

Abstract
To evaluate the effect of genetic background on oxygen (O2) toxicity, nine genetically diverse mouse strains (129/SvIm, A/J, BALB/cJ, BTBR+(T)/tf/tf, CAST/Ei, C3H/HeJ, C57BL/6J, DBA/2J, and FVB/NJ) were exposed to >99% O2 for 72 hours. Immediately following the hyperoxic challenge, the mouse strains demonstrated distinct pathophysiologic responses. The BALB/cJ and CAST/Ei strains, which were the only strains to demonstrate mortality from the hyperoxic challenges, were also the only strains to display significant neutrophil infiltration into their lower respiratory tract. In addition, the O2-challenged BALB/cJ and CAST/Ei mice were among six strains (A/J, BALB/cJ, CAST/Ei, BTBR+(T)/tf/tf, DBA/2J and C3H/HeJ) that had significantly increased IL-6 concentrations in the whole lung lavage fluid and were among all but one strain that had large increases in lung permeability compared to air-exposed controls. By contrast, the DBA/2J strain was the only strain not to have any significant alterations in lung permeability following hyperoxic challenge. The expression of the extracellular matrix (ECM) proteins, including collagens I, III and IV, fibronectin-I, and tenascin-C, also varied markedly among the mouse strains, as did the activities of total superoxide dismutase (SOD) and manganese-SOD (Mn-SOD or SOD2). These data suggest that the response to O2 depends, in part, on the genetic background and that some of the strains analyzed could be used to identify specific loci and genes underlying the response to O2.
Keywords: Hyperoxia, oxygen toxicity, inflammation, superoxide dismutase, extracellular matrix
Introduction
Exposure to high concentrations of O2 is toxic for the lung and is often accompanied by the onset of acute lung injury (ALI). This injury is thought to be initiated by the local generation of reactive oxygen species (ROS), and as it evolves, pulmonary structural damage becomes extensive, accompanied by airway inflammation, alveolar and interstitial edema, and reduced lung function (Kapanci and Weibel, 1969; Crapo, 1986). The components of the inflammatory response include increased levels of pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1beta and IL-6 (Johnston et al. 1997), and the recruitment of neutrophils to the distal airspaces (Johnston et al. 1997; Keeney et al. 1995; Fracica et al. 1991; Bureau et al. 1985). The roles of neutrophils in hyperoxic lung injury remain unclear, but it has been suggested that the neutrophils in the airways release oxidants that contribute to tissue damage (Keeney et al. 1995).
As hyperoxic lung injury progresses, damage to the ultrastructure and extracellular matrix (ECM) of the lung occurs, which further impairs the physiological and biological function of the lung (Durr et al. 1987). For instance, endothelial injury and cell death are major features in the pathogenesis of oxidant injury (Adamson et al. 1970, 1990; Maniscalco et al. 1995). In addition, the matrix is degraded and basement membranes, which serve as structural support for cells, are denuded. In attempts to restore the lung to homeostasis following hyperoxia, the lung begins a process of repair, with recruitment and proliferation of alveolar type II pneumocytes and interstitial fibroblasts at the sites of injury (Crapo et al. 1978, 1980; Thet et al. 1983). Additionally, there is increased synthesis and secretion of various types of extracellular components into the lung parenchyma. Among them, collagen, fibronectin, and tenascin contribute to the repair of hyperoxic lung injury.
The human lung has developed several lines of defense against O2-enriched environments. The endogenous antioxidant enzymes detoxify ROS including superoxide anion (O2−) and hydrogen peroxide (H2O2), which form, for instance, during respiration. This prevents or limits the formation of other strong oxidants such as hydroxyl radical (•OH) and peroxynitrite anion (ONOO−). Therefore, hyperoxic injury can be directly related to the generation of ROS and subsequent oxidation of macromolecules including proteins, lipids and nucleic acids. One of the most important protective antioxidant enzymes in the distal airways and alveolar region is superoxide dismutase (SOD), of which there are three isoforms; copper/zinc SOD (SOD1), manganese SOD (SOD2), and extracellular SOD (SOD3). SOD2 has been shown to be protective against hyperoxia-induced lung injury (Wispe et al. 1992).
Among humans, the severity of injury in the development of hyperoxic lung injury is multifactorial. Important variables in individual susceptibility to O2 injury include the patient’s health, age and gender (Denke and Fanburg; 1980; Keeney et al. 1995; Capellier et al. 1999). Genetic factors are also thought to play a fundamental role. For instance, there is a strong genetic predisposition to the harmful effects of O2 toxicity in infants (Clark and Clark, 2005; Nickerson and Taussig, 1980; Hallman and Haataja; 2003). In addition, mice and rats also vary considerably in their susceptibility. C3H/HeJ mice are more sensitive to oxidant injury than C57BL/6J mice (Cho et al. 2002; Hudak et al. 1992), and Fisher 344 rats are more sensitive than Sprague-Dawley rats (He et al. 1990; Stenzel et al. 1992). Although studies such as these have established that genetic factors play a role, further refinement of the O2 response lung phenotype in multiple, genetically diverse inbred mouse strains is needed to inform and direct further efforts to identify O2 response genes.
In the present investigation, we characterized physiological and biological responses of nine genetically diverse inbred strains (129/SvIm, A/J, BALB/cJ, BTBR+(T)/tf/tf, C3H/HeJ, C57BL/6J, CAST/Ei, DBA/2J, FVB/NJ) following an acute exposure to hyperoxia conditions. Further, we also evaluated changes in lung structure and ECM and the total and SOD2 activity as indicators of lung injury. Our results suggest that the response to O2 depends, in part, on the genetic background and that some of the strains analyzed might be used to identify specific loci and genes underlying the response to O2.
Materials and Methods
Animals
Twenty-four male mice (6–8 weeks) from 7 strains (129/SvIm, A/J, BTBR+(T)/tf/tf, C3H/HeJ, C57BL/6J, DBA/2J, and FVB/NJ) and 48 mice (age-matched) from 2 strains (BALB/cJ and CAST/Ei) were obtained commercially (Jackson laboratories). The animals were housed in shoebox type cages under pathogen-free conditions prior to the challenges and water and mouse-chow were provided ad libitum. The study protocol was in accordance with guidelines set forth by the Duke University Animal Care and Use committee.
Exposures
Twenty-four mice from each strain were randomly divided into two groups: Hyperoxic- (n=12) and Control-group (n=12). The hyperoxic-mice of each strain were exposed continuously to >99% O2 (flow rate of 10 L/min; CO2<0.1%; temp=25–26°C; and humidity<40%) in polystyrene chambers for 72 hours as described previously (Que et al. 1998; Folz et al. 1999), except for 10 min/day when the cages were opened to room air for general maintenance. The air-exposed control mice from each strain were housed in the same chambers and exposed to room air under identical flow, temperature, and humidity conditions for the same time period. The concentration of O2 in the chambers was monitored continuously with a Servomex O2 analyzer (model 572; Sybron, Norwood, MA, USA). The mice were allowed ad libitum access to water and mouse chow throughout the exposures.
Immediately following the exposures, the mice were euthanized by intraperitoneal injections of sodium pentobarbital (150 mg/kg). Six hyperoxic and control mice from each strain were randomly selected for assessment of inflammation in the airways and expression of ECM genes, and the remaining six mice from each strain were used for wet/dry ratios and analysis of total SOD and SOD2 activity.
