

Abstract
The effects of TCDD (2,3,7,8-Tetrachlorodibenzo-p-dioxin) on action potential and afterdepolarizations were studied in rat ventricular myocytes using nystatin-perforated whole-cell patch-clamp technique. TCDD treatment, in the concentration range of 1 to 100 nM, significantly prolonged action potential duration measured at 90% of repolarization (APD90). The triggered delayed-afterdepolarizations (DADs) was observed in 6 out of 8 cells after exposure of TCDD (10 nM). In the presence of isoproterenol (ISO, 10 nM) or Bay K 8644 (1 μM), TCDD (10 nM) markedly augmented the amplitude and frequency of the arrhythmogenic DADs and triggered sustained spontaneous firings in ventricular myocytes. Voltage-clamp data indicated that TCDD (10 nM) exposure significantly enhanced the transient inward current (Iti). The triggered early-afterdepolarizations (EADs) were evoked only in cells simultaneously exposed to TCDD (10 nM) and ISO (or Bay K 8644). Further study indicated that TCDD treatment increased L-type Ca2+ current. These results indicate that activation of TCDD signaling pathway can prolong action potential duration and cause abnormal triggered afterdepolarizations. These effects may lead to clinically relevant ventricular arrhythmia especially when susceptible individuals are under elevated sympathetic stress or suffering from other myocardiopathies coincided with Ca2+-overload.
Keywords: TCDD, cardiac action potential, triggered activity, afterdepolarizations, transient outward current, ventricular arrhythmia
Introduction
Dioxin TCDD (2,3,7,8-Tetrachlorodibenzo-p-dioxin) and its related halogenated aromatic hydrocarbons (HAHs) continue to be of concern to the public because of their persistence and bioaccumulation in the environment and because low-level exposure is associated with subtle but significant cardiovascular, behavioral, and neurological deficits [1–5]. As the prototype toxin for the diverse HAH chemicals, TCDD was classified as a human carcinogen [4] and has the potential to disrupt multiple signaling pathways [6–10]. Humans are generally exposed to such compounds via consuming polluted food, water, and air [5,10]. The major toxic responses to TCDD are believed to be mediated by activating aryl hydrocarbon receptor (AhR) that dimerizes with AhR nuclear translocator (ARNT) [10]. The complex binds to downstream dioxin-responsive element (DRE, a specific DNA sequence, 5′-GCGTG-3′) and induces the target gene (CYP1A1 or CYP1A2 or CYP1B1) expression [11,12]. However, the lack of a direct correlation between the induction of cytochrome P450 enzymes and the diverse toxic responses of TCDD in various animal species suggests that additional signaling mechanisms may be involved in TCDD-associated toxicities [9,13]. Previous reports indeed demonstrated that TCDD by activating AhR, independent of the induction of gene expression, rapidly modulates PKC, PKA, and protein tyrosine kinases in vitro as well as in vivo [8–9, 13–15].
The heart, like the liver and thymus, has the AhR-ARNT-DRE system [2, 16] and been considered as one of the target organs of TCDD toxicity [11–12, 17–22]. Although it was claimed that TCDD or related aryl hydrocarbons did not increase the incidence of cardiovascular diseases [23, 24], studies did reveal a strong dose-dependent relation between mortality due to heart diseases and exposure to dioxin even at very low doses in both human beings and experimental animals [1, 25–26]. Many cardiac histological and pathological changes associated with TCDD exposure have been defined in animal models, including ventricular dilatation and edema [27], myocardial degeneration [28] and defects [29], slowed heart rate [30], altered cardiac contractility [16, 31], decreased sensitivity of myocardium to β–adrenergic agonist stimulation and disrupted intracellular calcium homeostasis [18], altered responses in chicken embryo hearts to antiarrhythmic agent flecainide [19], as well as increased incidence of ischemic heart disease [26] and cardiomyopathy [21].
In this study we examined TCDD-caused abnormal afterdepolariztions, i.e., early afterdepolarizations (EADs) and delayed afterdepolariztions (DADs) in freshly isolated rat ventricular myocytes. EAD usually arises at the plateau phase of action potential and appears to relate to time- and voltage-dependent reactivation of L-type Ca2+ currents [32]. DAD, defined as the "oscillations” in membrane potential after completion of the action potential [33], is believed to be due to intracellular Ca2+ overload and oscillation of Ca2+ release from the sarcoplasmic reticulum (SR) [32, 34]. Both EADs and DADs play important roles in triggering deteriorative or life-threaten cardiac arrhythmias [34, 35]. We found that TCDD prolonged action potential duration, induced abnormal triggered-afterdepolarizations and spontaneous action potentials, especially when the cells are challenged with either β–adrenergic agonist isoproterenol (ISO) or L-type Ca2+ channel opener BAY K8644. This finding may help to define the TCDD vulnerable populations like children, elderly people or patients with predisposed pro-arrhythmic cardiac conditions.
Methods
Materials
The stock solution of TCDD (100 μM in DMSO) was prepared at the Environmental Toxicology Program of National Institute of Environmental Health Sciences. All other chemicals were purchased from Sigma Chemical Corp. (St. Louis, MO) except type II collagenase (Worthington Chemical Corp. Lakewood NJ). Isoproterenol (1 mM) was dissolved in aqueous solution containing 3 mM ascorbic acid to prevent its oxidation. BAY K 8644 and nystatin was dissolved in dimethylsulfoxide (DMSO).
Ventricular myocyte isolation
Single ventricular cells were isolated by enzymatic digestion of female Sprague-Dawley rat hearts. In brief, the rats were anesthetized with pentobarbital sodium (50 mg/kg) by intraperitoneal injection. The heart was excised and perfused through the aorta with oxygenated Tyrode solution containing (in mM): 140 NaCl, 5.4 KCl, 1.5 MgCl2, 1.8 CaCl2, 0.33 NaH2PO4, 5.5 D-(+)-Glucose, 5.5 HEPES (pH=7.4 with NaOH). After a 5–10 min nominally Ca2+-free Tyrode solution perfusion, the heart was digested by type II collagenase (0.4 mg/ml, ~300 units/mg) with 1.6 mg/ml bovine serum albumin (BSA, V). Then, ventricles were minced, incubated, and shaken at 36.5ºC for 5–20 min in the enzyme solution. Individual cells were collected, washed, and stored at room temperature (22 ± 0.5 ºC) in a recovery Tyrode solution containing 0.1 mM Ca2+ for later use. Animal use was in accordance with the protocols approved by the Institutional Animal Care and Use Committee.
Electophysiology
Action potential and transmembrane current were recorded under nystatin-perforated whole-cell configuration of patch-clamp technique. Bath solution was a modified Tyrode’s solution containing same components as specified above except reducing MgCl2 to 0.53 mM. Patch pipettes were filled with a solution containing (in mM): 140 KCl, 1 MgCl2, 0.5 CaCl2, 10 Glucose, 10 Hepes (pH adjusted to 7.2 with KOH) and had a resistance range of 4 to 7 MΩ. Nystatin (~8.5 nM) was added to the pipette solution freshly before experiments. Records were low-pass filtered with a cutoff frequency of 2.9 kHz, digitized at 2 kHz and acquired with an EPC9 amplifier/Pusle software system (HEKA Instrutech Corp. Lambrecht, Germany). Action potentials were elicited by current pulses delivered through the patch pipette with strength of 1.2–1.5 fold of the threshold level and a duration of 10 ms at a frequency of 0.25~2 Hz. DADs were evoked by 2 Hz pacing or by combination of pacing with pharmacological stimulation. To facilitate the development of Iti, repeated trains of 20 voltage-clamp conditioning steps (from −80 to +40 mV and in a duration of 300 msec) were applied at a frequency of 2 Hz [36]. All data were acquired at 35 ± 1 ºC. The action potential duration was measured at 90% of repolarization (APD90). The amplitude of DADs or Iti was measured as the difference between the peak of the "oscillation" and the basal membrane potential or current. In case of multiple oscillations, the largest one was taken for analysis.
