

Abstract
Liver insufficiency and damage is a major cause of death and disease worldwide and may result from exposure to environmental toxicants, specific combinations or dosages of pharmaceuticals and microbial metabolites. The generation of reactive intermediates, in particular 4-hydroxynonenal (4-HNE), is a common event in liver damage caused by a variety of hepatotoxic drugs and solvents. The peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that are involved in the transcriptional regulation of lipid metabolism as well as other biological functions. Importantly, we have observed that the PPARβ/δ−/− mouse is more susceptible to chemically-induced hepatotoxicity than its wildtype counterpart, and our objective in this study was to elucidate the mechanism(s) by which PPARβ/δ confers protection to hepatocytes. We hypothesized that PPARβ/δ plays a protective role by responding to toxic lipids and altering gene expression accordingly. In support, oxidized-VLDL and constituents including 13-S-hydroxyoctadeca-dienoic acid (13(S)-HODE) and 4-HNE are PPARβ/δ ligands. A structure-activity relationship was established where 4-HNE and 4-hydroperoxynonenal (4-HpNE) enhanced the activity of the PPARβ/δ subtype while 4-hyroxy-hexenal (4-HHE), 4-oxo-2-Nonenal (4-ONE), and trans-4,5-epoxy-2(E)-decenal did not activate this receptor. Increasing PPARβ/δ activity with a synthetic agonist decreased sensitivity of hepatocytes to 4-HNE and other toxic agents, whereas inhibition of this receptor had the opposite result. Gene expression microarray analysis identified several important PPARβ/δ-regulated detoxification enzymes involved in 4-HNE metabolism that are regulated at the transcript level. This research established 4-HNE as an endogenous modulator of PPARβ/δ activity and raises the possibility that agonists of this nuclear receptor may be utilized to prevent or treat liver disease associated with oxidative damage.
INTRODUCTION
Greater than 2.2 million hospitalized Americans suffer adverse drug reactions each year, with liver toxicity presenting as the most common adverse effect, and approximately 100,000 individuals die unintentionally from administration of medications [1, 2]. Many additional cases of liver failure occur due to acute, chronic and degenerative disease processes, including those related to acetaminophen overdose, alcohol consumption and solvent exposures. Reactive oxygen intermediates (ROI) elicit oxidative decomposition of polyunsaturated fatty acids (i.e, lipid peroxidation), leading to the formation of a complex mixture of aldehydic end-products, including malondialdehyde (MDA), 4-HNE, and other alkenals [3]. These aldehydic molecules have been considered the ultimate mediators of toxic effects elicited by oxidative stress but may also affect cellular function at non-toxic levels via signal transduction, gene expression and cell proliferation. Although the overt toxicity caused by aldehydic end-products is due primarily to covalent binding to cellular macromolecules, the effects on signal transduction are not well-characterized. Since millions of individuals suffer adverse drug reactions each year it is important to understand how the cell responds to intracellular insults such as production of ROI and 4-HNE.
The peroxisome proliferator-activated receptors (PPARs) are nuclear receptors that exist as three subtypes (α, β/δ and γ), which exhibit tissue-specific expression, preferential ligand recognition, and distinct biological functions [4–9]. Although important as targets of pharmaceutical intervention, there is increasing evidence that the biological niche occupied by the PPARs is that of a receptor for fatty acid and their metabolites. Of the three PPAR genes (α, β/δ, γ), the PPARβ/δ isoform is the least well-studied in terms of its biological functions and endogenous ligands. PPARβ/δ plays an important role in differentiation of epithelial tissues, fatty acid catabolism in skeletal muscle, improvement of insulin sensitivity, attenuated weight gain and elevated HDL levels [10]. Emerging evidence suggests the presence of this receptor is important in ameliorating the effects of hepatotoxicants. For example, histological examination of liver and analysis of markers of overt damage to this organ (serum GPT) after treatment with the xenobiotics azoxymethane (AOM), arsenic or carbon tetrachloride demonstrated that the extent of liver toxicity in PPARβ/δ-null mice was more severe than in wild-type mice1. While it is remotely possible that the metabolic fate of these hepatotoxicants could be influenced by PPARβ/δ, it is more likely that regulation of oxidative stress underlies the protective role of this receptor in liver. These chemicals share a common mechanism of overt toxicity via production of ROI and oxidized lipid intermediates. For example, CCl4 affects eicosanoid pathways [11, 12] and increases circulating prostaglandin E2 (PGE2) levels [13] and 4-HNE and 4-HNE-protein adducts [3, 14, 15]. The purpose of this study was to determine the extent to which oxidized lipids and their metabolites interact with PPARβ/δ and influence gene regulation. We hypothesized that PPARβ/δ acts as an oxidative stress sensor in hepatocytes and, upon interaction with products of lipid peroxidation, regulates detoxification genes accordingly to ameliorate the toxic insult.
One possible explanation for the increased susceptibility of PPARβ/δ−/− mice to hepatotoxicity is that oxidative damage increases the production of an endogenous ligand for PPARβ/δ. This putative agonist which would in turn stimulate lipid metabolism and degradation of lipid peroxidation intermediates. PPARs are well-recognized as transcriptional regulators of lipid metabolism, transport, storage and other activities [16]. In the absence of PPARβ/δ the signaling cascade would be disrupted and accumulation of toxic lipids such as 4-HNE would result. If our hypothesis were correct, endogenous ligands of PPARβ/δ should include oxidized lipids, in particular those derived from fatty acids. In support, we discovered that oxidized-VLDL and constituents including 13(S)-HODE and 4-HNE are PPARβ/δ agonists. In addition, modulating PPARβ/δ activity, either by activation with synthetic PPARβ/δ-selective agonist tetradecylthioacetic acid (TTA) or inhibition with PPAR pan-antagonist GW9662[17], affects the sensitivity of hepatocytes to 4-HNE and other toxic agents. This research raises the possibility that PPARβ/δ agonists may be utilized to prevent or treat liver disease associated with the generation of ROIs.
MATERIALS AND METHODS
Reagents
VLDL (human plasma) was purchased from Calbiochem (La Jolla, CA), LPL was purchased from Sigma (lyophilized powder) and reconstituted in PBS (10 mg/mL), and 13(S)-HODE, 13(S)-HpODE, 4-HHE, 4-ONE, trans-4,5-epoxy-2E-decenal, 4-HpNE, and 4-HNE were purchased from Cayman Chemical (Ann Arbor, MI) and used without further purification. The semi-enzymatic synthesis and purification of some of the linoleic and arachidonic acid oxidation products such as 9-HODE, 12-HpODE, 5-HETE, 9-HETE, 12-HETE, 15-HETE, 5-HpETE, 15-HpETE, 5,15-diHpETE and 5,6-diHETE were performed as described [18]. The authenticity of each of the lipid mediators was confirmed using co-chromatography as well as gas chromatography-mass spectrometric experiments. UV-Visible spectroscopy was used to determine their respective concentrations, and the compounds were reconstituted in anhydrous ethanol to the desired concentration. The compounds were used within 30 min of reconstitution in ethanol.
Plasmids
Plasmids used in reporter assays including pM/mPPAR-α, -β, and –γ, pBK/mPPAR-α, -β, and –γ (murine), ACO Luciferase and pFR-luciferase (Promega) have been described elsewhere [19].