Whole-lung lavage and cell preparation
Whole-lung lavage was performed immediately following euthanasia in the mice that were selected for assessment of inflammation in the airways and gene expression in the lungs. Briefly, the chest of each animal was opened and the trachea was exposed and intubated via PE-90 tubing (0.86mm I.D., 1.27mm O.D.). Sterile pyrogen-free saline was infused into the lungs to total lung capacity (TLC), which was defined at a pressure of 25 cmH2O, and then collected. A total of 6-mls of saline was infused into the lungs and the total volume of retrieved fluid was recorded. The cells were then isolated from the whole-lung lavage fluid by centrifugation at 5,000 X g for 10 min. The supernatant was stored at −80°C for later assessment of total protein and cytokine (TNF-α, IL-1β, and IL-6) levels. The cells were resuspended in 1.0 ml sterile Hank’s balanced salt solution and counted using a hemacytometer. A 100 μl sample of the suspension from each animal was also centrifuged (model Cytospin-3, Shandon Inc., Pittsburgh, PA) to extract cells onto cytoslide (Shandon Inc., Pittsburgh, PA) preparations. The cells were then stained with hematoxylin and eosin (Hema 3, Wright-Giemsa stain, Biochemical Sciences Inc., Swedesboro, NJ). Differential cell counts of pulmonary leukocytes (alveolar macrophages, neutrophils, lymphocytes, and eosinophils) were determined using standard morphological criteria, and then the number and percentage of neutrophils per milliliter of lavage fluid was calculated.
Cytokine Analysis
Concentrations of IL-6 in the whole-lung lavage fluid were measured using a commercial enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN) following the manufacture’s instructions. The minimum detectable dose for the IL-6 ELISA kit was 1.6 pg/ml.
Total Protein
Total protein in the whole-lung lavage fluid was measured using a detergent compatible (DC) protein assay kit (Bio-Rad Laboratories, Hercules, CA), similar to methods as described previously (Lowry et al. 1951). The assay kit was employed according to the manufacturer’s protocol. The minimum detectable dose was 5 μg/ml.
Lung RNA Preparation
Following the whole-lung lavage, leukocytes and red blood cells were cleared from the vasculature of both lungs. A catheter was inserted into the right ventricle to the pulmonary artery and isotonic saline was infused through the pulmonary blood vessels at 25 cm H2O. Approximately 5.0 ml of saline was perfused through the heart to clear the lungs. The right and left lung lobes were then excised and flash frozen in liquid N2. Total RNA was isolated from individual O2-exposed or control mouse lungs by homogenization in 1.4 ml TRIzol Reagent (Invirogen Corporation, Carlsbed, CA) using a tissue homogenizer (Biospec Products, Racine, WI). Homogenates were then centrifuged at 15,000 x g for 10 minutes. 300 μl of chloroform was added to the cleared homogenates and the samples were centrifuged again for another 15 minutes. 700 μl of isopropyl alcohol was added to the upper aqueous phase containing the RNA, and then the RNA was collected by centrifugation at 15,000 x g for 20 minutes. The RNA pellets were washed with 75% ethyl alcohol and resuspended in DEPC-treated water at a concentration of 1.0 μg/μl. The RNA was stored at −80°C to be used later for quantitative assessment of mRNA expression.
Real-Time (RT) Quantitative PCR for mRNA expression
Total RNA (50–500 ng) was reversed-transcribed into cDNA in a volume of 25 μl, containing 12.5 μl 2X SYBR Green buffer (Applied Biosystems), 12.5 U reverse transcriptase, 20 U RNase inhibitor, and 0.5 μM oligo (dT) primers (Table 1). The primers were specific for mouse procollagen (Col1α2, Col3α1, Col4α3), FN-1, and ten-C, which are key components of the ECM. 18s rRNA and TFIID primers were used as an internal control. Each primer set spanned two intron/exon boundaries and had melting temperatures (Tm) between 58 and 60°C, which each Tm of the corresponding primer sets not varying more than 1°C. The RT reaction was carried out in 1 cycle at 50°C for 30 min and 95°C at 10 min. The PCR reaction was carried out by a two-step amplification cycle (40 cycles): denaturizing at 95°C for 15 sec and annealing/extending at 60–62°C (Table 1) for 2 min. A dissociation curve was carried out in three steps for 1 cycle as follows: 95°C for 15 sec, 58°C for 20 sec, and 95°C for 15 sec. Each sample was run in triplicate for each primer set and the mean value was normalized to the internal controls.
Table 1.
Primer sequences for RT-PCR
| cDNAs | Primer Sequences | Amount RNA (ng) | Annealing Temp | |
|---|---|---|---|---|
| Collagen I, alpha 2
(Col1a2) |
Forward
Reverse |
AACACTGGTAGAGATGGTGCTCGT
CACCACGTTCACCAGGGCTA |
200 | 60 |
| Collagen III, alpha 1
(Col3a1) |
Forward
Reverse |
CCCTGGTCCACAAGGATTACA
CTCCAGGTGCACCAGAATCA |
50 | 60 |
| Collagen IV, alpha 3
(Col4a3) |
Forward
Reverse |
ATGTTCGCAATGACTGGGAAA
CTCTGAGAAGCGGGCCATACT |
50 | 60 |
| Fibronectin 1
(Fn1) |
Forward
Reverse |
AGACAATGCCGTGGTCCTAACA
GAGTTGGCGGTGATATCAGAAGA |
50 | 62 |
| Tenascin C
(Tnc) |
Forward
Reverse |
TAGATGTTCCAAAGAGCCAGCAA
CGTAAGTCCTTGGGTGCATCA |
50 | 62 |
| 18s rRNA | Forward
Reverse |
CGGCTACCACATCCAAGGAA
GCTGGAATTACCGCGGCT |
50 | 58–62 |
| TFIID | Forward
Reverse |
ACGGACAACTGCGTTGATTTT
ACTTAGCTGGGAAGCCCAAC |
50 | 58–62 |
Wet to Dry Weight Ratio
After euthanasia of the hyperoxic- and control-mice that were selected for wet-to-dry ratios and the activity of total SOD and SOD2, the chest of each animal was opened and the right main stem bronchus was clamped. The right lung lobes were dissected from the bronchus, blotted dry, and weighed. The lungs were then dried at 60°C until the weight was stable. The dried weights of the lungs were measured and the wet-to-dry weight ratios of the lungs were calculated. The wet-to-dry ratio was used as a marker of pulmonary edema.
The Activity of Total Superoxide Dismutase and SOD2
The left lung lobes were homogenized in 1 mL buffer containing 50 mM potassium phosphate (pH 7.4) with 0.3 M KBR, and antiproteolytic agents including 0.5 mM phenyl methylsulfonyl fluoride, 3mM diethylenetriaminepentaacetic acid, 0.09 mg aprotinin, 0.01 mg pepstatin, 0.01 mg chymostatin, and 0.01 mg leupeptin. The homogenates were centrifuged at 15,000 x g for 20 minutes at 4°C.
Total SOD activity was measured for the left lobes (U/left lobes) using a commercial SOD assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s protocol. The detection limit of the kit ranged from 0.025–0.25 units/ml SOD. In a separate assay, the lung homogenates were centrifuged at 15,000 x g for 20 minutes at 4°C to precipitate the mitochondria and separate Mn-SOD from the other two isoforms (Mattiazz et al. 2002). Potassium cyanide (2 mMol) was added to each sample to inhibit any remaining activity of SOD1 and SOD3 (MacMillan-Crow et al. 1996). The activity of the latter SOD isoforms together was estimated by subtracting the activity of SOD2 from the total SOD activity.
Statistical Analysis
All results were presented as means ± standard error (SEM). Analysis of variance (ANOVA) was used with Bonferroni post-test correction to determine the levels of difference among all groups. Statistical significance was set at ρ < 0.05.
Results
Animal Survival
Fifty percent (6/12) of the BALB/cJ mice and forty-two percent (5/12) of the CAST/Ei mice died during or immediately following the initial hyperoxic exposure. To maintain 12 viable mice for the post challenge evaluation, additional BALB/cJ and CAST/Ei mice were exposed to the same hyperoxic conditions. None of the animals that died were included in the data analysis; therefore, only the most resistant mice of the BALB/cJ and CAST/Ei strains were included in the biological results. All mice from the remaining seven strains survived the duration of the hyperoxic exposure. In addition, all mice of the nine strains survived the duration of the air-only (controls) exposure.