TCDD exposure
Two types of exposure schemes were carried out. In the first scheme, TCDD was applied to the cells after patch-clamp recordings had been established, which allowed us to assess the baseline electrophysiological parameters of the cells and their immediate (within 30 sec) responses to TCDD exposure. In the second scheme, to measure the effects of relatively prolonged TCDD exposure (from 30 min to 4 hrs), the cells were pre-incubated with various concentrations of TCDD before starting the electrophysiological recording. The TCDD-induced responses occurred in 5~30 min following exposure and lasted for more than 2 hrs in cells pre-incubated with TCDD. TCDD exposure did not elicit rapid responses (<1 min) and there was no significant difference between the two exposure schemes when the response reached the steady-state. Thus, the data collected from the two exposure schemes were pooled together for analysis.
Data analysis
All values were expressed as mean ± SE. Differences between group-means and non-treatment versus treatment interventions were considered statistically significant at p<0.05. For multinomial experiments in which the data is classified with respect to two factors, the data were analyzed with the Chi-Square Test (Analysis of Contingency Tables). Two-population (independent) t-test or one-way analysis of variance (ANOVA) was used to analyze the quantitative data followed by Student-Newman-Keul’s post hoc test for multigroup comparison if applicable.
Results
This study characterized the effects of acute exposure to TCDD on action potential parameters, triggered after-depolarization activity, and the transient inward current in freshly isolated rat ventricular myocytes.
TCDD prolonged action potential duration (APD)
To avoid disrupting the intracellular milieu, all the action potentials were recorded under nystatin-perforated configuration, which greatly prolonged the stead-state recording time and minimized the inconsistency in the shape of action potentials between cells. After exposed to TCDD through bath perfusion or pre-incubation, APD90 was significantly prolonged (figure 1A), mainly by lengthening the plateau phase. The prolongation effect was concentration-dependent and measurable when the cells were exposed to 1 nM TCDD (figure 1). TCDD exposure did not alter the resting membrane potential, the rate of phase 0 depolarization, and the amplitude of action potentials. On average, TCDD at 10 nM concentration prolonged APD90 by 43.3% (p<0.05). Figure 1B summarizes the concentration-dependent effect of TCDD on APD90.
Figure 1.
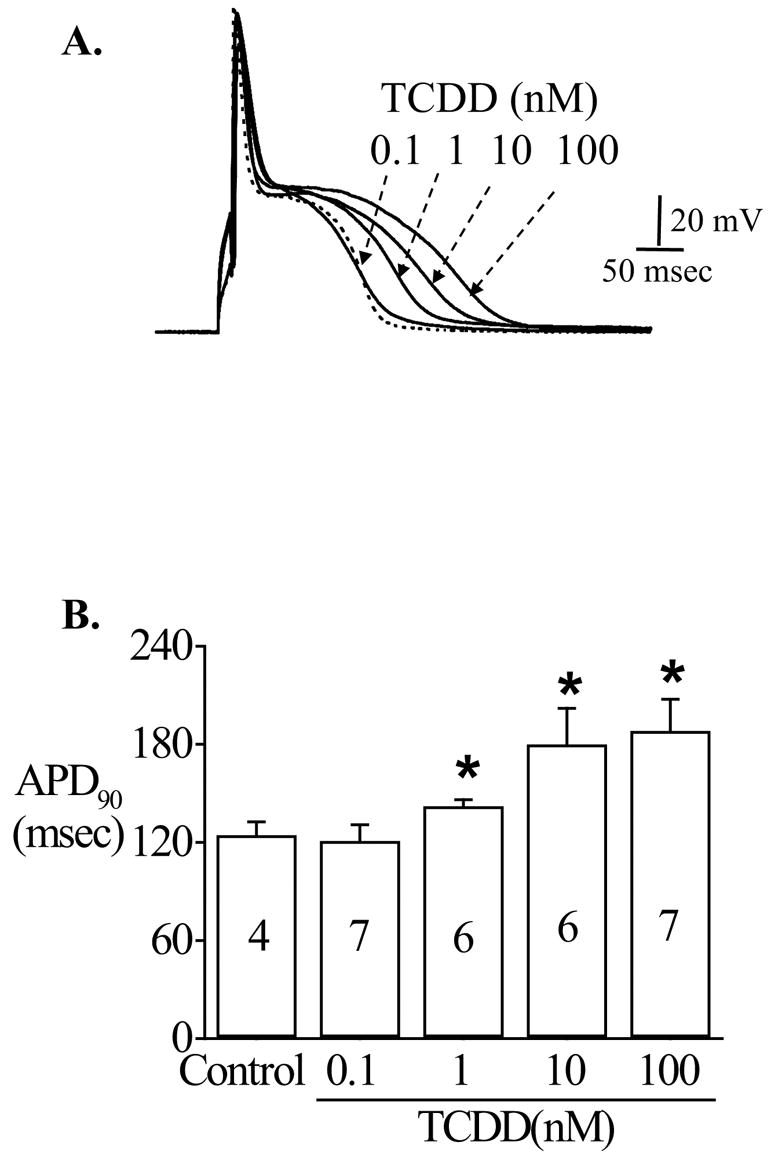
Effect of TCDD exposure on action potentials. Action potentials were recorded from freshly isolated rat ventricular myocytes under nystatin-perforated configuration. The injected current (10 msec duration) was adjusted to 1.2~1.5 fold of the threshold current needed to excite a cell without producing artifact overshoots. A. A typical alteration in action potential shape by different concentrations of TCDD (0.1, 1, 10, 100 nM). The dashed line is the action potential recorded before TCDD exposure (DMSO control). B. The average change in APD90 for experiments shown in panel A. * indicates statistic significance at p<0.05 level.
TCDD treatment increased delayed-afterdepolarizations (DADs)
When the cells were driven for 15 sec by 2 Hz pacing pulses, DADs were elicited only in one of eighth control ventricular cells. However, in the presence of TCDD (10 nM), 75% of cells (6/8) responded with multiple DADs immediately following the termination of driving pulses (figure 2). The average amplitude of DADs recorded in TCDD-treated cells was 2.1±0.7 mV (figure 2B). This magnitude of after-depolarization could contribute to trigger conductible arrhythmogenic action potentials as shown in figure 3B and 4B.
Figure 2.