Cell culture
3T3-L1 preadipocyte cells were grown in standard high-glucose Dulbecco’s Modified Eagle Medium (DMEM) containing 10% FBS at 37 °C and 5% CO2. MuSH cells were grown in standard α-MEM containing 10% FBS. Cells were treated as described in Fig. legends.
Transient transfection and treatment
Cells were counted using a hemocytometer following staining with Trypan Blue Solution (0.4%) (Sigma) and plated onto 10-cm dishes at densities between 600,000–800,000 cells/dish for transfection. Transfections were performed at 37 °C for 6 h using 12 μg of the appropriate Gal4-PPAR ligand binding domain (LBD) plasmid (pM/mPPAR-α, -β, or –γ) containing a Luciferase reporter, and 24 μg Lipofectamine (Invitrogen). Transfection media was then replaced with fresh high-glucose DMEM after washing the dishes with PBS and the cells were allowed to recover overnight. Cells were re-seeded into 96-well plates (Costar, Corning, NY) 24 h following transfection and allowed to sit undisturbed at 37 °C for 2–3 h to adhere to the well bottom. The media was removed and 100 μL of fresh media containing the compound of interest at the desired concentration was added to the cells. Treatments were performed for 12 h at 37 °C and 5% CO2.
VLDL Experiments
VLDL control experiments used DMSO (the vehicle for positive controls ciprofibrate, tetradecylthioacetic acid (TTA) and rosiglitazone for PPARα, -β/δ and –γ, respectively) only in a volume equal to the highest volume used for positive controls, as well as a CuSO4 control at 10 μM. All solutions were prepared in a sterile environment using high-glucose DMEM and were allowed to incubate at 37 °C for 1 h prior to treatment. Cells received VLDL and oxVLDL at 10 μg/mL. VLDL and oxVLDL were treated with 1 μL each of Lipoprotein lipase (LpL, 715 units/μL) and allowed to incubate at 37 °C without shaking for 1 h prior to treatment.
13(S)-HODE and 4-HNE experiments
Fatty acid treatment experiments used ethanol (EtOH; the vehicle for 13(S)-HODE and other fatty acids and their metabolites) only in a volume equal to the highest volume used for 13(S)-HODE or 4-HNE. Cells received 13(S)-HODE and other fatty acids at 50 μM and 4-HNE at 25 μM, except where indicated.
Oxidation of VLDL and 13(S)-HODE with Cu2+
Ten-μM copper solutions were prepared fresh for every experiment from a stock solution of aqueous CuSO4 at 20 mM. VLDL and 13(S)-HODE solutions were treated with an equal volume of aqueous CuSO4 and allowed to incubate at 37 °C in an incubator/shaker for a period of 72 h, unless otherwise indicated.
Temperature-sensitive SV40 virus preparation
CV-1 cells were grown to confluence in T-75 flasks at 37°C in αMEM-4% FBS. Cells were incubated with stock temperature sensitive SV40 virus[20] at 37°C and agitated every 15 minutes for 2 h. Media was changed to αMEM-8% FBS, and the cells were incubated at 34°C for seven days. Virus-containing media was removed upon gross cell pathology (nuclear inclusion, cytoplasmic vacuolation and lysis) and stored in liquid nitrogen. Quantification of virus titer was not performed.
Isolation, maintenance and infection of primary hepatocytes
The isolation and infection of hepatocytes was adapted from a previously described method [21]. Purebred wild-type and PPARβ-null mice on a SV/129 background were used and have been previously described [22, 23]. Primary hepatocytes were isolated from two male wild-type mice (10 day old) and two male PPARβ-null mice (5 day old). Mice were euthanized by overexposure to carbon dioxide and whole liver was removed and incubated at 37°C with 1% collagenase in Hanks buffered saline solution (HBSS) for 5 minutes. Hepatocytes were isolated by centrifugation in 10 % Percoll (Amersham, Piscataway, NJ) in HBSS at 1000 rpm for 20 minutes at 4°C. The cells were washed with 10% fetal bovine serum (FBS) in αMEM, centrifuged and resuspended in αMEM-10% FBS. Each liver was separated into three populations to be infected separately; individual clones were not selected. Cells were cultured with αMEM, 4% FBS, dexamethasone (dex) and 1% penicillin/streptomycin and allowed to grow to approximately 75% confluency at 34°C. The cells were then washed with αMEM-10% FBS, overlayed with 1mL of αMEM-10% FBS and infected with 200 μL of stock virus per well. Cells were incubated at 34°C for 2–3 hours and gently agitated by hand. Virus-containing media was then aspirated, and the cells were given αMEM-4% FBS-0.1 μM dex. Media was changed twice weekly for approximately six weeks. Resultant colonies were isolated as mixed populations (Murine SV40-immortalized Hepatocytes (MuSH)) or as individual clones and are designated MuSH WT (wild-type) or MuSHβ/δ−/− (PPARβ/δ null).
Messenger RNA examination
MuSH WT and MuSH β/δ−/− cells were grown in αMEM containing 10% FBS, 100 units/mL penicillin and 100 μg/mL streptomycin. MuSH cells were grown to 75% confluency and treated overnight with the compound of interest. Total RNA was isolated using Tri-Reagent. Quantitative reverse transcriptase polymerase chain reaction (RT-PCR) was performed to measure β-actin, and adipose differentiation related protein (ADRP) mRNA levels. The following ADRP gene-specific primers were synthesized and used: 5′-AGTGGAAGAGAAGCATCGGCT-3′ (forward) and 5′-TCGATGTGCTCAACACAGTGG-3′ (reverse); β-actin primers have been described elsewhere [19].
Mouse Oligonucleotide Arrays
The Mouse Genome Oligo Set Version 1 was purchased from Operon (Alameda, CA) and contains 6800 optimized 70-mers plus 24 controls, melting temperature normalized to 78°C. Sequences were optimized by the manufacturer using BLAST against all known mouse genes to minimize cross-hybridization. Oligonucleotides were printed onto glass slides using GeneMachines Omnigrid (San Carlos, California) with additional controls obtained from Stratagene (SpotReport system, La Jolla, CA) at the Penn State University microarray core facility.
Microarray Analysis
Total RNA was isolated by TriReagent (Sigma) and further purified with RNAEasy (Qiagen) according the manufacturers’ instructions. Labeling and hybridization was performed as discussed previously [19, 21] In the present experiments, co-hybridization was performed with cDNA from three separate experiments (all listed as Cy5/Cy3-labeled): MuSHWT / MuSHβ/δ−/−; MuSH WT cells treated with TTA (50μM) compared to DMSO 6 h after treatment (TTA/DMSO), and MuSH WT cells treated with 4-HNE (25μM) compared to DMSO 6 h after treatment (4-HNE/DMSO). Statistical analysis was performed using a Student t-test with a p-value of 0.05 with the additional criteria of being either 2-fold increased or decreased by PPARβ/δ activation. Results are given as mean and p-value and was performed in GeneSpring (7.3, Agilent Technologies).
13(S)-HODE Enzyme-linked immunosorbent assay
One hundred fifty-μL of VLDL was oxidized as described previously and incubated with 2 μL LpL at 37 °C for 1h. Detection of 13(S)-HODE was achieved by using a 13(S)-HODE Correlate™ enzyme immunoassay kit (Assay Designs).