Pulmonary Edema
Wet-to-dry lung ratios were determined to assess whether interstitial and alveolar edema fluid attenuated in the lungs following the hyperoxic exposure. Seven of the nine strains (129/SvIm, BALB/cJ, BTBR+(T)/tf/tf, CAST/Ei, C3H/HeJ, C57BL/6J, and FVB/NJ) had significantly elevated fluid in the lungs compared to the strain-matched mice challenged with room air (Fig. 1). The C57BL/6J and C3H/HeJ strains demonstrated the greatest magnitude of pulmonary edema, with increases in wet-to-dry lung ratios of 1.6 and 1.5 fold, respectively. By contrast, the wet-to-dry lung ratios of the DBA/2J and A/J strains were not significantly increased following hyperoxic challenge.
Fig. 1.
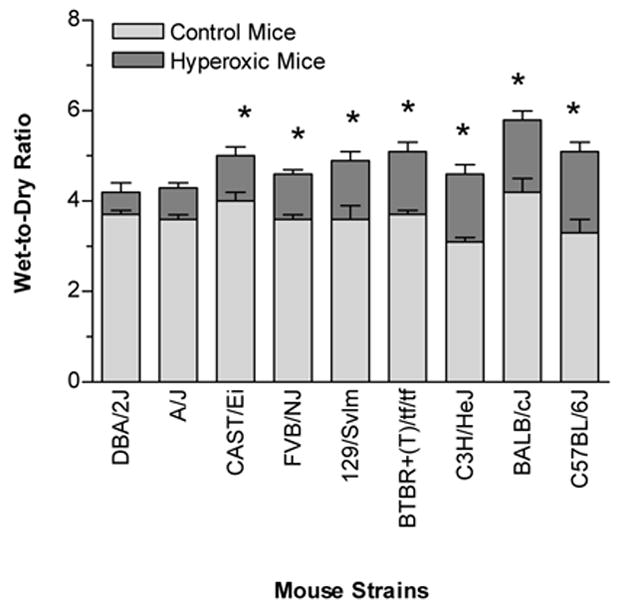
Wet-to-dry ratios of the right lungs from inbred mouse strains that were either exposed to >99% O2 or room air (control) for 72 hours. The strains were ranked according to the differences of wet-to-dry ratios between the hyperoxic and control mice. The wet-to-dry ratios are used as a marker of pulmonary edema. Data are presented as the mean ± SEM for n = 6 per group. * Significantly different (P ≤ 0.05) from control group (One-way ANOVA).
Total Protein in Whole-Lung Lavage Fluid
The total protein concentrations in the lavage fluid were used as an estimate of lung permeability and/or injury elicited by hyperoxia. The total protein in the lavage fluid was significantly elevated following O2 challenge in seven of the nine strains (129/SvIm, A/J, BALB/cJ, BTBR+(T)/tf/tf, CAST/Ei, C3H/HeJ, and C57BL/6J) (Fig. 2). Of these, the BALB/cJ and BTBR+(T)/tf/tf strains demonstrated the greatest increases in airway permeability; 6.3- and 6.2-fold, respectively. By contrast, the DBA/2J and FVB/NJ strains showed the least change in permeability following the hyperoxic exposure. Total protein in the lavage fluid of these strains only increased 2.4- and 3.2-fold, respectively, and neither value reached statistical significance.
Fig. 2.
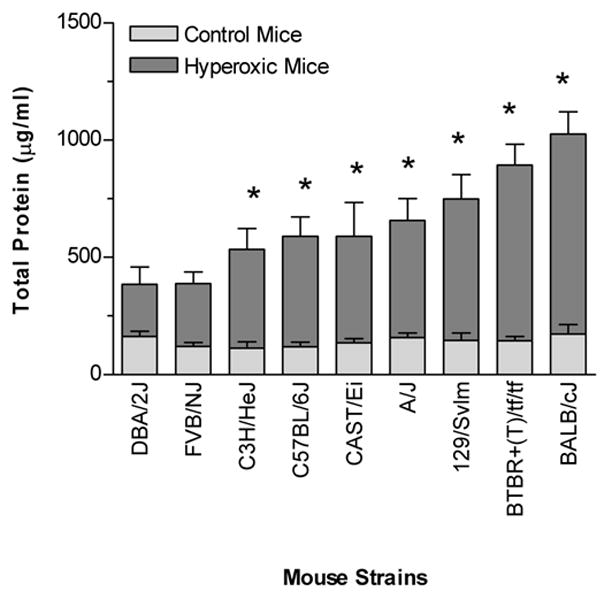
The concentration of total protein in the whole-lung lavage fluid from inbred mouse strains that were either exposed to >99% O2 or room air (control) for 72 hours. An increase in total protein was used as an indicator of lung permeability elicited by hyperoxia. The strains were ranked according to the increase of total protein in the lavage fluid from the hyperoxic mice of each strain. Data are presented as the mean ± SEM for n = 6 per group. * Significantly different (P ≤ 0.05) from control group (One-way ANOVA).
Inflammatory Response
Whole-lung lavage fluid was collected and the concentration of leukocytes was determined for both the O2- and air-exposed mice from each of the 9 strains. Most of the O2-challenged strains did not have significant increases in total leukocytes in the airway compared to unchallenged or air-treated mice (Fig. 3). By contrast, the BALB/cJ and CAST/Ei strains did have significant increases in the number of leukocytes in the airway. Differential analysis of the airway cells in these mice revealed that neutrophils were the primary cellular infiltrate (Fig. 4).
Fig. 3.
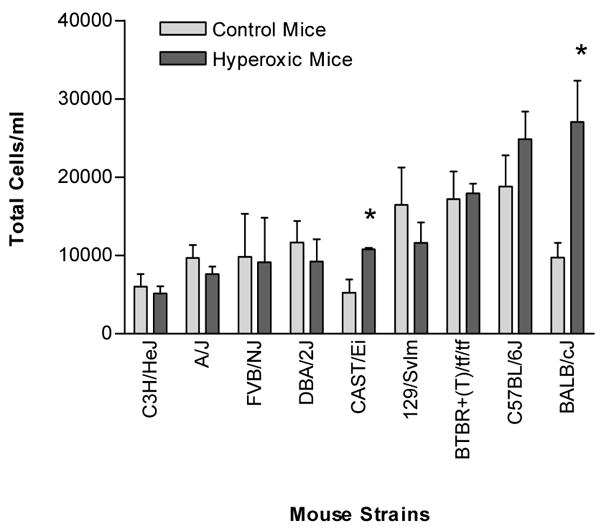
Number of total cells in the whole-lung lavage fluid from various strains of mice that were either exposed to >99% O2 or room air (control) for 72 hours. Data are presented as the mean ± SEM for n = 6 per group. * Significantly different (P ≤ 0.05) from control group (One-way ANOVA).
Fig. 4.