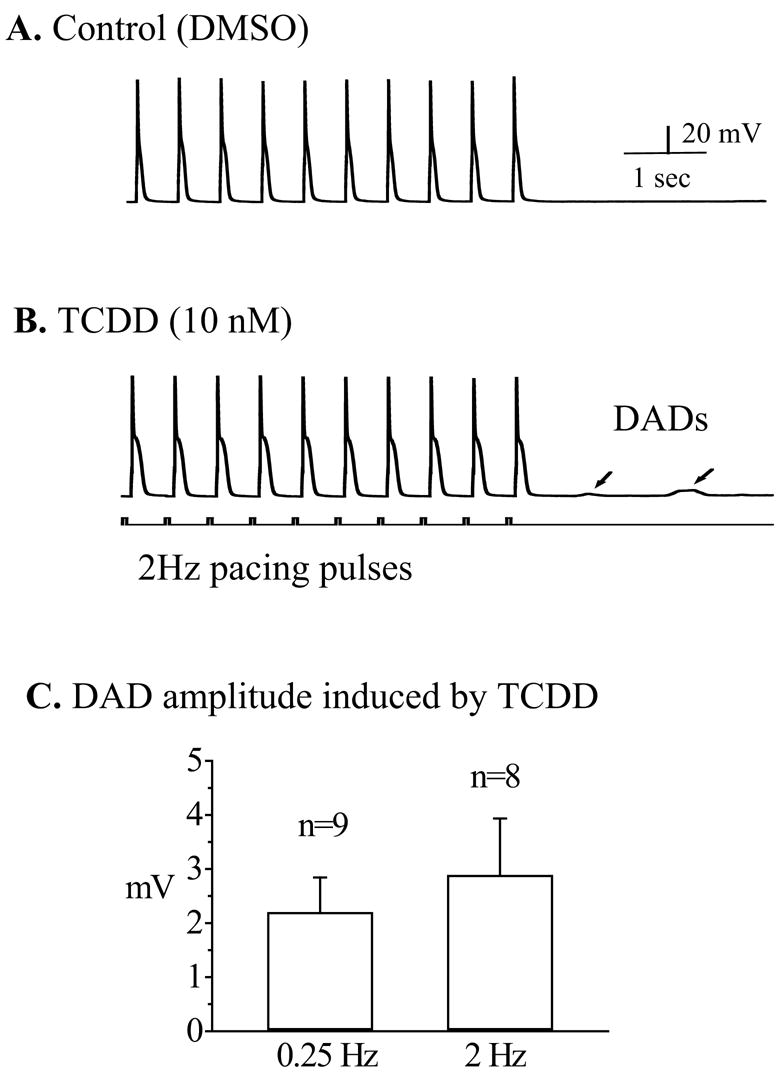
TCDD induced delayed-afterdepolarizations (DADs). A train of action potentials (30 in total) was elicited by a 15 sec overdriving stimulation at a frequency of 2 Hz. Only last 10 action potential traces were shown. Recordings were carried out after either DMSO or TCDD (10 nM) application. A. A typical response of a ventricular mycyte treated with DMSO at 2 Hz pacing. Note that there was no after-depolarization activity when the pacing was terminated. B. In another myocyte treated with TCDD (10 nM), DADs (as indicated by arrows) were generated by the same stimulation protocol. Calibration marks apply to both panel A and B. Note, if there were multiple DADs in a single recording, the largest DAD was chosen to measure the amplitude of the DADs for the given cell.
Figure 3.
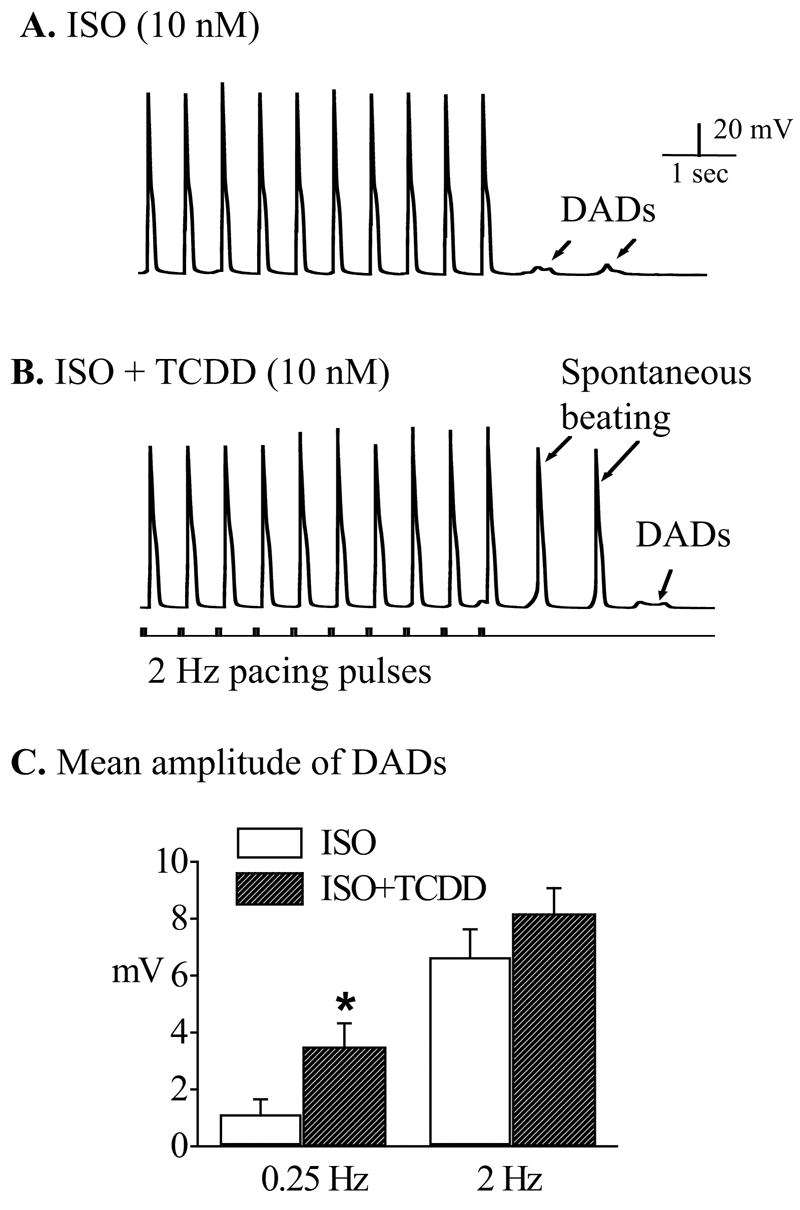
TCDD promoted ISO-induced DADs into spontaneous action potentials. Recordings were carried out after treating the cells with ISO (10 nM) alone or TCDD (10 nM) plus ISO (10 nM). A. A typical response of a ventricular mycyte treated with ISO (10 nM) to 2 Hz pacing. Note that there were after-depolarization activities when the pacing was terminated. B. In another myocyte treated with TCDD (10 nM) plus ISO (10 nM), DADs (as indicated by arrows) were developed into conductible spontaneous action potentials by the same stimulation protocol. Calibration marks apply to both A and B. C. The average amplitudes of DADs induced by ISO alone and ISO plus TCDD at two pacing frequencies (0.25 and 2 Hz). * indicates statistic significance at p<0.05 level.
Figure 4.