Expression of mPPARβ/δ LBD and 4-HNE-Capture Western immunoblot analysis
Mouse PPARβ/δ ligand binding domain (LBD) was expressed as a His-tagged fusion in bacteria using standard approaches. The mPPARβ/δLBD-His protein, thus purified, was determined to be of >60 % purity. To test the nature of interaction of 4-HNE with PPARβ/δ, we incubated the PPARβ/δ (containing domains D,E/F) with 100 μM 4-HNE for 24 h in the dark. The mixture was resolved by PAGE, transferred to a polyvinyl pyrrolidone (PVDF) membrane, and probed with a goat polyclonal antibody raised against 4-HNE mixed protein adducts (Northwest Life Sciences, Vancouver, WA) or PPARβ/δ (Santa Cruz). Appropriate secondary antibodies conjugated with HRP were used followed by autoradiography.
Molecular Modeling
Molecular modeling employed Macromodel v. 8.6 and the coordinates of PPARβ/δ bound to eicosapentaenoic acid (PDB ID: 3GWX). The carbonyl of the bound fatty acid was superimposed with the carbonyl of 4-(S)-hydroxynonenal. Ligand and protein residues within 6 Å were minimized to convergence using the OPLS 2003 force field.
RESULTS
We hypothesized that endogenous ligands for PPARβ/δ would include oxidized lipids, such as those derived from VLDL. The VLDL particle is a mixture of free and esterified cholesterols, triglycerides (formed from various fatty acids) and apolipoproteins. As seen in Fig. 1A, the oxidation of VLDL with CuSO4 (oxVLDL) and subsequent incubation with Lipoprotein Lipase (LpL) greatly increased PPARβ/δ activity in a dose-dependent manner (Fig. 1B). To determine which potential products released by oxVLDL are playing a role in the enhancement of PPARβ/δ activity, we conducted a screen of fatty acids and their CuSO4 oxidation products. In Fig. 1C we show that, while 13(S)-HODE alone activates PPARβ/δ, its oxidation product increases activity roughly two-fold higher; 15-HETE was also a PPARβ/δ agonist. Both 13(S)-HODE and its hydroperoxy derivative, 13(S)-HpODE, were equally potent in their ability to activate PPARβ/δ (data not shown). Furthermore, we hypothesized that 13(S)-HODE is a component of the oxVLDL particle which is hydrolyzed upon incubation with LpL and ultimately induces PPARβ/δ activity. Our results show that the release of 13(S)-HODE increased correspondingly with the amount of oxVLDL used (Fig. 1C, Inset).
Fig 1.
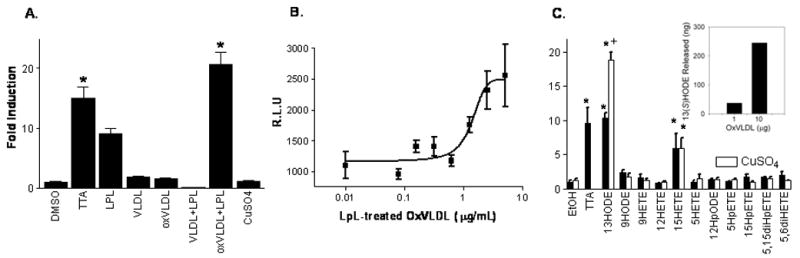
(A) Oxidation of VLDL leads to increased activation of PPARβ/δ. VLDL (10μg/ml) was oxidized with CuSO4 (10μM) for 72 h, then pretreated with lipoprotein lipase (LpL) for 1 h at 37°C, and the mixture was added to 3T3-L1 cells transfected with GAL4-PPARβ/δ-LBD and the UAS-luciferase constructs. Following treatment the cells were lysed and relative luciferase activity was determined. Results were standardized to the average DMSO-treated samples (n=8, representative of two independent experiments). (B) Dose-dependent activation of PPARβ/δ by LPL-treated oxVLDL. 3T3-L1 preadipocytes were transfected with Gal4-LBD-mPPARβ and treated overnight with oxidized VLDL incubated for 1 h with LpL. The cells were lysed and relative luciferase activity was determined. (n = 8, representative of at least three independent experiments). (C). Oxidized 13(S)-HODE is a strong PPARβ/δ agonist. Chemicals were obtained as described in Methods. UV-Visible spectroscopy was used to determine their respective concentrations, and the original solvent was evaporated under a stream of N2 and the chemical reconstituted in ethanol to 50 μM. Oxidation products were obtained via incubation with CuSO4 (10 μM) for 72 h. 3T3-L1 preadipocytes were transfected with Gal4-LBD-mPPARβ/δ and UAS-luciferase constructs before being treated with compound overnight. The cells were lysed and relative luciferase activity was determined. Results were normalized to EtOH/H2O. N = 3. *P<0.05 Significantly different than the EtOH control and +is significantly different from the unoxidized counterpart; (Inset) Oxidation of VLDL releases 13(S)-HODE. VLDL was oxidized using 10 μM CuSO4 for 72 h. Oxidized VLDL was then incubated with LpL for 1 h. An ELISA specific for 13(S)-HODE (Assay Designs) was performed to quantify the amount hydrolyzed from oxVLDL. Samples were prepared by diluting 1, and 10 μg of oxVLDL with standard diluent to a final volume of 100 μL (n = 2).
Based on the increased activity observed following CuSO4 treatment of lipids, we suspected that 4-HNE or other aldehydic oxidation products formed via a Hock Cleavage mechanism from 13(S)-HODE or 13(S)-HpODE were PPARβ/δ ligands. A structure-activity relationship was established using a representative compound from each family of lipid peroxidation product, 2-alkenals, 4-hydroxy-2-alkenals, and ketoaldehydes (Fig. 2A). Reporter assays clearly showed that 4-HNE enhanced the activity of the PPARβ/δ subtype while 4-HHE, 4-ONE, and trans-4,5-epoxy-2(E)-decenal did not activate this receptor (Fig. 2B). Periods of oxidative stress initiate free-radical mediated oxidative damage of lipids, such as 13(S)-HODE. 4-ONE results in lipid peroxidation and damage, but did not activate this receptor, showing that an indirect oxidative stress response is not responsible for activating PPARβ/δ. The breakdown pathway from lipid to 4-HNE includes a hydroperoxy-derivative, 4-hydroperoxynonenal (4-HpNE), which is rapidly reduced to 4-HNE by cellular peroxidases. In fact, 4-HNE activated PPARβ/δ to a greater extent than its precursor (Fig. 2C) and did so in a dose-dependent manner (Fig. 2D, EC5016 μM); while cellular toxicity was observed at concentrations higher than 30 μM.
Fig 2.