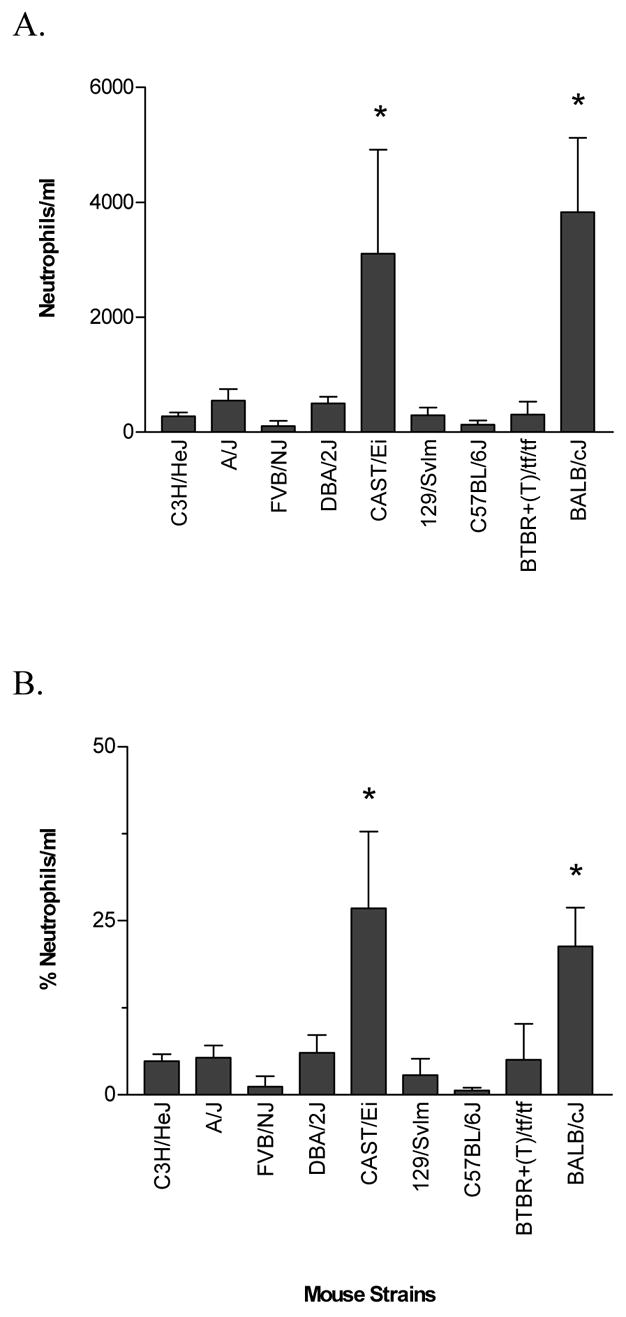
Number (a) and percentage (b) of neutrophils in the whole-lung lavage fluid from various strains of mice that were exposed to >99% O2 for 72 hours. The numbers and percentages of neutrophils from the air-exposed (control) mice of each strain were < 50.0 neutrophils/ml and < 2.0 %, respectively. Data are presented as the mean ± SEM for n = 6 per group. * Significantly different (P ≤ 0.05) from control group (One-way ANOVA).
Cytokines in Whole-lung Lavage Fluid
We next evaluated the concentration of IL-6 in whole-lung lavage fluid as a measurement of inflammatory responses in O2-challenged mice. IL-6 levels in the airways varied markedly among the 9 inbred mouse strains following the hyperoxic challenge (Fig. 5). Three strains, FVB/NJ, C57BL/6J and 129/SvIm, did not have significant increases in airway concentrations of IL-6 following the hyperoxic challenge. However, the other six strains (A/J, BALB/cJ, BTBR+(T)/tf/tf, CAST/Ei, C3H/HeJ and DBA/2J) did have significant increases in IL-6 following the challenge, with A/J, BALB/cJ, CAST/Ei and BTBR+(T)/tf/tf having the highest increases.
Fig. 5.
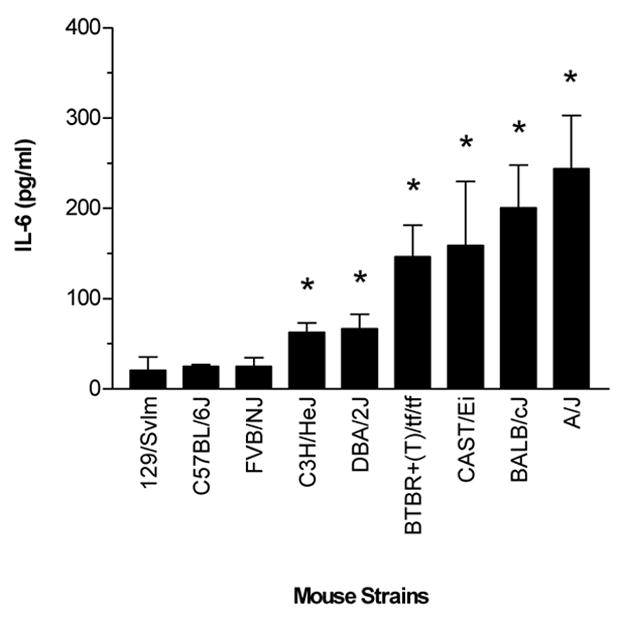
IL-6 concentrations in the whole-lung lavage of various strains of mice following exposure to >99% O2 for 72 hours. Data are presented as the mean ± SEM for n = 6 per group. The concentrations of IL-6 for the air-exposed (control) mice of each strain were < 5.0 pg/ml. * Significantly different (P ≤ 0.05) from control group.
Expression of Extra Cellular Matrix Proteins
To further evaluate the severity of hyperoxic injury immediately after the O2 and room-air challenges, we measured the concentration of mRNAs corresponding to three types of procollagen, (Col1α2, Col3α1, Col4α3), FN-1 and ten-C (Fig. 6 and 7), which are known contributors to the repair of lung injury. Concentrations of these mRNAs varied markedly among the various strains studied. The BTBR+(T)/tf/tf, 129/SvIm and A/J strains were the only strains to have increased expression of each of these genes. Interestingly, these three strains were also among the strains having the highest protein concentrations in the airways (Fig. 2), but they were not among the strains having the greatest increase in lung edema (Fig. 1) or airway inflammation (Fig. 4 and 5). The DBA/2J strain had the lowest expression of these ECM-related genes.
Fig. 6.
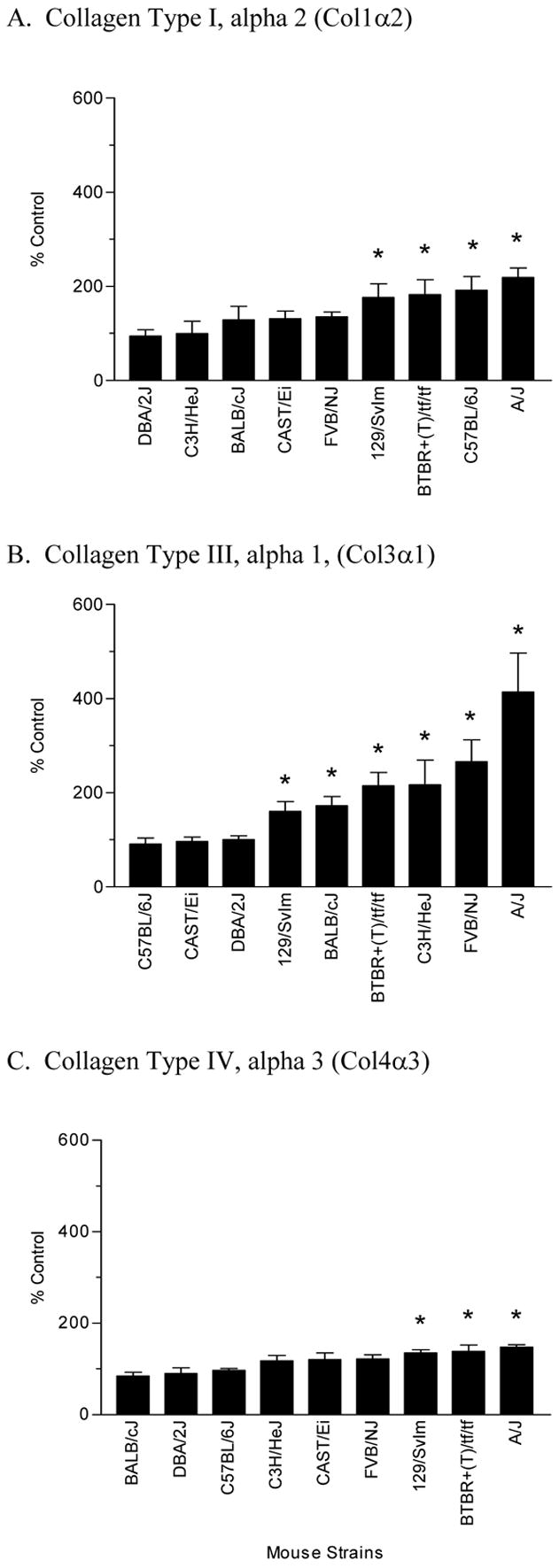
Expression of procollagens I (a), III (b) and IV (c) in the lungs of various strains of mice exposed to >99% O2 for 72 hours. For each strain, data represent the percent change of expression between a minimum of four hyperoxic-treated and air-exposed (control) mice. * Significantly different (P ≤ 0.05) from control group.