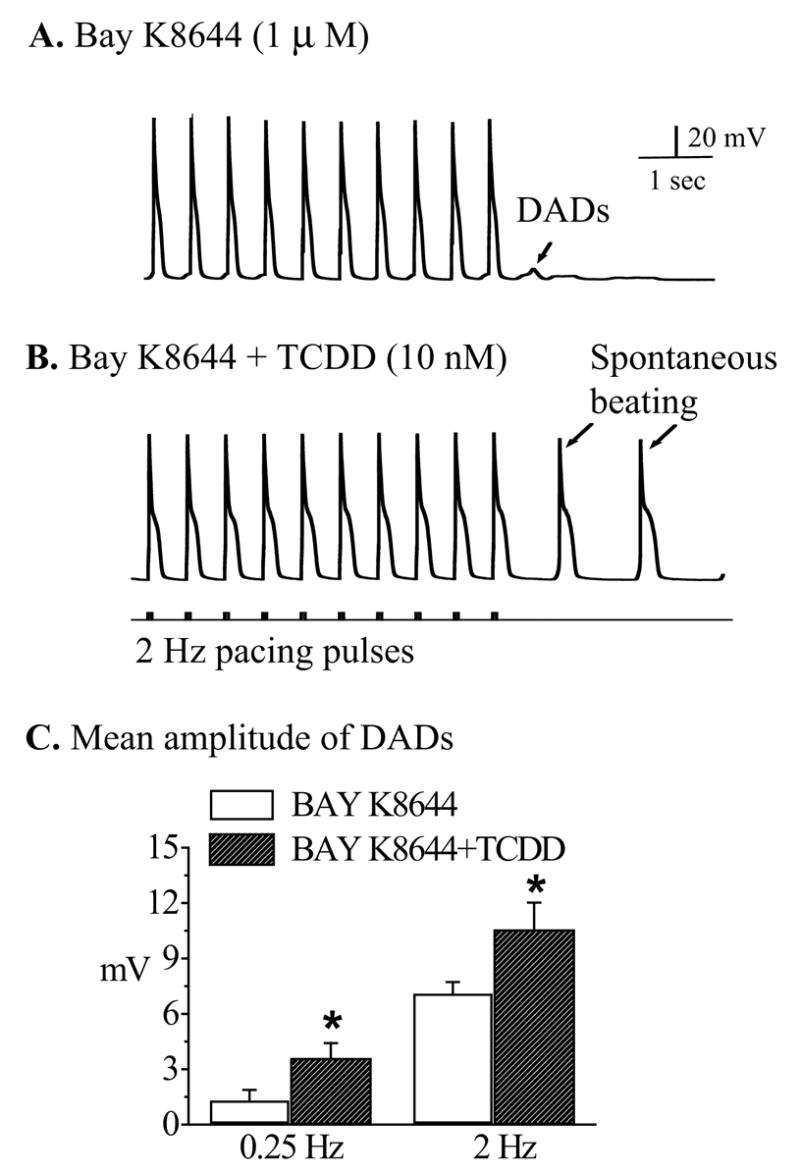
TCDD promoted Bay K 8644-induced DADs into spontaneous action potentials. Recordings were carried out after treating the cells with Bay K (1 μM) alone or TCDD (10 nM) plus Bay K (1 μM). A. A typical response of a ventricular mycyte treated with Bay K (1 μM) to 2 Hz pacing. B. In another myocyte treated with TCDD (10 nM) plus Bay K (1 μM) DADs were developed into conductible spontaneous action potentials following the same stimulation protocol. Calibration marks apply to both A and B. C. The average amplitudes of DADs induced by Bay K alone and Bay K plus TCDD at two pacing frequencies (0.25 and 2 Hz). * indicates statistic significance at p<0.05 level.
TCDD enhanced β-adrenergic agonist-triggered DADs
β-adrenergic stimulation facilitates afterdepolarizations in cardiomyocytes by increasing L-type Ca2+ current. It is possible that TCDD may alter the triggered responses of myocytes to sympathetic stimulation. By titrating the concentrations of isoproterenol (ISO, a β-adrenergic agonist), we found that application of ISO (10 nM) alone facilitated DADs in all the ventricular cells over-driven by 2 Hz current pulses (figure 3A) and few cells (less than one-thirds) developed conductible spontaneous action potentials (no shown). In comparison, when TCDD (10 nM) was added on the top of ISO challenge, not only was the amplitude of the DADs augmented, but nine of ten cells (90%) transpired bursting spontaneous action potentials (figure 3B). When the cells were driven by a slow pacing rate (0.25 Hz), the average amplitudes of DADs from the cells treated with TCDD (10 nM) plus ISO (10 nM) was significantly greater than that from the cells treated with ISO alone (figure 3C).
TCDD enhanced Bay K 8644-triggered DADs
Dihydropyridine analogue, Bay K 8644 directly stimulates L-type Ca2+-channels and increases Ca2+ influx and facilitates the triggered afterdepolarizations in myocytes. We expected that TCDD exposure would also enhance Bay K-caused Ca2+ overload, therefore augment Bay K 8644-triggered DADs. Bay K (1 μM) increased DAD activities in most ventricular cells (figure 4A). In spite of that, only 1 out of 8 cells (12.5%) developed conductible spontaneous action potentials. When TCDD (10 nM) was applied on top of Bay K (1 μM), the amplitude of the DADs was augmented and 7 out of 8 cells (87.5%) developed DAD-triggered spontaneous action potentials (figure 4B). When the cells were paced at a slow rate (0.25 Hz), the average amplitude of DADs in cells exposed to TCDD (10 nM) and Bay K (1 μM) was significantly augmented (figure 4C).
TCDD increased transient inward current (Iti)
It is known that the occurrence of DADs is due to instigation of a transient inward transmembrane current (Iti). We predicted that TCDD-caused DADs should also be correlated with an increase in Iti. As shown in figure 5, TCDD (10 nM) treatment did induce Iti at the end of conditioning voltage clamp pulses. The average magnitude of TCDD-induced Iti was 36.8±4.8 pA (n=7) in the cells paced at a frequency of 2 Hz for 15 sec.
Figure 5.
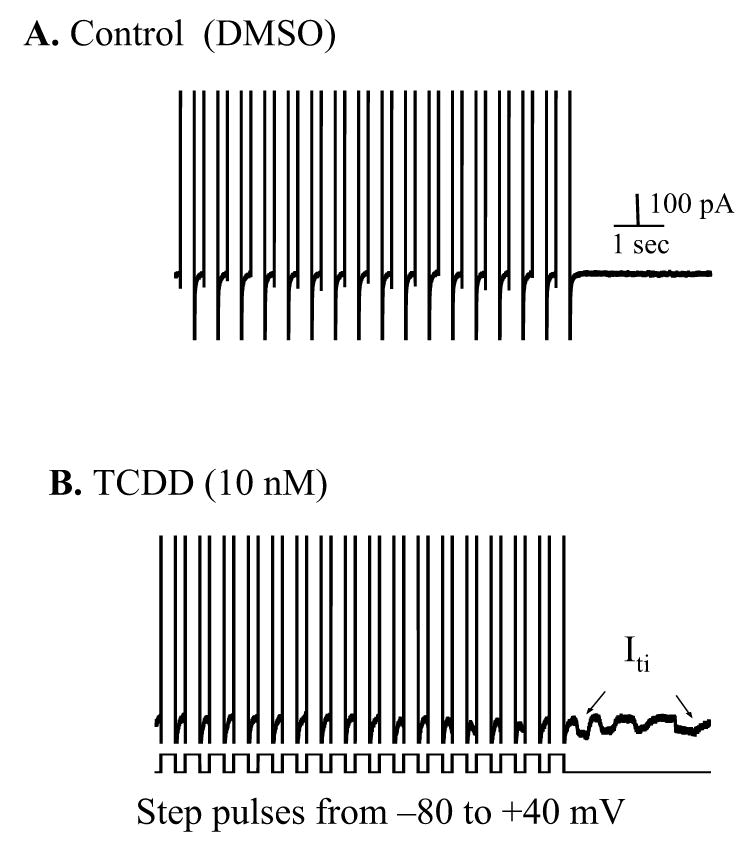
TCDD induced a transient inward current (Iti). The transient inward current (Iti) was recorded at −80 mV holding potential following a 10 sec train of 300 msec depolarizing pulses (from −80 to +40 mV at 2 Hz) in a control (A) and a TCDD-treated (B) cell, respectively. The traces show partial of the step pulses and a 3 sec length of membrane current after ending the conditioning depolarization. Iti was indicated by arrow. Calibration marks apply to both panel A and B.
TCDD augmented ISO- and Bay K 8644-stimulated Iti
When ventricular cells were pre-stimulated to induce Iti by either β-adrengergic agonist ISO or Ca2+ channel opener Bay K 8644, bath application of TCDD (10 nM) further increased the amplitude and the multiplicity of Iti activities (figure 6.). On average, the magnitude of Iti was increased from 37.6±5.2 pA in ISO (or 38.4±2.4 in Bay K) alone to 60.1±10.4 pA in ISO (or 60.8±8.5 in Bay K) plus TCDD. The TCDD-induced net increase was sufficient to depolarize the membrane potential to its excitation threshold and trigger conductible spontaneous action potentials (as shown in figure 3B and 4B), which might form the material basis for arising into ectopic ventricular foci.
Figure 6.