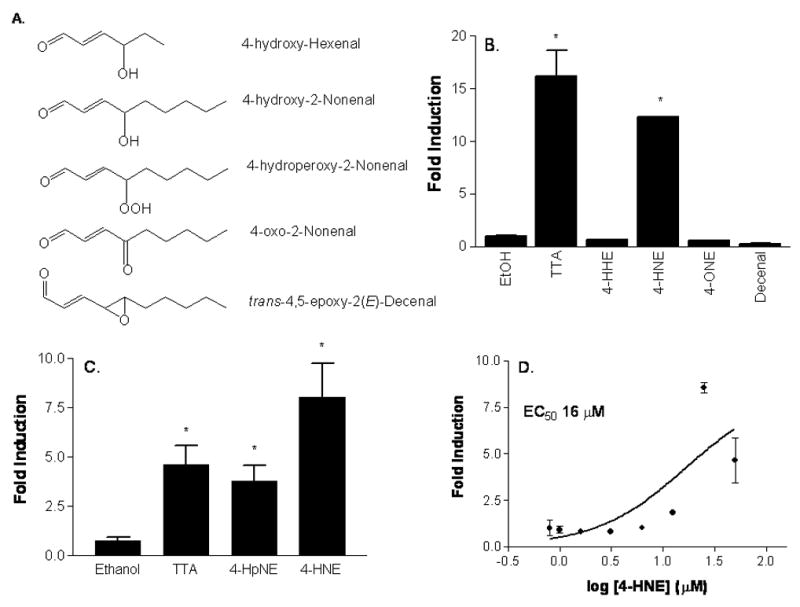
(A) Structures of 4-HNE and other test molecules examined. (B) PPARβ/δ is activated by 4-HNE. 3T3-L1 preadipocytes were transfected with Gal4-LBD-mPPARβ/δ and UAS-luciferase constructs followed by treatment with each of the lipid peroxidation products or vehicle control for 12 h. Following the treatment, cells were lysed and luciferase activity was determined and normalized to EtOH. Treatment was with 25 μM 4-HHE, 4-HNE, 4-ONE and trans-4,5-epoxy-2E-decenal (Decenal; Cayman) overnight. *P<0.05 compared to vehicle control. (C) HpNE activates PPARβ/δ. Cells were treated with 25 μM 4-HpNE or 4-HNE for 12 h as described above. *P<0.05 compared to vehicle control. (D) Dose-response of PPARβ/δ activation by 4-HNE. PPARβ/δ activity is increased with addition of 25 μM 4-HNE, while cellular toxicity is observed at higher concentrations. n = 8. *P<0.05 compared to vehicle control.
In order to analyze if 4-HNE affected gene expression or toxicity in a PPARβ/δ-dependent manner, studies were performed using murine SV40-immortalized hepatocytes prepared from wild-type (MuSH WT) and PPARβ/δ−/− (MuSHβ/δ−/−) mice [21]. The expression of a PPARβ/δ-regulated gene, adipose differentiation-related protein (ADRP [16]), was up-regulated in a PPARβ/δ-dependent manner in cells treated with 13(S)-HODE and 4-HNE (Fig. 3A). Importantly, the cells lacking PPARβ/δ were more susceptible to 4-HNE cytotoxicity than the wild-type cells (Fig. 3B). The EC50 value for MuSH WT cells (68 μM) was approximately 10-times higher than that observed for the MuSHβ/δ−/− cells (5 μM). Also, the MuSHβ/δ−/− cells showed overt toxicity at lower concentrations of sodium arsenite than their wildtype counterpart (Fig. 3C) thus confirming the increased hepatotoxicity observed in vivo in the PPARβ/δ−/− mouse model (data not shown). To further examine the role of PPARβ/δ activity on conferring resistance to 4-HNE cytotoxicity, immortalized hepatocytes (MuSH WT) were pretreated with either a PPARβ/δ agonist tetradecylthioacetic acid (TTA) or PPAR antagonist GW9662 (at concentrations known to inhibit PPARβ/δ [17]) prior to 4-HNE administration. Pretreating hepatocytes with TTA resulted in a significant protection from 4-HNE-depdendent toxicity (Fig. 3D). Conversely, treating MuSH WT cells with GW9662 increased sensitivity to this toxic lipid (Fig. 3E). This data clearly shows a role for PPARβ/δ in cytoprotection against at least one aldehydic toxic lipid (4-HNE) and a xenobiotic which mediates its toxicity at least in part via oxidative mediators (As). Also, this raises the possibility that one can augment PPARβ/δ activity in order to protect cells from the damaging affects of 4-HNE.
Fig. 3.
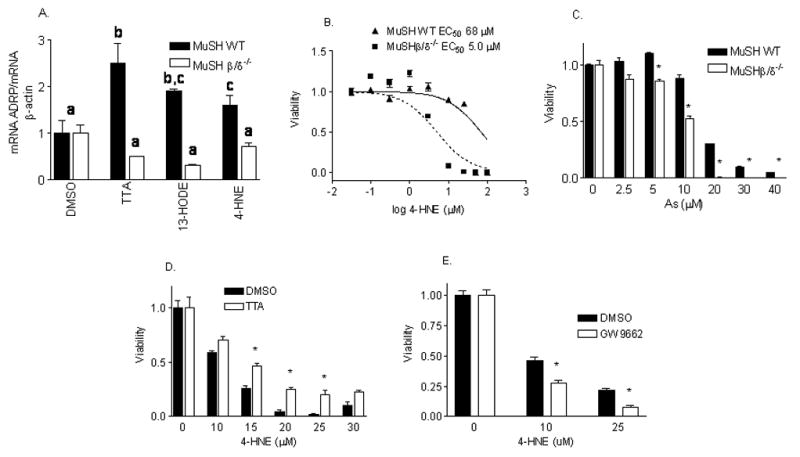
(A) 4-HNE regulates gene expression in a PPARβ/δ-dependent manner. MuSH WT and MuSHβ/δ−/− cells were treated with 13(S)-HODE and 4-HNE overnight and the cells were lysed. Total RNA was extracted using Tri-Reagent and RT-PCR performed for ADRP and corrected for β-actin expression. N = 3. ANOVA with Fishers multiple comparisons test was performed in Minitab (State College, PA). Bars with different letters are significantly different (P<0.05). (B) PPARβ/δ is crucial in ameliorating the toxic effects of 4-HNE. MuSHWT and MuSHβ/δ−/− hepatocytes were treated with 0, 0.1, 0.3, 1, 3, 10 or 25 μM 4-HNE for 24 h. Hepatocytes treated with concentrations of 50 μM or higher 4-HNE do not survive, regardless of genotype. The data is normalized for % viable cells, shown on the y-axis. N = 8. Representative of six independent experiments. (C). PPARβ/δ−/− cells are more sensitive to arsenic induced toxicity. MuSHWT and MuSHβ/δ−/− hepatocytes were treated graded concentrations of sodium arsenite for 24 h. The data is normalized for % viable cells, shown on the y-axis. n = 5. Representative of two independent experiments. *P<0.05 compared with MuSH PPARβ/δ−/− hepatocytes. (D). Activation of PPARβ/δ leads to protection from oxidative damage. MuSHWT cells were pre-treated with vehicle or TTA (25 μM) for 6 h prior to treatment with 4-HNE. n= 5. *P<0.05 compared to vehicle-treated counterpart. (E). Inhibition of PPARβ/δ leads to increased susceptibility to oxidative damage. MuSHWT cells were pre-treated with vehicle or GW9662 (10 μM) for 6 h prior to treatment with 4-HNE. n = 5. *P<0.05 compared to vehicle-treated counterpart.