Fig. 7.
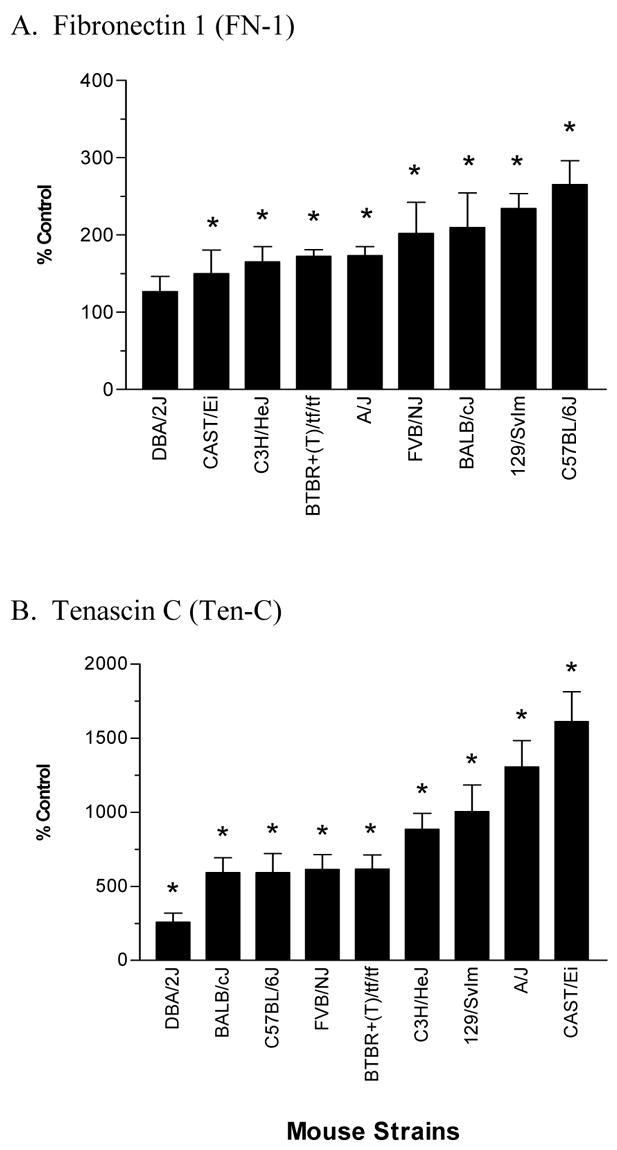
Expression of Fn I (a) and Ten C (b) in the lungs of various strains of mice exposed to >99% O2 for 72 hours. For each strain, data represent the percent change of expression between a minimum of four hyperoxic-treated and air-exposed (control) mice. * Significantly different (P ≤ 0.05) from control group.
Rankings of the susceptibility of the mouse strains following hyperoxia
To evaluate the comparative vulnerability of the 9 mouse strains to high concentrations of O2, we ranked the mouse strains for several phenotypes explored in this study (Table 2). For mortality, we attributed a score of a ‘zero’ for the mouse strains that had 100% survivability following the 72 hour O2 challenge and then numbered the remaining strains, CAST/Ei and BALB/cJ, from lowest (starting at 1) to highest for the number of mouse deaths that did occur. In addition, we also attributed a score for each strain in terms of lung permeability (pulmonary edema and protein leakage), inflammation (airway neutrophils and IL-6 concentrations), and ECM protein expression (Col1α2, Col3α1, Col4α3, FN-1 and ten-C), as well as we tallied a total score for each strain for all the phenotypes. Once again, a mouse strain was assigned a ‘zero’ for any phenotype in which hyperoxia did not induce significant changes. The remaining strains in which there were significant differences were rated from lowest to highest depending upon the extent of increase induced by the hyperoxic conditions. Based upon this scoring system we were able to identify the DBA/2J strain as the most resistant strain and the A/J and BALB/cJ strains as the most susceptible to hyperoxia.
Table 2.
Rankings of the susceptibility of the mouse strains following hyperoxia
| Mouse Strains | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DBA/2 | FVB/N | C3H/He | CAST/Ei | C57BL/6 | 129/SvIm | BTBR | BALB/c | A/J | ||
| Mortality | Total | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 |
|
|
||||||||||
| Lung | Edema | 0 | 2 | 5 | 1 | 7 | 3 | 4 | 6 | 0 |
| Permeability | Tot. Protein | 0 | 0 | 1 | 3 | 2 | 5 | 6 | 7 | 4 |
|
|
||||||||||
| Total | 0 | 2 | 6 | 4 | 9 | 8 | 10 | 13 | 4 | |
| Inflammation | Neutrophils | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 |
| IL-6 | 2 | 0 | 1 | 4 | 0 | 0 | 3 | 5 | 6 | |
|
|
||||||||||
| Total | 2 | 0 | 1 | 5 | 0 | 0 | 3 | 7 | 6 | |
| ECM | Col-1a | 0 | 0 | 0 | 0 | 3 | 1 | 2 | 0 | 4 |
| Expression | Col-3a | 0 | 5 | 4 | 0 | 0 | 1 | 3 | 2 | 6 |
| Col-4a | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 0 | 3 | |
| FN-1 | 0 | 5 | 2 | 1 | 8 | 7 | 3 | 6 | 4 | |
| Ten-C | 1 | 4 | 6 | 9 | 3 | 7 | 5 | 2 | 8 | |
|
|
||||||||||
| Total | 1 | 14 | 12 | 10 | 14 | 17 | 15 | 10 | 25 | |
|
| ||||||||||
| ALL | TOTAL | 3 | 16 | 19 | 20 | 23 | 25 | 28 | 32 | 35 |
A rating of a “zero” was given to mouse strains in which no deaths occurred due to the hyperoxic exposures and in which there were no significant changes for the phenotypes of lung permeability (lung edema (wet-to-dry-ratios) and protein levels), airway inflammation (neutrophil infiltration in the airways and IL-6 concentrations in lavage fluid) and the expression of ECM proteins (procollagens I (Col-1a), III (COl-3a) and IV (Col-4a), Fibronectin-1 (FN-1), and tenascin C (Ten-C). The mouse strains in which there were deaths and significant changes were ranked from lowest, starting at 1, to the highest. Mouse strains are ranked according to the tallied score for all phenotypes.
The activity of total SOD and SOD2
To gain insight into the extent of the pulmonary SOD response to the oxidative stress in O2-and air-challenged mice, we measured total SOD and SOD2 activity. The total SOD activity among the O2-challenged mouse strains ranged from 18.1 to 29.6 U/lung (Fig. 8a-b). The BALB/cJ strain was the only strain that had significantly increased total SOD activity compared to the air challenged controls, with an increase of 38.9%. The CAST/Ei strain had the lowest total SOD activity among the strains.
Fig. 8.

Total SOD activity (a) and SOD2 (Mn-SOD) activity (b) in the lungs of various strains of mice exposed to >99% O2 for 72 hours, and the percent differences between the hyperoxic- and air-exposed (control) mice (n = 6 per group). The % differences of total and SOD2 activity between the hyperoxic and control mice were determined by ((hyperoxic – control/control)* 100). The mouse strains were ranked according to the activity for that group. * Significantly different (P ≤ 0.05) from control group.
The SOD2 activity of the hyperoxic mice of the various mouse strains ranged from 11.6 to 23.2 U/lung. There were no significant differences in SOD2 activity between the O2-exposed and air-control mice for any of the tested strains. Interestingly, the BALB/cJ, BTBR+(T)/tf/tf, and 129/SvIm strains, which ranked among the most susceptible strains (Table 2), had the highest percentage increase in SOD2 activity between the O2-exposed and air-control mice.