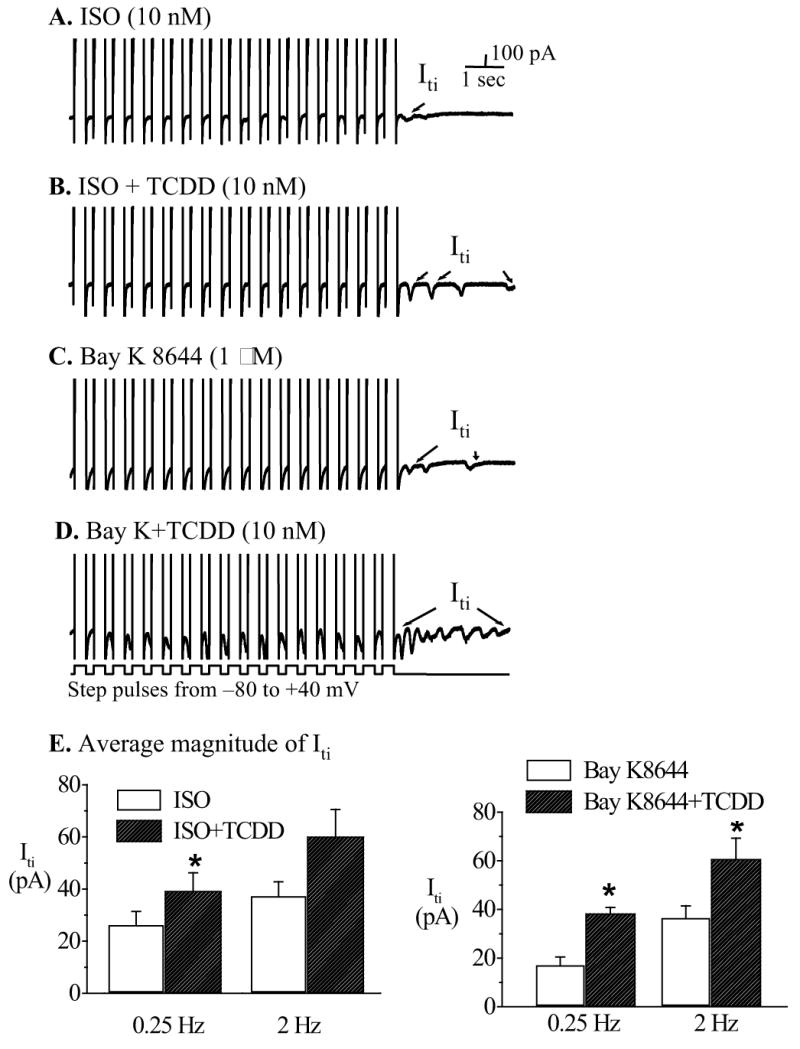
TCDD enhanced ISO and Bay K 8644-induced Iti. Iti was recorded under the same condition as in Figure 5. A. ISO-induced Iti. B. Addition of TCDD further increased the incidence and amplitude of Iti. C. Bay K-induced Iti. D. Effect of TCDD plus Bay K. E. Average amplitude of Iti measured at control and indicated treatments. * indicates statistic significance at p<0.05 level.
TCDD induced EADs in cells pretreated with BAY K 8644 or ISO
The early afterdepolarizations (EADs) occuring during the action potential [33] are thought to be associated with life-threatening ventricular arrhythmias, such as long-QT syndrome and polymorphic ventricular tachycardia (Torsade de Pointes) [37, 38]. The occurrence of early-afterdepolarizations (EADs) is also related to cellular Ca2+ overload [39]. In our study, although EADs were occasionally observed in cells treated with either TCDD, or ISO, or Bay K 8644 alone, application of TCDD (10 nM) in combination with BAY K (1μM) greatly increased the likelihood of triggering EADs in cells paced at slow frequency (0.25 Hz, figure 7A). More often, polymorphic EADs and DADs were concurrently elicited in these cells when challenged with Bay K plus TCDD (figure 7B).
Figure 7.
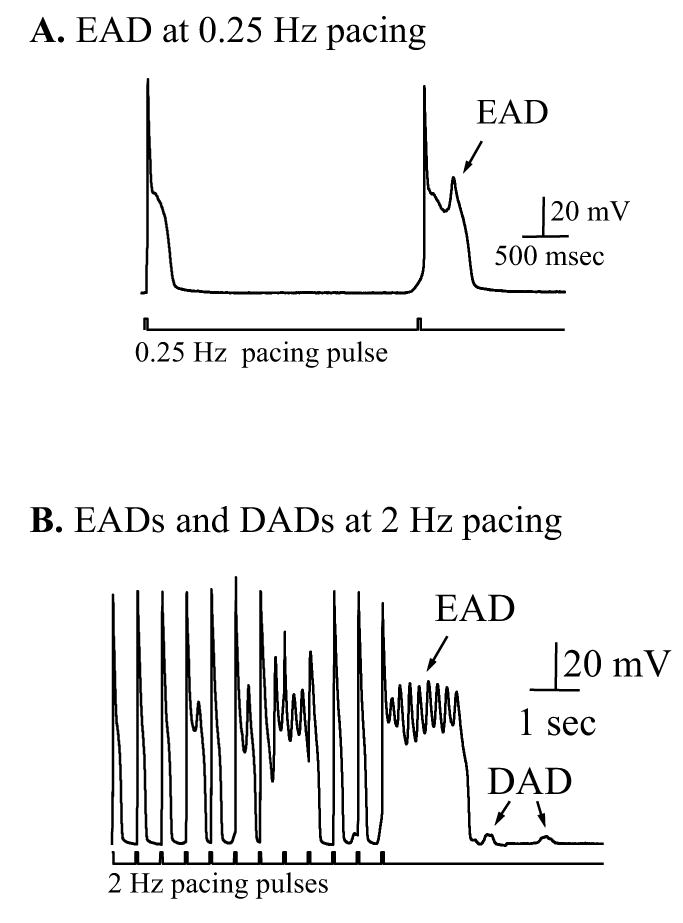
EADs triggered by TCDD in combination with Bay K 8644. A. TCDD triggered EADs in a cell paced by 0.25 Hz current pulses in the presence of 1 μM Bay K 8644. Similar response was observed in a separate experiment. B. Concurrent occurrence of EADs and DADs in a cell treated as in panel A but paced at 2 Hz. The same results were obtained from 3 other cells. TCDD facilitated the firing of EADs as indicated by the arrows.
Effect of TCDD on L-type Ca2+ current
To directly evaluate the role of Ca2+ influx through L-type Ca2+ channels in TCDD-caused intracellular Ca2+ overload, we used voltage-clamp mode to study the alteration of L-type Ca2+ current (ICa,L). The peak inward current elicited by voltage step from −40 mV to 0 mV was significantly increased by TCDD exposure (figure 8A). The stimulated current was sensitive to dihydropyridine Ca2+ channel blocker nifedipine (figure 8A inset). TCDD did neither alter the holding current (at −80 mV or −40 mV) nor the outward potassium currents measured at the end of the 300 msec depolarizing pulses. On average, 10 nM TCDD increased the peak ICa.L by 24.7%, i.e., from 8.1 ± 0.4 of control to 10.1 ± 0.6 pA/pF. (p<0.05, n=8).
Figure 8.