Gene expression affected by PPARβ/δ activation was examined by gene expression microarray. Of the 6800 unique genes on the arrays, 1001 gave a reliable signal in all three slides. The determination of statistically significant regulated genes was examined as we have previously reported [19, 21]. Using this conservative measure of significant (p<0.05 and a twofold change in gene expression), 41 genes were regulated in a PPARβ/δ manner in MuSH WT cells, in this case all were increased in levels relative to controls (Table 1). The biotransformation of 4-HNE to less toxic intermediates can be accomplished by oxidative (aldehyde dehydrogenase; ALDH), reductive (alcohol dehydrogenase; ADH), and conjugative (glutathione S-transferase; GST) pathways [24]. Following gene expression microarray experiments (see Table 1), we found activation of PPARβ/δ increased mRNA for Aldh3a1 (22-fold increase), Gstm3 (2.3-fold increase) and Gsto1 (2.0-fold increase). This suggests that PPARβ/δ activation is capable of increasing the detoxification of 4-HNE, although this will require formal confirmation.
Table 1.
Microarray analysis of gene expression: Genes statistically regulated by PPARβ/δ in MuSH hepatocytes
| Systematic | Genbank | Common | Description | Ratio | P-value |
|---|---|---|---|---|---|
| M001480_01 | U57368 | Ddx26 | DEAD/H (Asp-Glu-Ala-Asp/His) box polypeptide 26 | 24.60 | 0.03 |
| M001508_01 | U12785 | Aldh3a1 | aldehyde dehydrogenase family 3, subfamily A1 | 22.00 | 0.04 |
| M001584_01 | Z49916 | Clcn4-2 | chloride channel 4-2 | 7.95 | 0.04 |
| M002685_01 | L09737 | Gch | GTP cyclohydrolase 1 | 7.61 | 0.04 |
| M001176_01 | L29468 | Cfl2 | cofilin 2, muscle | 5.10 | 0.03 |
| M001458_01 | U10094 | Klra7 | killer cell lectin-like receptor, subfamily A, member 7 | 4.95 | 0.04 |
| M003715_01 | K02108 | Krt2-6a | keratin complex 2, basic, gene 6a | 4.92 | 0.03 |
| M004670_01 | AF144628 | Slit2 | slit homolog 2 (Drosophila) | 4.39 | 0.03 |
| M001078_01 | L43326 | Ktn1 | kinectin 1 | 4.37 | 0.03 |
| M006400_01 | AF233333 | Otor; Fdp; MIA; MIAL; CDRAP | OTOR; Mus musculus otoraplin mRNA, complete cds. | 3.98 | 0.02 |
| M006224_01 | AF155045 | Htr3b | 5-hydroxytryptamine (serotonin) receptor 3B | 3.77 | 0.02 |
| M000902_01 | AB016424 | Rbm3 | RNA binding motif protein 3 | 3.50 | 0.01 |
| M003862_01 | AF142672 | B4galt4 | UDP-Gal:betaGlcNAc beta 1,4-galactosyltransferase, polypeptide 4 | 3.24 | 0.04 |
| M006356_01 | AJ243502 | Facl4 | fatty acid-Coenzyme A ligase, long chain 4 | 2.82 | 0.02 |
| M005257_01 | AL078630 | Mouse DNA sequence from clone CT7-BM573K1 on chromosome 17, complete sequence. | 2.81 | 0.03 | |
| M006846_01 | U83174 | Gt(ROSA)26Sor | gene trap ROSA 26, Philippe Soriano | 2.74 | 0.04 |
| M002404_01 | L11065 | Oprk1 | opioid receptor, kappa 1 | 2.72 | 0.04 |
| M001253_01 | NM_010921 | Nkx3-1 | NK-3 transcription factor, locus 1 (Drosophila) | 2.56 | 0.04 |
| M005466_01 | AF071180 | Fpr-rs2 | FPRL2; Mus musculus N-formylpeptide receptor-like 2 gene, complete cds. | 2.47 | 0.04 |
| M001767_01 | Y00769 | Itgb1 | integrin beta 1 (fibronectin receptor beta) | 2.33 | 0.01 |
| M003686_01 | X17459 | Rbpsuh | recombining binding protein suppressor of hairless (Drosophila) | 2.32 | 0.04 |
| M001250_01 | U96701 | Spi15 | serine protease inhibitor 15 | 2.32 | 0.01 |
| M004771_01 | J03953 | Gstm3 | glutathione S-transferase, mu 3 | 2.31 | 0.00 |
| M001752_01 | X14194 | Nid1 | nidogen 1 | 2.25 | 0.05 |
| M001676_01 | M27073 | Ppp1cb | protein phosphatase 1, catalytic subunit, beta isoform | 2.25 | 0.01 |
| M004895_01 | AB032602 | Cntn6 | contactin 6 | 2.23 | 0.02 |
| M001746_01 | L08266 | Fancc | Fanconi anemia, complementation group C | 2.23 | 0.02 |
| M000365_01 | J03168 | Irf2 | interferon regulatory factor 2 | 2.22 | 0.00 |
| M002136_01 | M19911 | Gm1067 | precursor; Mouse Ig rearranged kappa-chain mRNA, clone AN08K. | 2.18 | 0.05 |
| M000886_01 | L31783 | Umpk | uridine monophosphate kinase | 2.17 | 0.02 |
| M002704_01 | U97327 | Cacybp | calcyclin binding protein | 2.17 | 0.03 |
| M001065_01 | X97042 | Ube2l3 | ubiquitin-conjugating enzyme E2L 3 | 2.15 | 0.02 |
| M000012_01 | M75953 | Tlx2 | T-cell leukemia, homeobox 2 | 2.13 | 0.02 |
| M003072_01 | X59379 | App | amyloid beta (A4) precursor protein | 2.11 | 0.02 |
| M003454_01 | AF111172 | Cln2 | ceroid-lipofuscinosis, neuronal 2 | 2.10 | 0.01 |
| M006504_01 | AF096288 | Ucp2 | Mus musculus uncoupling protein-2 (Ucp2) gene, nuclear gene encoding mitochondrial protein, complete cds. | 2.10 | 0.02 |
| M006374_01 | AF257520 | Raet1d | retinoic acid early transcript delta | 2.10 | 0.01 |
| M003956_01 | U47008 | Nab1 | Ngfi-A binding protein 1 | 2.07 | 0.05 |
| M000086_01 | U80819 | Gsto1 | glutathione S-transferase omega 1 | 2.05 | 0.02 |
| M006838_01 | L11316 | Ect2 | ect2 oncogene | 2.02 | 0.04 |
| M004779_01 | AB030279 | Pign | phosphatidylinositol glycan, class N | 2.02 | 0.03 |
An interaction with a nuclear receptor and 4-HNE has not been described previously and was examined in more detail using molecular modeling and the coordinates of PPARβ/δ bound to eicosapentaenoic acid (Fig. 4A [25]). PPARβ/δ bound to (S)-4-HNE (CPK model) in a conformation similar to the natural ligand (Fig. 4B). However, a putative hydrogen-bonding interaction between His413 (stick model) and the 4-hydroxyl group of the 4-HNE ligand (CPK model) occurs (Fig. 4C). As described above, PPARβ/δ was activated by 4-HpNE and 4-HNE but not 4-ONE, 4-HHE or decanol. Thus, the association between the hydroxyl group of 4-HNE and His413 is necessary for optimal receptor activation by this class of compound. In data not shown, the fatty acids nonenol and EPA activated PPARβ/δ to a similar extent as 4-HNE. This suggests that the interaction of carboxylic acids with PPARβ/δ proceeds by a different mechanism, one that would not require His413, but is still capable of activating the receptor. Several attempts were made at mutating His413 to either alanine or phenylalanine for a formal testing of the molecular modeling. However, the basal activity or that seen in the presence of PPARβ/δ ligands by these mutants was very low, suggesting that the His residue is critical for proper expression or folding. Although 4-HNE forms covalent adducts with cysteinyl, lysyl, and histidyl of target proteins, we were unable to show covalent interactions between 4-HNE and PPARβ/δ (Fig. 4D and E), strongly suggesting that the association with PPARβ/δ is reversible.