Comparison of Total and SOD2 with the Susceptibility of Mouse Strains to Hyperoxia
To further explore the relationships between the activity of SOD (total and SOD2) and lung injury among the nine strains of mice, co-segregation plots were generated. The SOD activity for each strain was plotted against the rating of susceptibility of the mouse strains following oxidant injury, as ranked by mortality, lung permeability (pulmonary edema and protein leakage), inflammation (airway neutrophils and IL-6 concentrations), and ECM protein expression (Col1α2, Col3α1, Col4α3, FN-1 and ten-C) (Table 2). Both plots had similar results, and indicated an association between O2-induced injury and the amount of both total and SOD2 activity, respectively (Fig. 9a and b). For instance, the A/J, BALB/cJ, BTBR+(T)/tf/tf, and 129/SvIm strains, which were rated as more sensitive among the strains following the O2 challenge (Table 2), also had the highest total and SOD2 activity. In addition, the DBA/2J and FVB/NJ strains, which were among the strains with the least amount of injury (Table 2), were among the strains with the least amount of total and SOD2 activity. The severity of injury for the CAST/Ei strain did not necessarily correlate with the total and SOD2 activities, and the severity for the C57BL/6 strain did not necessarily correlate with the total SOD activity.
Fig. 9.
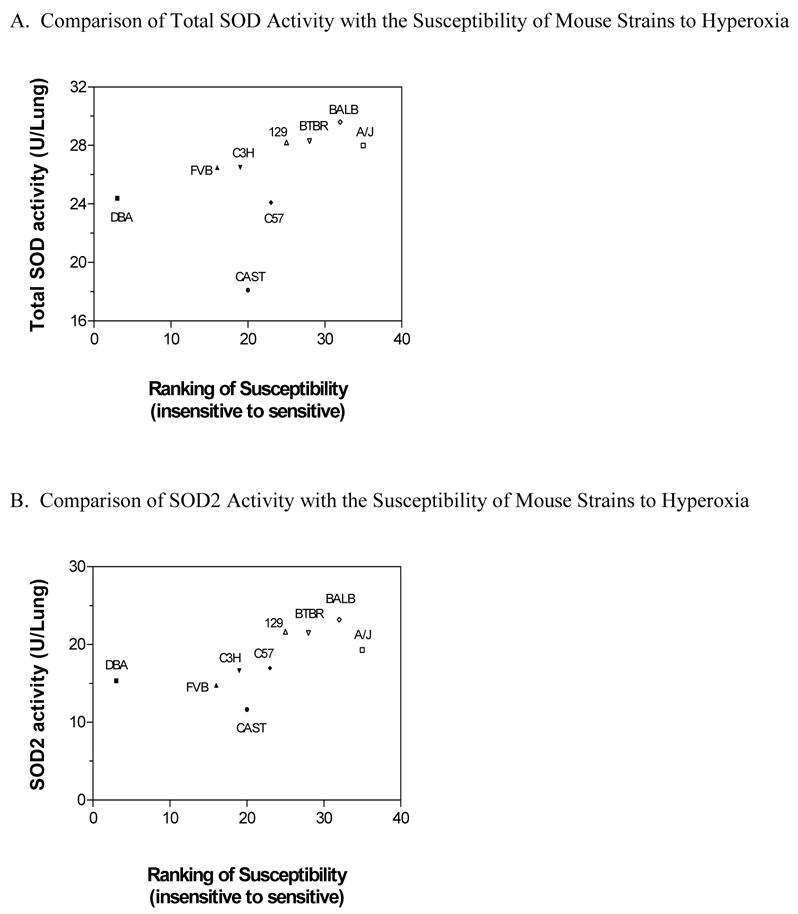
Cosegregation plots for the activity of Total SOD (a) and SOD2 (Mn-SOD) (b) in the lungs of various strains of mice exposed to >99% O2 for 72 hours versus our ranking of susceptibility of the mouse strains following oxidant injury (Table 2). A low score of susceptibility determined a mouse strain relatively insensitive (i.e. DBA/2J) and a high score determined a mouse strain sensitive (i.e. A/J). Note the scales for the y-axis are different for each Figure. For each strain, total SOD and SOD2 data are presented as the mean values of samples from 6 mice.
The activity of SOD1 plus SOD3
We investigated the SOD1 plus SOD3 activity in the various strains of mice after hyperoxia (Fig. 10). Interestingly, the hyperoxic mice of the BALB/cJ strain, which was the most susceptible strain, had the highest increase of SOD1 plus SOD3 activity, and the hyperoxic mice of the DBA/2J strain, which was the least susceptible strain, had the lowest decrease in activity.
Fig. 10.
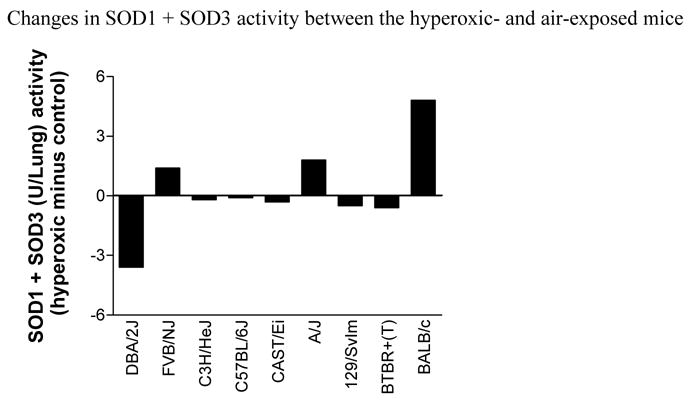
Changes of SOD1 (Cu/Zn SOD) plus SOD3 (EC-SOD) activity (total SOD activity minus the SOD2 activity) between mice exposed to O2 (hyperoxic) and room air (control) for 72 hours. For each strain, a mean value of samples from at least four hyperoxic and control mice were used to obtain the mean SOD1 + SOD3 activity. Note that the DBA/2J strain, which is the most resistant strain to hyperoxia injury, has the greatest decrease in activity; and the BALB/cJ strain, which is one of the most susceptible, has the greatest increase in activity.
Discussion
These novel results demonstrate that genetically distinct inbred mouse strains exhibit different responses to pulmonary oxygen toxicity, as measured by mortality, lung permeability, airway inflammation, and expression of genes that contribute to airway remodeling. However, in many cases, these phenotypes were not concordant, indicating that each hyperoxia-induced phenotype is controlled by distinct genes and biological pathways. Moreover, it appears SOD activity has a complex participation in the acute response to hyperoxia. Although other investigators have demonstrated that various mouse strains respond differently to acute O2 exposure (Takeda et al. 2000; Johnston et al 1998; Hudak et al. 1992), limited phenotypes were examined and the association with SOD activity was not explored. Taken together, these results demonstrate that the genetic background contributes to the susceptibility of acute hyperoxic lung injury, and that the phenotypically divergent strains may provide excellent reagents to further pursue the genetics and biology of hyperoxic lung injury.
Interestingly, there were some differences and similarities between our results and those of other investigators. For instance, Hudak et al. observed that the C3H/HeJ strain had greater alterations of lung permeability following a 72-hour hyperoxia challenge than did the DBA/2J strain, and the C57BL/6J strain had greater susceptibility than the BALB/c strain (Hudak et al. 1992). In addition, other investigators found that C57BL/6J mice develop airway inflammation following a 72-hour challenge, whereas in our hands, the C57BL/6 mice did not. These different responses might result from the slightly different exposure conditions (i.e. >99% O2 vs. >95%), post-exposure times of follow up, and/or the different methods to determine lung permeability. However, despite the differences, our findings generally support previous findings and confirm that the DBA/2J and C3H/HeJ strains are relatively insensitive and the BALB/cJ and C57BL/6J strains relatively sensitive to O2 challenge.