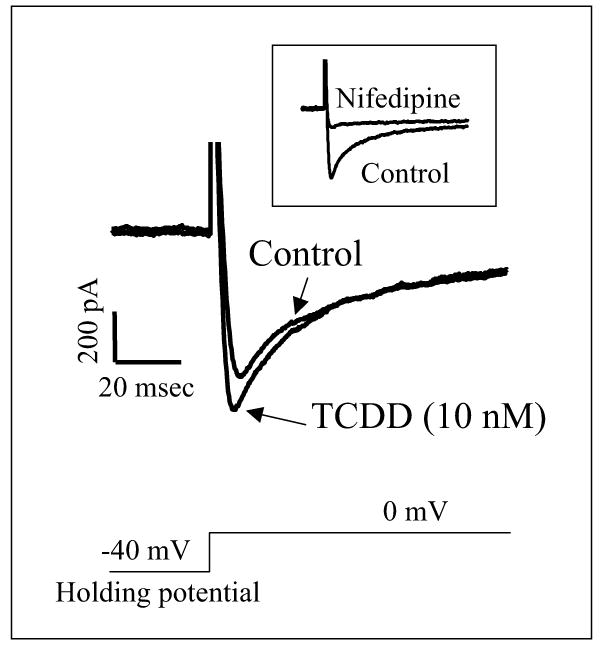
TCDD increase L-type Ca2+ currents (ICa,L). Transmembrane currents recorded before and 20 min after TCDD (10 nM) treatment. Macroscopic currents were elicited by 300-ms depolarizing pulses from −40 (holding potential) to 0 mV. The first 120 msec data were depicted.
Discussion
The results of this study demonstrate that TCDD concentration-dependently prolongs action potential duration (APD), induces afterdepolarizations and increases the transient inward current. Those effects of TCDD are more pronounced in cells challenged with pro-arrhythmogenic stimulations such as high frequency pacing, activation of β-adrenergic receptors, or augmentation of Ca2+ influx directly by Ca2+ channel opener. Although in theory a prolongation of APD will lead to an increase in the effective refractory period, which is anti-arrhythmic, the prolongation of ventricular APD is also found to be pro-arrhythmic, especially when triggered activity was developed from early afterdepolarizations (EADs) or delayed afterdepolarizations (DADs). For example, prolongation of APD can increase the switch speed of the time- and voltage-dependent L-type Ca2+ channels from inactivated to closed states so that facilitate the reopening of these channels and cause EADs arising near the plateau phase of action potentials [32]. Considering that afterdepolarizations play a pivotal role in the initiation of ventricular tachycardia, ventricular fibrillation, and Torsade de pointes [38, 40–42], the current findings suggest that TCDD exposure may result in clinically significant cardiac arrhythmia, especially in those patients with conditions such as pre-deposited myocardial Ca2+ overload.
Usually, arrhythmogenic DADs occurs at high heart rates or are provoked by overdriven pacing pulses under experimental condition [43, 44]. Under our perforated-recording conditions, we found that using a pacing frequency greater than 2 Hz could induce DADs in many cells, whereas when the pacing rate was ≤ 2 Hz, the chance of triggering DADs was greatly reduced. Therefore, to minimize basal DAD activities, we chose to use 2 Hz pacing rate to study the impact of TCDD on the triggered activities. The provoking DADs by a relatively slow pacing rate (from 0.25 to 2 Hz) in our experiments may be explained by two factors. First, our recordings were carried out under nystatin-perforated configuration that markedly reduced the variability of APD between cells. Indeed, we observed that the APD90 from perforated recordings was significantly longer than that from conventional recordings. Second, we lowered the external Mg2+ concentration (0.53 mM) to facilitate the occurrence of DADs and EADs [45].
The extra Ca2+ influx through L-type Ca2+ channels at the prolonged plateau phase can induce more Ca2+ releasing from SR stores [46, 47] and lead to intracellular Ca2+ overload, a condition known to enhance the triggering of DADs [34, 43]. The transient inward current (Iti) forms the ionic basis for generation of DADs [48]. Iti is a heterogeneous transmembrane current associated with 1) spontaneous Ca2+-release from the sarcoplasmic reticulum (SR) [43], 2) ions flowing through nonselective cation channels [49], 3) Na+/Ca2+-exchange current [50, 51], or 4) a Ca2+-activated Cl− current [52]. Canga et. al. reported that TCDD-treatment produced an intracellular Ca2+-overload in ventricular myocytes [17]. By demonstrating that TCDD triggers arrhythmogenic DADs and enhances Iti, we document a novel cardiac toxicity of the environmental pollutant, i.e., acute TCDD exposure may trigger ventricular arrhythmia. The ionic mechanism for TCDD-caused increase of Iti needs to be studied. The magnitude of TCDD-induced Iti could be critical of triggering arrhythmic activity in ventricular myocytes. The size of the Iti associated with TCDD exposure is sufficient to cause approximately 3–5 mV depolarization on the membrane potential, for the passive membrane resistance in ventricular myocytes was measured approximately 80~120 MΩ under the perforated recording conditions. The depolarizing fluctuation in the membrane potential may produce an ectopic excitation focus in the ventricular myocardium and cause ventricular premature contraction or more severe arrhythmic symptoms in susceptible individuals.
By activation of G-protein coupled β–adrenergic receptors, ISO can stimulate adenylyl cyclase activity and increase the level of the second messenger cAMP that activates protein kinase A. One major outcome of this well-defined signaling pathway in ventricular myocytes is to increase Ca2+ influx through phosphorylation-activated L-type Ca2+ channels [53]. The combination of pacing with ISO-stimulation is an established experimental protocol used to produce Ca2+ overload and trigger DADs, which mimics disruption of Ca2+ homeostasis under pathological conditions such as tachycardia or heart failure [36, 42]. Assessing the action of TCDD on DADs and Iti in the presence of ISO-stimulation enables us to evaluate the potential influence of the environmental toxin on the stressed myocytes. The additive effects of TCDD and ISO on DADs and Iti result in augmented "oscillations" of membrane potential, therefore facilitated the development of conductible arrhythmic pulses (figure 5B), which provides direct evidence that TCDD is a pro-arrhythmogenic environmental pollutant.
Activation of β–adrenergic receptor and PKA pathway is also associated with other cellular mechanisms such as cAMP-gated ion channel activity that may shorten action potential duration of ventricular myocytes, therefore, conceal the possibility of TCDD-induced EADs [54, 55]. In order to assess the role of activation of L-type Ca2+ channel alone in triggering afterdepolarizations and Iti, we used dihydropyridine analogue Bay K 8644 to keep the L-type Ca2+ channels under an activated state [56], and evaluated the effects of TCDD on DADs and Iti as well as EADs in the absence of cAMP-induced other ion currents. The results shown in figures 5 and 6 demonstrate that TCDD enhances Bay K-induced DADs and Iti. More interestingly, addition of TCDD promotes Bay K-induced prolongation of APD into EADs (figure 7) that may result in a dispersion of repolarization (a condition that provides a substrate to initiate a life-threatening polymorphic ventricular tachycardia known as Torsade de Pointes). This finding may have important implication to individuals who suffer from heart failure or someone who is under therapy with drugs such as phenothiazine antipsychotics that prolong QT-interval. As we know, in heart failure patients, Ca2+ overload and prolongation of APD are the characteristic alteration of ventricular cells [57, 58], which may increase the susceptibility of those patients to dioxin exposure in terms of triggering EADs and Torsade de Pointes.
Although it was reported that reported that dioxin could stimulates Ca2+ uptake in rat hippocampal neurons within 40 sec of exposure [59], we did not observe such rapid response in ventricular myocytes. In addition, the increase of ICa,L itself directly contributes to intracellular Ca2+ overload (figure 8), however, at this point, we do not know the significance of the mildly stimulated ICa,L by TCDD in its pro-arrhythmogenic impact on cardiomyocytes. We expect to have a clear picture on this issue after defining the action of TCDD on Ca2+-release from sarcoplasmic reticulum (SR) and on Na+/Ca2+-exchange current.
Finally, animal models contribute much to our understanding of the cardiac toxicity of TCDD. The patch-clamp technique is a highly sensitive and state-of-the-art in studying the potential impacts of environmental pollutant to the heart. The experimental conditions used in this study, however, may not reflect the actual human responses to dioxin exposure. Nevertheless, the findings of this study provide strong evidence that acute exposure to TCDD dioxin may induce arrhythmic cardiotoxicity.