Fig. 4.
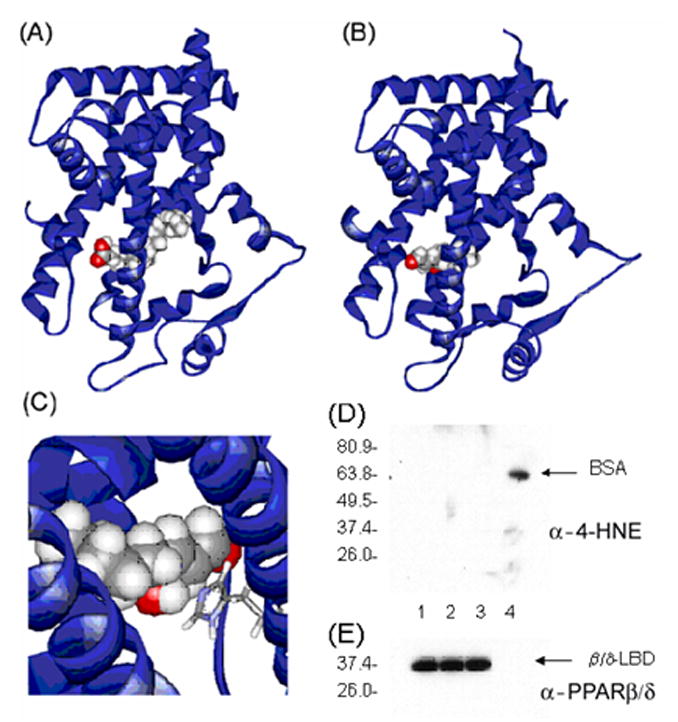
(A) X-ray structure of PPARβ/δ bound to eicosapentaenoic acid. Model is based on coordinates from [25]. (B) A molecular model of PPARβ/δ bound to (S)-4-hydroxynonenal (CPK model) in a conformation similar to the natural ligand. (C) Visualization of the putative hydrogen-bonding interaction between His413 (stick model) and the 4-hydroxyl group of the ligand (CPK model). (D) 4-HNE activates PPARβ/δ via a direct and non-covalent interaction. Mouse His-tagged PPARβ/δ LBD was expressed in bacteria and purified using a Nickel column and incubated with 4-HNE. Immunoblot analysis was used to determine 4-HNE-protein adducts. Lane 1 PPARβ/δ-LBD alone; Lane 2, PPARβ/δ-LBD plus EtOH; Lane 3, PPARβ/δ-LBD plus 4-HNE (100 μM); Lane 4, BSA plus 4-HNE (100 μM). (E) Identical incubation as performed above with immunoblot analysis of PPARβ/δ.
DISCUSSION
The PPARs are ligand-activated transcription factors that sense a variety of lipophilic molecules and control gene expression to maintain cellular homeostasis. Fatty acids and their metabolites are known endogenous agonists of all PPARs, with PPARβ/δ exhibiting similar structural and geometric preference as PPARα, whereas PPARγ tends to prefer long-chain polyunsaturated fatty acids [26]. PPARβ/δ agonists include linoleic acid, oleic acid, arachidonic acid and EPA, which has been co-crystallized within the ligand binding domain of this nuclear receptor [25]. Prostaglandin A1 (PGA1), PGD2 and PGD1 can activate PPARβ/δ in reporter assays [27]. Similar to PPARα and γ, incubation of triglyceride rich lipoproteins with LPL results in the production of PPARβ ligands [28, 29]. We have shown here that several molecules present in oxVLDL including 15-HETE, 13(S)-HODE and 4-HNE are agonists of PPARβ/δ (Fig. 1 and 2). Activation of PPARα and PPARγ by 13(S)-HODE has been observed previously [30], although this molecule has been reported to be an inhibitor of PPARβ/δ in colon epithelial cells [31]. Our data indicating 13(S)-HODE being a PPARβ/δ agonist in preadipocytes may suggest cell-type specific phenomena (Fig. 2). We have further examined the products of 13(S)-HODE, via the Hock Cleavage mechanism, which forms such alkenals as HpNE and HNE, to activate PPARβ/δ. We clearly demonstrate that of the several lipid peroxidation products, only 4-HNE, and to a lesser extent, its hydroperoxy-derivative, are capable of activating the PPARβ/δ receptor in transient transfection reporter assays. However, HpNE may be a relatively unstable molecule since it can be reduced to the corresponding alcohol by cellular peroxidases, including the selenium-dependent and independent GSH-peroxidases. Although it has been demonstrated that 4-HNE modulates up-regulation of PPARγ expression by influencing upstream kinases and troglitazone-dependent activity [32], this is the first instance of the lipid peroxidation product, 4-HNE, being described as a PPARβ/δ agonist. Coupled with the fact that PPARβ/δ is expressed virtually ubiquitously and is known to be a regulatory of several important physiological pathways makes this observation intriguing. Although the concentration of 4-HNE used in this investigation is higher than the “physiological” concentrations in several tissues and cells [33], it is very likely that high localized concentrations of 4-HNE may be formed within cells [34, 35] that could be sufficient to activate this nuclear receptor.
Examination of the PPARβ/δ null mice has shown a role for this receptor in several biological niches [36]. There is mounting evidence that PPARβ/δ plays a role in ameliorating the hepatotoxic effects of a wide range of xenobiotics including arsenic and acetaminophen (APAP). Interestingly, a similar hepatoprotective role has been noted for PPARα [37–39] and PPARγ [40] agonists, although the mechanism of this response is not clear. Studies performed with hepatocytes from wild type and PPARβ/δ−/− mice clearly demonstrate that PPARβ/δ plays a pivotal role in ameliorating oxidative stress induced by the hepatoxicant arsenic (Fig. 3C) which originates through free radical reactions and subsequent initiation of lipid peroxidation (reviewed in [11, 41]). This data supports our hypothesis that activation of PPARβ/δ by endogenous ligand such as 4-HNE may play a major role in the prevention of pathogenesis of both acute and chronic liver damage. This is further illustrated in Fig. 3D,E, where 4-HNE addition to TTA-activated MuSH wild-type cells are more resistant to oxidative stress compared to the ones that are treated with the antagonist. This is particularly relevant in since the enzymatic oxygenation of arachidonic acid by the cyclooxygenase (COX), an enzyme commonly induced by hepatotoxicants, may cause the release of reactive oxygen species (ROS) during the enzymatic reduction of hydroperoxides. The release of ROS may initiate a chain of responses that results in lipid mediators including 4-HNE with subsequent protein and DNA damage. In addition to formation of ROS and lipid peroxides, COX- and lipoxygenase (LOX)-mediated fatty acid metabolism leads to the formation of various prostaglandins and other lipid mediators depending on the various isomerases and reductases present in the in a tissue-specific manner. These molecules are potent bioactive compounds in vivo [11] and may contribute directly or indirectly to liver damage. The amount of lipid substrate, the activity of phospholipase A2 (PLA2), COX, LOX, CYP as well as various antioxidant defense mechanisms are all dynamically regulated and will ultimately affect the damage caused by a hepatotoxicant.