Exposure to high concentrations of O2 is toxic for the lung and often leads to acute lung injury. We observed that different strains of mice were phenotypically unique in their response to high oxygen tensions, and that many of the phenotypes investigated among the various mouse strains were discordant (Table 2). Our results suggest that the pathogenic response to O2 is complex and involves multiple biological pathways. Furthermore, our results also suggest that independent biological pathways are being initiated as the various strains of mice are in different stages of injury, and that the biological responses are controlled by unique genetics of each strain. However, this is not unexpected since the response to oxygen represents a complex interaction of many genes involved in airway inflammation, fibrinolysis, extracellular matrix repair, and detoxifying ROS (Wagenaar et al. 2004).
Lung permeability, together with increased airspace inflammation, is a key feature of hyperoxia injury (Keeny et al. 1995; Shasby et al. 1980). In fact, if we relied simply on these phenotypes, we could distinguish the resistant strains from the sensitive strains. For instance, as demonstrated in table 2, the DBA/2J and FVB/NJ strains, which were ranked as the two least sensitive strains to oxidant stress, showed the least change in permeability (pulmonary edema (wet-dry ratios) and protein concentrations in the lavage fluid) following the hyperoxic exposure and had only minimal increases of airway inflammation (airway neutrophils and IL-6 concentrations). In contrast, the remaining strains demonstrated significant elevations in either lung permeability or airway inflammation, or both. A qualification of whether significant changes occurred was not particularly helpful in distinguishing sensitive from resistant strains of mice.
Interestingly, we also found a relationship between the recruitment of neutrophils to the airspaces and the severity of injury, which has also been found by other investigators (Shasby et al. 1980). For instance, we found that the BALB/cJ and CAST/Ei strains, which were the only strains to have increased neutrophils in the lavage due to exposure to hyperoxia, were the only strains to have hyperoxia-induced mortality (Table 2). However, other mouse strains had significant increases in lung permeability without neutrophil infiltration. Therefore, these data do not discern the contribution of the neutrophil to the lung injury.
Fibronectin and tenascin are also major components of the ECM and may play roles in the pathology and repair of the lungs (Hynes, 1985; Grinnell et al. 1987; Kaarteenaho-Wiik et al. 1996, 2002). FN depositions have been observed in the lungs of patients with pulmonary fibrosis and are also observed in patients and experimental animals following exposure to toxic levels of O2 (Torikata et al. 1985; Davis et al. 1983; Sinkin et al. 1992). Tenascin is increased in patients with fibrotic lung disorders including ALI and bronchopulmonary dysphasia (Kaarteenaho-Wiik et al. 1996, 2002). Furthermore, ten-C is one isoform of the tenascins that has been shown to contribute to the development of pulmonary fibrosis (Kaminski et al. 2002). Our finding of which FN and Ten-C expression are increased in nearly all the O2–challenged strains further supports a role for these genes in the repair process following oxidant injury.
Antioxidant enzymes, including superoxide dismutases, participate in protecting the lung against pulmonary O2 toxicity (Comhair and Erzurum, 2002). However, studies with transgenic and gene knockout mice show that neither SOD1 nor glutathione peroxidase-I can prevent lung injury from oxygen exposure. Thus, other cellular antioxidant enzymes are likely more important in defense against hyperoxia, supported especially by studies involving SOD2 and SOD3 (Tsan, 2001). Normally SOD2 accounts for about 20% of the pulmonary SOD activity, but here we found that about half of the total SOD activity in mouse lung partitioned into the mitochondrial pellet, which raises an important question about the differential value of the simple assay method. Nevertheless, the total and SOD2 assay of the lungs of O2-exposed mice revealed that both SOD activities were highest in the strains that were the most sensitive (Fig. 9). Many of the pulmonary SOD changes, including increases in SOD2 activity, did not reach statistical significance, but the total SOD activity did increase significantly in one of the most susceptible strains, BALB/cJ. Although lung SOD activity appears to increase in relation to the extent of lung injury, the data do not allow us to draw any conclusions about whether it protects or contributes differentially to the damage in the tested strains.
Taken together, our results demonstrate that genetically diverse strains of mice manifest responses to hyperoxia that are quantitatively different. The divergent strains can be further pursued at various times following exposure to high O2 levels to identify the individual genes that are responsible for each of these distinct lung phenotypes.
Acknowledgments
This research was sponsored by NIH awards: ES07498, ES09607, HL62628, HL66611, HL66604, and HL62641.
Footnotes
This study was supported by grants from the National Institutes of Health (ES07498, ES 09607, ES012496, ES11375, ES11961, HL66611, and HL66604) and the Department of Veterans’ Affairs (Merit Review).
References
- Adamson IYR, Bowden DH, Wyatt JP. Oxygen poisoning in mice. Ultrastructural and surfactant studies during exposure and recovery. Arch Pathol. 1970;90:463–472. [PubMed] [Google Scholar]
- Adamson IY, Hedgecock C, Bowden DH. Epithelial cell-fibroblast interactions in lung injury and repair. Am J Pathol. 1990;137:385–92. [PMC free article] [PubMed] [Google Scholar]
- Bureau M, Sourouille P, Brun-Pascaud M, Fey M, Pocidalo JJ. Hyperoxia effects on lung vascular circulating and marginated leukocytes in the rat. Bull Eur Physiopathol Respir. 1985;21:325–9. [PubMed] [Google Scholar]
- Capellier G, Maupoil V, Boussat S, Laurent E, Neidhardt A. Oxygen toxicity and tolerance (Review) Minerva Anestesiol. 1999;65:388–92. [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Reddy SP, Zhang LY, Kensler TW, Kleeberger SR. Linkage analysis of susceptibility to hyperoxia: Nrf2 is a candidate gene. Am J Respir Cell Mol Biol. 2002;26:42–51. doi: 10.1165/ajrcmb.26.1.4536. [DOI] [PubMed] [Google Scholar]
- Clark H, Clark LS. The genetics of neonatal respiratory disease. Semin Fetal Neonatal Med. 2005;10:271–82. doi: 10.1016/j.siny.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Crapo JD, Peters-Golden M, Marsh-Salin J, Shelburne JS. Pathologic changes in the lungs of oxygen-adapted rats: A morphometric analysis. Lab Invest. 1978;39:640–53. [PubMed] [Google Scholar]
- Crapo JD, Barry BE, Foscue HA, Shelburne J. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis. 1980;122:123–43. doi: 10.1164/arrd.1980.122.1.123. [DOI] [PubMed] [Google Scholar]
- Crapo JD. Morphologic changes in pulmonary oxygen toxicity (Review) Annu Rev Physiol. 1986;48:721–31. doi: 10.1146/annurev.ph.48.030186.003445. [DOI] [PubMed] [Google Scholar]
- Davis WB, Rennard SI, Bitterman PB, Crystal RG. Pulmonary oxygen toxicity: Early reversible changes in human alveolar structures induced by hyperoxia. N Engl J Med. 1983;309:878–83. doi: 10.1056/NEJM198310133091502. [DOI] [PubMed] [Google Scholar]