Footnotes
The work was supported by the TIP grant (1K22ES000367-01) from the National Institute of Environmental Health Sciences (NIH). N.J.W. is supported by the Intramural Research Program of the NIEHS, NIH.
References
- 1.Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- 3.Gilday D, Gannon M, Yutzey K, Bader D, Rifkind AB. Molecular cloning and expression of two novel avian cytochrome P450 1A enzymes induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem. 1996;271(51):33054–33059. doi: 10.1074/jbc.271.51.33054. [DOI] [PubMed] [Google Scholar]
- 4.Cole P, Trichopoulus D, Pastides H, Starr T, Mandel J. Dioxin and cancer: a critical review. Regul Toxicol Pharmacol. 2003;38:378–388. doi: 10.1016/j.yrtph.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Schecter A, Birnbaum L, Ryan JJ, Constable JD. Dioxins: An overview. Environ Res. 2006 Jan 27; doi: 10.1016/j.envres.2005.12.003. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 6.Grassman JA, Masten SA, Walker NJ, Lucier GW. Animal models of human response to dioxins. Environ Health Perspect. 1998;106(Suppl 2):761–775. doi: 10.1289/ehp.98106761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Safe SH. Modulation of gene expression and endocrine response pathways by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Pharmacol Ther. 1995;67(2):247–281. doi: 10.1016/0163-7258(95)00017-b. [DOI] [PubMed] [Google Scholar]
- 8.Long WP, Perdew GH. Lack of an absolute requirement for the native aryl hydrocarbon receptor (AhR) and AhR nuclear translocator transactivation domains in protein kinase C-mediated modulation of the AhR pathway. Arch Biochem Biophys. 1999;371(2):246–259. doi: 10.1006/abbi.1999.1452. [DOI] [PubMed] [Google Scholar]
- 9.Kim SY, Yang JH. Neurotoxic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin in cerebellar granule cells. Exp Mol Med. 2005;37(1):58–64. doi: 10.1038/emm.2005.8. [DOI] [PubMed] [Google Scholar]
- 10.Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol [B] 2005;175(4):221–230. doi: 10.1007/s00360-005-0483-3. [DOI] [PubMed] [Google Scholar]
- 11.Gannon M, Gilday D, Rifkind AB. TCDD induces CYP1A4 and CYP1A5 in chick liver and kidney and only CYP1A4, an enzyme lacking arachidonic acid epoxygenase activity, in myocardium and vascular endothelium. Toxicol Appl Pharmacol. 2000;164(1):24–37. doi: 10.1006/taap.1999.8864. [DOI] [PubMed] [Google Scholar]
- 12.Abbott BD, Probst MR. Developmental expression of two members of a new class of transcription factors: II. Expression of aryl hydrocarbon receptor nuclear translocator in the C57BL/6N mouse embryo. Dev Dyn. 1995;204(2):144–155. doi: 10.1002/aja.1002040205. [DOI] [PubMed] [Google Scholar]
- 13.Matsumura F. How important is the protein phosphorylation pathway in the toxic expression of dioxin-type chemicals? Biochem Pharmacol. 1994;48(2):215–224. doi: 10.1016/0006-2952(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 14.Bombick DW, Jankun J, Tullis K, Matsumura F. 2,3,7,8-Tetrachlorodibenzo-p-dioxin causes increases in expression of c-erb-A and levels of protein-tyrosine kinases in selected tissues of responsive mouse strains. Proc Natl Acad Sci U S A. 1988;85(12):4128–4132. doi: 10.1073/pnas.85.12.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DePetrillo PB, Kurl RN. Stimulation of protein kinase C by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rat thymocytes. Toxicol Lett. 1993;69(1):31–36. doi: 10.1016/0378-4274(93)90142-k. [DOI] [PubMed] [Google Scholar]
- 16.Kelling CK, Menahan LA, Peterson RE. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment on mechanical function of the rat heart. Toxicol Appl Pharmacol. 1987;91(3):497–501. doi: 10.1016/0041-008x(87)90072-x. [DOI] [PubMed] [Google Scholar]
- 17.Canga L, Paroli L, Blanck TJ, Silver RB, Rifkind AB. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases cardiac myocyte intracellular calcium and progressively impairs ventricular contractile responses to isoproterenol and to calcium in chick embryo hearts. Mol Pharmacol. 1993;44(6):1142–1151. [PubMed] [Google Scholar]
- 18.Canga L, Levi R, Rifkind AB. Heart as a target organ in 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity: decreased beta-adrenergic responsiveness and evidence of increased intracellular calcium. Proc Natl Acad Sci U S A. 1988;85(3):905–909. doi: 10.1073/pnas.85.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan L, Ovadia M, Friedman DM, Rifkind AB. Ventricular preexcitation sensitive to flecainide in late stage chick embryo ECGs: 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs inotropic but not chronotropic or dromotropic responses to isoproterenol and confers resistance to flecainide. Toxicol Appl Pharmacol. 2000;166(1):43–50. doi: 10.1006/taap.2000.8948. [DOI] [PubMed] [Google Scholar]
- 20.Fukunaga BN, Probst MR, Reisz-Porszasz S, Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J Biol Chem. 1995;270(49):29270–29278. doi: 10.1074/jbc.270.49.29270. [DOI] [PubMed] [Google Scholar]
- 21.Jokinen MP, Walker NJ, Brix AE, Sells DM, Haseman JK, Nyska A. Increase in cardiovascular pathology in female Sprague-Dawley rats following chronic treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin and 3,3′,4,4′,5-pentachlorobiphenyl. Cardiovasc Toxicol. 2003;3(4):299–310. doi: 10.1385/CT:3:4:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivnitski-Steele ID, Sanchez A, Walker MK. 2,3,7,8-tetrachlorodibenzo-p-dioxin reduces myocardial hypoxia and vascular endothelial growth factor expression during chick embryo development. Birth Defects Res A Clin Mol Teratol. 2004;70(2):51–58. doi: 10.1002/bdra.10151. [DOI] [PubMed] [Google Scholar]
- 23.Calvert GM, Wall DK, Sweeney MH, Fingerhut MA. Evaluation of cardiovascular outcomes among U.S. workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Environ Health Perspect. 1998;106(Suppl 2):635–643. doi: 10.1289/ehp.98106635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steenland K, Piacitelli L, Deddens J, Fingerhut M, Chang LI. Cancer, heart disease, and diabetes in workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Natl Cancer Inst. 1999;91(9):779–786. doi: 10.1093/jnci/91.9.779. [DOI] [PubMed] [Google Scholar]
- 25.Pesatori AC, Zocchetti C, Guercilena S, Consonni D, Turrini D, Bertazzi PA. Dioxin exposure and non-malignant health effects: a mortality study. Occup Environ Med. 1998;55(2):126–131. doi: 10.1136/oem.55.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dalton TP, Kerzee JK, Wang B, Miller M, Dieter MZ, Lorenz JN, Shertzer HG, Nerbert DW, Puga A. Dioxin exposure is an environmental risk factor for ischemic heart disease. Cardiovasc Toxicol. 2001;1(4):285–298. doi: 10.1385/ct:1:4:285. [DOI] [PubMed] [Google Scholar]
- 27.Firestone D. Etiology of chick edema disease. Environ Health Perspect. 1973;5:59–66. doi: 10.1289/ehp.730559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kociba RJ, Keyes DG, Beyer JE, Carreon RM, Wade CE, Dittenber DA, Kalnins RP, Frauson LE, Park CN, Barnard SD, Hummel RA, Humiston CG. Results of a two-year chronic toxicity and oncogenicity study of 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats. Toxicol Appl Pharmacol. 1978;46(2):279–303. doi: 10.1016/0041-008x(78)90075-3. [DOI] [PubMed] [Google Scholar]
