. DNA polymerase mutagenic bypass and proofreading of endogenous DNA lesions. Mutat Res. 1999;424:221–236. doi: 10.1016/s0027-5107(99)00021-4. [DOI] [PubMed] [Google Scholar]
- 43.Freisinger E, Grollman AP, Miller H, Kisker C. Lesion (in)tolerance reveals insights into DNA replication fidelity. EMBO J. 2004;23:1494–1505. doi: 10.1038/sj.emboj.7600158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hogg M, Wallace SS, Doublie S. Crystallographic snapshots of a replicative DNA polymerase encountering an abasic site. EMBO J. 2004;23:1483–1493. doi: 10.1038/sj.emboj.7600150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ling H, Boudsocq F, Woodgate R, Yang W. Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol Cell. 2004;13:751–762. doi: 10.1016/s1097-2765(04)00101-7. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Diaz M, Kunkel TA. Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem Sci. 2006;31:206–214. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 47.Abbotts J, Bebenek K, Kunkel TA, Wilson SH. Mechanism of HIV-1 reverse transcriptase: trmination of processive synthesis on a natural DNA template is influenced by the sequence of the template–primer stem. J Biol Chem. 1993;268:10312–10323. [PubMed] [Google Scholar]
- 48.Harfe BD, Jinks-Robertson S. DNA polymerase ζ introduces multiple mutations when bypassing spontaneous DNA damage in Saccharomyces cerevisiae. Mol Cell. 2000;6:1491–1499. doi: 10.1016/s1097-2765(00)00145-3. [DOI] [PubMed] [Google Scholar]
- 49.Efrati E, Tocco G, Eritja R, Wilson SH, Goodman MF. Abasic translesion synthesis by DNA polymerase beta violates the “A- rule”. Novel types of nucleotide incorporation by human DNA polymerase beta at an abasic lesion in different sequence contexts. J Biol Chem. 1997;272:2559–2569. doi: 10.1074/jbc.272.4.2559. [DOI] [PubMed] [Google Scholar]
- 50.Efrati E, Tocco G, Eritja R, Wilson SH, Goodman MF. “Action-at-a-distance” mutagenesis. 8-Oxo-7,8-dihydro-2′-deoxyguanosine causes base substitution errors at neighboring template sites when copied by DNA polymerase beta”. J Biol Chem. 1999;274:15920–15926. doi: 10.1074/jbc.274.22.15920. [DOI] [PubMed] [Google Scholar]
- 51.Bebenek K, Kunkel TA. Analyzing the fidelity of DNA polymerases. Methods Enzymol. 1995;262:217–232. doi: 10.1016/0076-6879(95)62020-6. [DOI] [PubMed] [Google Scholar]