- Deneke SM, Fanburg BL. Normobaric oxygen toxicity of the lung (Review) N Engl J Med. 1980;303:76–86. doi: 10.1056/NEJM198007103030204. [DOI] [PubMed] [Google Scholar]
- Durr RA, Dubaybo BA, Thet LA. Repair of chronic hyperoxic lung injury: Changes in lung ultrastructure and matrix. Exp Mol Pathol. 1987;47:219–40. doi: 10.1016/0014-4800(87)90077-3. [DOI] [PubMed] [Google Scholar]
- Comhair SA, Erzurum SC. Antioxidant responses to oxidant-mediated lung diseases (Review) Am J Physiol Lung Cell Mol Physiol. 2002;283:L246–55. doi: 10.1152/ajplung.00491.2001. [DOI] [PubMed] [Google Scholar]
- Folz RJ, Abushamaa AM, Suliman HB. Extracellular superoxide dismutase in the airways of transgenic mice reduces inflammation and attenuates lung toxicity following hyperoxia. J Clin Invest. 1999;103:1055–66. doi: 10.1172/JCI3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fracica PJ, Knapp MJ, Piantadosi CA, Takeda K, Fulkerson WJ, Coleman RE, Wolfe WG, Crapo JD. Responses of baboons to prolonged hyperoxia: physiology and qualitative pathology. J Appl Physiol. 1991;71:2352–62. doi: 10.1152/jappl.1991.71.6.2352. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Toda K, Takashima A. Activation of keratinocyte fibronectin receptor function during cutaneous wound healing (Review) J Cell Sci Suppl. 1987;8:199–209. doi: 10.1242/jcs.1987.supplement_8.11. [DOI] [PubMed] [Google Scholar]
- Hallman M, Haataja R. Genetic influences and neonatal lung disease (Review) Semin Neonatol. 2003;8:19–27. doi: 10.1016/s1084-2756(02)00196-3. [DOI] [PubMed] [Google Scholar]
- He LS, Chang SW, Ortiz de Montellano P, Burke TJ, Voelkel NF. Lung injury in Fischer but not Sprague-Dawley rats after short-term hyperoxia. Am J Physiol. 1990;259:L451–8. doi: 10.1152/ajplung.1990.259.6.L451. [DOI] [PubMed] [Google Scholar]
- Hudak BB, Zhang LY, Kleeberger SR. Inter-strain variation in susceptibility to hyperoxic injury of murine airways. Pharmacogenetics. 1992;3(3):135–43. doi: 10.1097/00008571-199306000-00003. [DOI] [PubMed] [Google Scholar]
- Hynes R. Molecular biology of fibronectin (Review) Annu Rev Cell Biol. 1985;1:67–90. doi: 10.1146/annurev.cb.01.110185.000435. [DOI] [PubMed] [Google Scholar]
- Johnston CJ, Wright TW, Reed CK, Finkelstein JN. Comparison of adult and newborn pulmonary cytokine mRNA expression after hyperoxia. Exp Lung Res. 1997;23:537–52. doi: 10.3109/01902149709039242. [DOI] [PubMed] [Google Scholar]
- Johnston CJ, Stripp BR, Piedbeouf B, Wright TW, Mango GW, Reed CK, Finkelstein JN. Inflammatory and epithelial responses in mouse strains that differ in sensitivity to hyperoxic injury. Exp Lung Res. 1998;24:189–202. doi: 10.3109/01902149809099582. [DOI] [PubMed] [Google Scholar]
- Kaarteenaho-Wiik R, Tani T, Sormunen R, Soini Y, Virtanen I, Paakko P. Tenascin immunoreactivity as a prognostic marker in usual interstitial pneumonia. Am J Respir Crit Care Med. 1996;154:511–18. doi: 10.1164/ajrccm.154.2.8756830. [DOI] [PubMed] [Google Scholar]
- Kaarteenaho-Wiik R, Kinnula VL, Herva R, Soini Y, Pollanen R, Paakko P. Tenascin-C is highly expressed in respiratory distress syndrome and bronchopulmonary dysplasia. J Histochem Cytochem. 2002;50:423–31. doi: 10.1177/002215540205000313. [DOI] [PubMed] [Google Scholar]
- Kaminski N, Zuo F, Cojocaro G, Yakhini Z, Ben-Dor A, Morris D, Sheppard D, Pardo A, Selman M, Heller RA. Use of oligonucleotide microarrays to analyze gene expression patterns in pulmonary fibrosis reveals distinct patterns of gene expression in mice and humans. Chest. 2002;121:31S–32S. [PubMed] [Google Scholar]
- Kapanci Y, Weibel ER, Kaplan HP, Robinson FR. Pathogenesis and reversibility of the pulmonary lesions of oxygen toxicity in monkeys. II. Ultrastructural and morphometric studies. Lab Invest. 1969;20:101–18. [PubMed] [Google Scholar]
- Keeney SE, Mathews MJ, Haque AK, Schmalstieg FC. Comparison of pulmonary neutrophils in the adult and neonatal rat after hyperoxia. Pediatr Res. 1995;38:857–63. doi: 10.1203/00006450-199512000-00006. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–75. [PubMed] [Google Scholar]
- MacMillan-Crow LA, Crow JP, Kerby JD, Beckman JS, Thompson JA. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci U S A. 1996;93:11853–58. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniscalco WM, Watkins RH, Finkelstein JN, Campbell MH. Vascular endothelial growth factor mRNA increases in alveolar epithelial cells during recovery from oxygen injury. Am J Respir Cell Mol Biol. 1995;13:377–86. doi: 10.1165/ajrcmb.13.4.7546767. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–33. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Que LG, Kantrow SP, Jenkinson CP, Piantadosi CA, Huang YC. Induction of arginase isoforms in the lung during hyperoxia. Am J Physiol. 1998;275:L96–102. doi: 10.1152/ajplung.1998.275.1.L96. [DOI] [PubMed] [Google Scholar]
- Shasby DM, Fox RB, Harada RN, Repine JE. Reduction of the edema of acute hyperoxic lung injury by granulocyte depletion. J Appl Physiol. 1982;52:1237–44. doi: 10.1152/jappl.1982.52.5.1237. [DOI] [PubMed] [Google Scholar]
- Sinkin RA, LoMonaco MB, Finkelstein JN, Watkins RH, Cox C, Horowitz S. Increased fibronectin mRNA in alveolar macrophages following in vivo hyperoxia. Am J Respir Cell Mol Biol. 1992;7:548–55. doi: 10.1165/ajrcmb/7.5.548. [DOI] [PubMed] [Google Scholar]
- Stenzel JD, Welty SE, Benzick AE, Smith EO, Smith CV, Hansen TN. Hyperoxic lung injury in Fischer-344 and Sprague-Dawley rats in vivo. Free Radic Biol Med. 1993;14:531–39. doi: 10.1016/0891-5849(93)90110-g. [DOI] [PubMed] [Google Scholar]
- Takeda K, Haczku A, Lee JJ, Irvin CG, Gelfand EW. Strain dependence of airway hyperresponsiveness reflects differences in eosinophil localization in the lung. Am J Physiol Lung Cell Mol Physiol. 2001;281:L394–402. doi: 10.1152/ajplung.2001.281.2.L394. [DOI] [PubMed] [Google Scholar]
- Nickerson BG, Taussig LM. Family history of asthma in infants with bronchopulmonary dysplasia. Pediatrics. 1980;65:1140–44. [PubMed] [Google Scholar]
- Thet LA, Wrobel DJ, Crapo JD, Shelburne JD. Morphologic aspects of the protection by endotoxin against acute and chronic oxygen-induced lung injury in adult rats. Lab Invest. 1983;48:448–57. [PubMed] [Google Scholar]
- Torikata C, Villiger B, Kuhn C, 3rd, McDonald JA. Ultrastructural distribution of fibronectin in normal and fibrotic human lung. Lab Invest. 1985;52:399–408. [PubMed] [Google Scholar]
- Tsan MF. Superoxide dismutase and pulmonary oxygen toxicity: lessons from transgenic and knockout mice (Review) Int J Mol Med. 2001;7:13–19. doi: 10.3892/ijmm.7.1.13. [DOI] [PubMed] [Google Scholar]
- Wagenaar GT, ter Horst SA, van Gastelen MA, Leijser LM, Mauad T, van der Velden PA, de Heer E, Hiemstra PS, Poorthuis BJ, Walther FJ. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic Biol Med. 2004;36:782–801. doi: 10.1016/j.freeradbiomed.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Wispe JR, Warner BB, Clark JC, Dey CR, Neuman J, Glasser SW, Crapo JD, Chang LY, Whitsett JA. Human Mn-superoxide dismutase in pulmonary epithelial cells of transgenic mice confers protection from oxygen injury. J Biol Chem. 1992;267:23937–41. [PubMed] [Google Scholar]