- 29.Walker MK, Pollenz RS, Smith SM. Expression of the aryl hydrocarbon receptor (AhR) and AhR nuclear translocator during chick cardiogenesis is consistent with 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced heart defects. Toxicol Appl Pharmacol. 1997;143(2):407–419. doi: 10.1006/taap.1996.8068. [DOI] [PubMed] [Google Scholar]
- 30.Hermansky SJ, Holcslaw TL, Murray WJ, Markin RS, Stohs SJ. Biochemical and functional effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the heart of female rats. Toxicol Appl Pharmacol. 1988;95(2):175–184. doi: 10.1016/0041-008x(88)90154-8. [DOI] [PubMed] [Google Scholar]
- 31.Brewster DW, Matsumura F, Akera T. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on guinea pig heart muscle. Toxicol Appl Pharmacol. 1987;89(3):408–417. doi: 10.1016/0041-008x(87)90160-8. [DOI] [PubMed] [Google Scholar]
- 32.January CT, Moscucci A. Cellular mechanisms of early afterdepolarizations. Ann N Y Acad Sci. 1992;644:23–32. doi: 10.1111/j.1749-6632.1992.tb30999.x. [DOI] [PubMed] [Google Scholar]
- 33.Cranefield PF. Action potentials, afterpotentials, and arrhythmias. Circ Res. 1977;41:415–423. doi: 10.1161/01.res.41.4.415. [DOI] [PubMed] [Google Scholar]
- 34.January C, Fozzard HA. Delayed afterdepolarizations in heart muscle: mechanisms and relevance. Pharmacol Rev. 1988;40(3):219–227. [PubMed] [Google Scholar]
- 35.Fozzard HA. After depolarizations and triggered activity. Basic Res Cardiol. 1992;87(Suppl 2):105–113. doi: 10.1007/978-3-642-72477-0_10. [DOI] [PubMed] [Google Scholar]
- 36.Song Y, Belardinelli L. ATP promotes development of afterdepolarizations and triggered activity in cardiac myocytes. Am J Physiol. 1994;267(5 Pt 2):H2005–2011. doi: 10.1152/ajpheart.1994.267.5.H2005. [DOI] [PubMed] [Google Scholar]
- 37.Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels AP, Wellens HJ, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res. 2000;46(3):376–392. doi: 10.1016/s0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- 38.Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J Physiol. 2002;543(Pt 2):615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volders PG, Kulcsar A, Vos MA, Sipido KR, Wellens HJ, Lazzara R, Szabo B. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res. 1997;34(2):348–359. doi: 10.1016/s0008-6363(96)00270-2. [DOI] [PubMed] [Google Scholar]
- 40.Dessertenne F. Ventricular tachycardia with 2 variable opposing foci. Arch Mal Coeur Vaiss. 1966;59(2):263–272. [PubMed] [Google Scholar]
- 41.Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 1998;98(22):2404–2414. doi: 10.1161/01.cir.98.22.2404. [DOI] [PubMed] [Google Scholar]
- 42.Verkerk AO, Veldkamp MW, Baartscheer A, Schumacher CA, Klopping C, van Ginneken AC, Ravesloot JH. Ionic mechanism of delayed afterdepolarizations in ventricular cells isolated from human end-stage failing hearts. Circulation. 2001;104(22):2728–2733. doi: 10.1161/hc4701.099577. [DOI] [PubMed] [Google Scholar]
- 43.Wit AL, Rosen MR. Afterdepolarizations and triggered activity: distinction from automaticity as an arrhythmogenic mechanism. In: Fozzard HA, Haber E, Jennings RB, et al., editors. The Heart and Cardiovascular System. 2. New York, NY: Raven Press; 1992. pp. 2113–2163. [Google Scholar]
- 44.Tatsukawa Y, Arita M, Kiyosue T, Mikuriya Y, Nasu M. A comparative study of effects of isoproterenol and dihydroouabain on calcium transients and contraction in cultured rat ventricular cells. J Mol Cell Cardiol. 1993;25(6):707–720. doi: 10.1006/jmcc.1993.1083. [DOI] [PubMed] [Google Scholar]
- 45.Aomine M, Tatsukawa Y, Yamato T, Yamasaki S. Antiarrhythmic effects of magnesium on rat papillary muscle and guinea pig ventricular myocytes. Gen Pharmacol. 1999;32(1):107–114. doi: 10.1016/s0306-3623(98)00094-9. [DOI] [PubMed] [Google Scholar]
- 46.Sah R, Ramirez RJ, Kaprielian R, Backx PH. Alterations in action potential profile enhance excitation-contraction coupling in rat cardiac myocytes. J Physiol. 2001;533(Pt 1):201–214. doi: 10.1111/j.1469-7793.2001.0201b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Isenberg G, Han S. Gradation of Ca2+-induced Ca2+ release by voltage-clamp pulse duration in potentiated guinea-pig ventricular myocytes. J Physiol. 1994;480(Pt 3):423–438. doi: 10.1113/jphysiol.1994.sp020372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lederer WJ, Tsien RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J Physiol. 1976;263(2):73–100. doi: 10.1113/jphysiol.1976.sp011622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kass RS, Tsien RW, Weingart R. Ionic basis of transient inward current induced by strophanthidin in cardiac Purkinje fibres. J Physiol. 1978;281:209–226. doi: 10.1113/jphysiol.1978.sp012417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bers DM, Ziolo MT. When is cAMP not cAMP? Effects of compartmentalization. Circ Res. 2001;89(5):373–375. [PubMed] [Google Scholar]
- 51.Karagueuzian HS, Chen PS. Cellular mechanism of reentry induced by a strong electrical stimulus: implications for fibrillation and defibrillation. Cardiovasc Res. 2001;50(2):251–262. doi: 10.1016/s0008-6363(00)00298-4. [DOI] [PubMed] [Google Scholar]
- 52.Zygmunt AC. Intracellular calcium activates a chloride current in canine ventricular myocytes. Am J Physiol. 1994;267(5 Pt 2):H1984–1995. doi: 10.1152/ajpheart.1994.267.5.H1984. [DOI] [PubMed] [Google Scholar]
- 53.Tiaho F, Piot C, Nargeot J, Richard S. Regulation of the frequency-dependent facilitation of L-type Ca2+ currents in rat ventricular myocytes. J Physiol. 1994;477(Pt 2):237–251. doi: 10.1113/jphysiol.1994.sp020187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bennett PB, Begenisich TB. Catecholamines modulate the delayed rectifying potassium current (IK) in guinea pig ventricular myocytes. Pflugers Arch. 1987 Sep;410(1–2):217–219. doi: 10.1007/BF00581919. [DOI] [PubMed] [Google Scholar]
- 55.Harvey RD, Clark CD, Hume JR. Chloride current in mammalian cardiac myocytes. Novel mechanism for autonomic regulation of action potential duration and resting membrane potential. J Gen Physiol. 1990 Jun;95(6):1077–1102. doi: 10.1085/jgp.95.6.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schramm M, Thomas G, Towart R, Franckowiak G. Novel dihydropyridines with positive inotropic action through activation of Ca2+ channels. Nature. 1983;303(5917):535–537. doi: 10.1038/303535a0. [DOI] [PubMed] [Google Scholar]
- 57.Swynghedauw B. Remodeling of the heart in chronic pressure overload. Basic Res Cardiol. 1991;86(Suppl 1):99–105. [PubMed] [Google Scholar]
- 58.Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. [Na+]i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. 2003;57(4):986–995. doi: 10.1016/s0008-6363(02)00848-9. [DOI] [PubMed] [Google Scholar]
- 59.Hanneman WH, Legare ME, Barhoumi R, Burghardt RC, Safe S, Tiffany-Castiglioni E. Stimulation of calcium uptake in cultured rat hippocampal neurons by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology. 1996;112(1):19–28. doi: 10.1016/0300-483x(96)03346-x. [DOI] [PubMed] [Google Scholar]