Thus, there are many means by which PPARβ/δ may be protecting the cell from oxidative damage, either by inhibiting the production of ROS and lipid intermediates or by increasing degradation of these molecules. Based on gene expression data performed herein and elsewhere [16], it appears that the most likely candidate would be PPARβ/δ-dependent metabolism and detoxification of lipid intermediates. The biotransformation of 4-HNE to less toxic intermediates can be accomplished aldehyde and alcohol dehydrogenases, glutathione S-transferases and perhaps other pathways [24]. Following gene expression microarray experiments, we found activation of PPARβ/δ increased mRNA for Aldh3a1, Gstm3 and Gsto1. Other microarray experiments have identified GSTA1 and ALDH9A1 as potential PPARβ/δ targets as well [16]. The induction of Aldh3a1 and GSTo1 are particularly intriguing due to their known role in protection against lipid peroxidation and toxicity [24]. Similar to the PPARβ/δ−/− mouse, mice that lack the transcription factor Nrf2 are more sensitive to the cytotoxic and genotoxic effects of foreign chemicals due to attenuated induction of Gsta1, Gstm1 and Gstm3 [42]. Thus, it is possible that these two transcription factors are sharing a common protective mechanism of action. However, in addition to being directly involved in the regulation of expression for these detoxification enzymes, PPARβ/δ is also sensing, or responding to, the intracellular levels of the toxic intermediate via a feed-back control mechanism.
There is a wealth of information on 4-HNE as being a marker of lipid peroxidation as well as a mediator of hepatotoxicity and a variety of other pathophysiological conditions including cancer, Alzheimer’s disease and atherosclerosis [35]. Target proteins that are inhibited by 4-HNE include the glucose transporter GLUT3, tau, the proteasome and IκB kinase [35] and 4-HNE causes a high frequency of G-to-T mutations in codon 249 in the p53 gene [43]. Based on the molecular modeling and Western immunoblot analyses, the covalent association of 4-HNE with cysteinyl, lysyl, and histidyl residues in PPARβ/δ appears to be largely reversible (Fig. 4), which is unique since 4-HNE is known to covalently modify these residues via Michael addition chemistry. In summary, we have shown that the lipid peroxidation product 4-HNE, which was regarded to be a mutagen and toxic end-product for all these years, has now been demonstrated to have a nuclear receptor modulatory activity. This receptor, PPARβ/δ, when activated provides a feed-back regulation of gene expression that ameliorates toxicity to the liver. The discovery of 4-HNE as an endogenous ligand reveals a crosstalk of PPARβ/δ in cellular events involved in inflammation and energy homeostasis. Given the large number of individuals hospitalized due to chemically-induced liver damage, it is attractive to speculate that this knowledge may be exploited for more effective means of protecting or treating hepatotoxicity as well as other diseases in which 4-HNE plays a causative role.
Acknowledgments
This research was supported by the Penn State University competitive grants program sponsored by the Huck Institute of Life Sciences and the Institutes of the Environment (to J.V.H., J.M.P and K.S.P.). Portions of this research were presented at the Society of Toxicology annual meeting in San Diego, CA.
Abbreviations
- 4-HNE
4-hydroxynonenal
- 13-S-HODE
13-S-hydroxyoctadecadienoic acid
- PPARs
peroxisome proliferator-activated receptors
- (ROI)
Reactive oxygen intermediates
- MDA
malondialdehyde
- LpL
Lipoprotein Lipase
Footnotes
Shan, W. et al manuscript in preparation
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee WM, Senior JR. Recognizing drug-induced liver injury: current problems, possible solutions. Toxicol Pathol. 2005;33:155–164. doi: 10.1080/01926230590522356. [DOI] [PubMed] [Google Scholar]
- 2.Lee WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology. 2004;40:6–9. doi: 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- 3.Comporti M. Lipid peroxidation and biogenic aldehydes: from the identification of 4-hydroxynonenal to further achievements in biopathology. Free Radic Res. 1998;28:623–635. doi: 10.3109/10715769809065818. [DOI] [PubMed] [Google Scholar]
- 4.Vanden Heuvel JP. Diet, fatty acids, and regulation of genes important for heart disease. Curr Atheroscler Rep. 2004;6:432–440. doi: 10.1007/s11883-004-0083-9. [DOI] [PubMed] [Google Scholar]
- 5.Khan SA, Vanden Heuvel JP. Role of nuclear receptors in the regulation of gene expression by dietary fatty acids (review) J Nutr Biochem. 2003;14:554–567. doi: 10.1016/s0955-2863(03)00098-6. [DOI] [PubMed] [Google Scholar]
- 6.Peters JM, Vanden Heuvel JP. Peroxisomes, peroxisome proliferators and peroxisome proliferator-activated receptors (PPARs) In: Vanden Heuvel JP, Perdew GH, Mattes WB, Greenlee WF, editors. Cellular and Molecular Toxicology. Amsterdam, The Netherlands: Elsevier Science; 2002. pp. 133–158. [Google Scholar]
- 7.Vanden Heuvel JP, Peters JM. Biology of the PPAR family of receptors. Comments Toxicol. 2001;7:233–257. [Google Scholar]
- 8.Vanden Heuvel JP. Peroxisome proliferator-activated receptors (PPARS) and carcinogenesis. Toxicol Sci. 1999;47:1–8. doi: 10.1093/toxsci/47.1.1. [DOI] [PubMed] [Google Scholar]
- 9.Vanden Heuvel JP. Peroxisome proliferator-activated receptors: a critical link among fatty acids, gene expression and carcinogenesis. J Nutr. 1999;129:575S–580S. doi: 10.1093/jn/129.2.575S. [DOI] [PubMed] [Google Scholar]
- 10.Burdick AD, Kim DJ, Peraza MA, Gonzalez FJ, Peters JM. The role of peroxisome proliferator-activated receptor-beta/delta in epithelial cell growth and differentiation. Cell Signal. 2006;18:9–20. doi: 10.1016/j.cellsig.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 11.Basu S. Carbon tetrachloride-induced lipid peroxidation: eicosanoid formation and their regulation by antioxidant nutrients. Toxicology. 2003;189:113–127. doi: 10.1016/s0300-483x(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 12.Kawada N, Mizoguchi Y, Kobayashi K, Yamamoto S, Morisawa S. Arachidonic acid metabolites in carbon tetrachloride-induced liver injury. Gastroenterol Jpn. 1990;25:363–368. doi: 10.1007/BF02779452. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Parra M, Claria J, Planaguma A, Titos E, Masferrer JL, Woerner BM, Koki AT, Jimenez W, Altuna R, Arroyo V, Rivera F, Rodes J. Cyclooxygenase-1 derived prostaglandins are involved in the maintenance of renal function in rats with cirrhosis and ascites. Br J Pharmacol. 2002;135:891–900. doi: 10.1038/sj.bjp.0704528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartley DP, Kolaja KL, Reichard J, Petersen DR. 4-Hydroxynonenal and malondialdehyde hepatic protein adducts in rats treated with carbon tetrachloride: immunochemical detection and lobular localization. Toxicol Appl Pharmacol. 1999;161:23–33. doi: 10.1006/taap.1999.8788. [DOI] [PubMed] [Google Scholar]
- 15.Hartley DP, Kroll DJ, Petersen DR. Prooxidant-initiated lipid peroxidation in isolated rat hepatocytes: detection of 4-hydroxynonenal- and malondialdehyde-protein adducts. Chem Res Toxicol. 1997;10:895–905. doi: 10.1021/tx960181b. [DOI] [PubMed] [Google Scholar]
- 16.Tachibana K, Kobayashi Y, Tanaka T, Tagami M, Sugiyama A, Katayama T, Ueda C, Yamasaki D, Ishimoto K, Sumitomo M, Uchiyama Y, Kohro T, Sakai J, Hamakubo T, Kodama T, Doi T. Gene expression profiling of potential peroxisome proliferator-activated receptor (PPAR) target genes in human hepatoblastoma cell lines inducibly expressing different PPAR isoforms. Nucl Recept. 2005;3:3. doi: 10.1186/1478-1336-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seimandi M, Lemaire G, Pillon A, Perrin A, Carlavan I, Voegel JJ, Vignon F, Nicolas JC, Balaguer P. Differential responses of PPARalpha, PPARdelta, and PPARgamma reporter cell lines to selective PPAR synthetic ligands. Anal Biochem. 2005;344:8–15. doi: 10.1016/j.ab.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 18.Chang M, Rao MK, Reddanna P, Li CH, Tu CP, Corey EJ, Reddy CC. Specificity of the glutathione S-transferases in the conversion of leukotriene A4 to leukotriene C4. Arch Biochem Biophys. 1987;259:536–547. doi: 10.1016/0003-9861(87)90520-0. [DOI] [PubMed] [Google Scholar]
- 19.Tien ES, Davis JW, Vanden Heuvel JP. Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated receptor alpha coregulator. J Biol Chem. 2004;279:24053–24063. doi: 10.1074/jbc.M401489200. [DOI] [PubMed] [Google Scholar]
- 20.Chou JY. Establishment of rat fetal liver lines and characterization of their metabolic and hormonal properties: use of temperature-sensitive SV40 virus. Methods Enzymol. 1985;109:385–396. doi: 10.1016/0076-6879(85)09104-2. [DOI] [PubMed] [Google Scholar]
- 21.Tien ES, Gray JP, Peters JM, Vanden Heuvel JP. Comprehensive gene expression analysis of peroxisome proliferator-treated immortalized hepatocytes: identification of peroxisome proliferator-activated receptor alpha-dependent growth regulatory genes. Cancer Res. 2003;63:5767–5780. [PubMed] [Google Scholar]
- 22.Burdick AD, Kim DJ, Peraza MA, Gonzalez FJ, Peters JM. The role of peroxisome proliferator-activated receptor-beta/delta in epithelial cell growth and differentiation. Cell Signal. 2005 doi: 10.1016/j.cellsig.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Michalik L, Desvergne B, Tan NS, Basu-Modak S, Escher P, Rieusset J, Peters JM, Kaya G, Gonzalez FJ, Zakany J, Metzger D, Chambon P, Duboule D, Wahli W. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J Cell Biol. 2001;154:799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Awasthi YC, Sharma R, Cheng JZ, Yang Y, Sharma A, Singhal SS, Awasthi S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol Aspects Med. 2003;24:219–230. doi: 10.1016/s0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 25.Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, Kliewer SA, Milburn MV. Molecular recognition of fatty acids by peroxisome proliferator- activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- 26.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 27.Yu K, Bayona W, Kallen CB, Harding HP, Ravera CP, McMahon G, Brown M, Lazar MA. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J Biol Chem. 1995;270:23975–23983. doi: 10.1074/jbc.270.41.23975. [DOI] [PubMed] [Google Scholar]
- 28.Ziouzenkova O, Perrey S, Asatryan L, Hwang J, MacNaul KL, Moller DE, Rader DJ, Sevanian A, Zechner R, Hoefler G, Plutzky J. Lipolysis of triglyceride-rich lipoproteins generates PPAR ligands: evidence for an antiinflammatory role for lipoprotein lipase. Proc Natl Acad Sci U S A. 2003;100:2730–2735. doi: 10.1073/pnas.0538015100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chawla A, Lee CH, Barak Y, He W, Rosenfeld J, Liao D, Han J, Kang H, Evans RM. PPARdelta is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci U S A. 2003;100:1268–1273. doi: 10.1073/pnas.0337331100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 31.Shureiqi I, Jiang W, Zuo X, Wu Y, Stimmel JB, Leesnitzer LM, Morris JS, Fan HZ, Fischer SM, Lippman SM. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100:9968–9973. doi: 10.1073/pnas.1631086100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pizzimenti S, Laurora S, Briatore F, Ferretti C, Dianzani MU, Barrera G. Synergistic effect of 4-hydroxynonenal and PPAR ligands in controlling human leukemic cell growth and differentiation. Free Radic Biol Med. 2002;32:233–245. doi: 10.1016/s0891-5849(01)00798-5. [DOI] [PubMed] [Google Scholar]
- 33.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 34.Benedetti A, Comporti M, Fulceri R, Esterbauer H. Cytotoxic aldehydes originating from the peroxidation of liver microsomal lipids. Identification of 4,5-dihydroxydecenal. Biochim Biophys Acta. 1984;792:172–181. doi: 10.1016/0005-2760(84)90219-4. [DOI] [PubMed] [Google Scholar]
- 35.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 36.Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, Reitman ML, Hudson LD, Gonzalez FJ. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta) Mol Cell Biol. 2000;20:5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehendale HM. PPAR-alpha: a key to the mechanism of hepatoprotection by clofibrate. Toxicol Sci. 2000;57:187–190. doi: 10.1093/toxsci/57.2.187. [DOI] [PubMed] [Google Scholar]
- 38.Manautou JE, Hoivik DJ, Tveit A, Hart SG, Khairallah EA, Cohen SD. Clofibrate pretreatment diminishes acetaminophen’s selective covalent binding and hepatotoxicity. Toxicol Appl Pharmacol. 1994;129:252–263. doi: 10.1006/taap.1994.1250. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen KA, Carbone JM, Silva VM, Chen C, Hennig GE, Whiteley HE, Manautou JE. The PPAR activator docosahexaenoic acid prevents acetaminophen hepatotoxicity in male CD-1 mice. J Toxicol Environ Health A. 1999;58:171–186. doi: 10.1080/009841099157377. [DOI] [PubMed] [Google Scholar]
- 40.Kon K, Ikejima K, Hirose M, Yoshikawa M, Enomoto N, Kitamura T, Takei Y, Sato N. Pioglitazone prevents early-phase hepatic fibrogenesis caused by carbon tetrachloride. Biochem Biophys Res Commun. 2002;291:55–61. doi: 10.1006/bbrc.2002.6385. [DOI] [PubMed] [Google Scholar]
- 41.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003;31:1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 42.Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GJ, Itoh K, Yamamoto M, Hayes JD. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J. 2002;365:405–416. doi: 10.1042/BJ20020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu W, Feng Z, Eveleigh J, Iyer G, Pan J, Amin S, Chung FL, Tang MS. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis. 2002;23:1781–1789. doi: 10.1093/carcin/23.11.1781. [DOI] [PubMed] [Google Scholar]