

Abstract
Serum IL-18 responses to LPS mice increase after pretreatment with CpG-containing DNA. Compared to saline-pretreated controls, mice pretreated with CpG for two days produced 20-fold more serum IL-18 when challenged with lipopolysaccharide (LPS). In contrast, IFNγ-deficiency or anti-IFNγ pretreatment reduced CpG-expanded IL-18 responses to LPS by 67% and 83%, respectively. Mice pretreated with either IFNγ or CpG comparably increased LPS-inducible serum IL-18 responses. LPS, compared to challenge with other TLR agonists, was best able to trigger high serum IL-18 levels in CpG-pretreated mice and this response were TLR4-dependent. CpG, compared to pretreatment with other TLR agonists, optimally expanded LPS-induced IL-18 responses that correlated with higher levels of circulating IFNγ levels prior to LPS challenge. High-level serum IL-18 responses were caspase-1-dependent and P2X7 receptor-independent. We conclude that CpG promotes high-level IL-18 synthesis by an IFNγ-dependent and IFNγ-sufficient mechanism in vivo that is optimally triggered by LPS. (150 words)
Keywords: Interleukin-18, Interferon, type II, Toll-Like Receptor 9, Receptors, Purinergic P2, Mice
Introduction
IL-18 was initially identified as an IFNγ-inducing factor in serum that was produced in LPS-injected mice treated with heat-killed Propionibacterium acnes [1]. IL-18 is now known to mediate critical antimicrobial responses in animal models of bacterial, fungal, viral and parasitic infections [2–5]. IL-18 supports both innate and adaptive immune production of IFNγ, while separately promoting IFNγ-independent effects on polymorphonuclear cell function. IL-18 synthesis is triggered by innate immune recognition of microbial products, such as lipopolysaccharide, which activates IL-18 release from murine macrophages or Kupffer cells through TLR4-dependent and Myd88-independent signaling pathways [6]. Cytoplasmic stores of pro-IL-18 peptide are proteolytically modified by caspase-1 to generate bioactive cytokine exported from the cell by a nonclassical, Golgi-independent transport pathway. Signals other than LPS also trigger IL-18 synthesis. For example, C. pneumoniae-induced IL-18 is also produced by Myd88-dependent and TLR4/TLR2-independent pathways [7].
The magnitude of a toll receptor-driven IL-18 response is modulated by several factors. Triggering of the P2X7 receptor by soluble ATP greatly amplifies the amounts of IL-18 and IL-1β released by LPS-exposed macrophages [8, 9]. IL-18 synthetic capacity also progressively increases during repetitive or continuous inflammatory challenges with LPS [2], after pretreatment with Propionibacterium acnes [1] or after treatment with recombinant IL-12 [10]. Previously, we showed that pretreatment of mice with CpG oligonucleotide 1826 for at least two days also dramatically increased IL-18 serum responses in vivo. Levels of IL-18 elicited by LPS in CpG-pretreated mice were 10- to 20-fold greater than in saline-pretreated mice [11]. These expanded serum IL-18 responses subsequently mediated parallel increases in the LPS-induced IFNγ response.
Since CpG stimulates prolonged release of IFNγ in vivo prior to LPS challenge [11, 12] we hypothesized that CpG-induced IFNγ contributed to the expansion of LPS-inducible IL-18 production in vivo. Previously, IFNγ-dependent effects on IL-18 synthesis had been observed in P. acnes -pretreated mice [13]. We show that high level IL-18 production in CpG-pretreated mice not only requires prior IFNγ activity, but that IFNγ alone is sufficient to expand IL-18 production in vivo. We also show that high-level IL-18 responses in CpG-pretreated mice are optimally triggered by TLR4-dependent stimuli, require caspase-1 expression, but are independent of P2X7 receptor expression. These findings demonstrate that IFNγ produced during inflammation dynamically regulates the extent of IL-18 production. The restrictive requirements for CpG and LPS as modulatory and stimulating signals, respectively, for high-level IL-18 synthesis further suggest that sequential TLR9 and TLR4 activation may uniquely program innate cytokine responses to bacterial challenge.
Materials and Methods
Reagents
Salmonella enteriditis lipopolysaccharide was purchased from Sigma Chemical (St Louis, MO). Phosphorothioated CpG 1826 (TCCATGACGTTCCTGACGTT 5′ to 3′) was purchased from Oligos Etc (Wilsonville, OR) and proven free of LPS contamination by limulus lysate assay (E-Toxate, Sigma Chemical Co). Recombinant murine IFNγ was obtained from Peprotech Inc. (Rocky Hill, NJ) and neutralizing anti-mouse IFNγ (XMG1.2; rat IgG1) and irrelevant control rat IgG1 monoclonal antibodies were purchased from Bio Express (West Lebanon, NH). The bacterial lipopeptide, Pam3CysSerLys4 was purchased from EMC Microcollections (Tuebingen, Germany). The TLR7 agonist, R848, was purchased from PharmaTech (Shanghai, China) and poly(IC) was obtained from Amersham Corp (Piscataway, NJ).
Mice
Four to six-week old female C3H/HeN and C3H/HeJ mice were purchased from Charles River (Lexington, MA). C57BL/6 and C57BL/6 IFNγ−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME). C57BL/6 P2X7 receptor knockout (P2X7−/−) mice [14] were provided by Dr. Laurent Audoly (Pfizer, Groton, CT), backcrossed at least 6 generations onto C57BL/6 and maintained at Taconic until shipment. Caspase-1 deficient C57BL/6 mice (ICE KO) were obtained from Dr. Richard Flavell (Yale University, New Haven CT). All mice were housed in the Case Western Reserve University animal facilities under specific pathogen-free conditions. For pretreatment with toll agonists, mice were injected i.p. daily for two days with 0.2 ml of PBS alone or PBS containing 50 ug of CpG OD 1826, 50 ug of Pam3CysSerLys4 or 20 ug of R848. At 72 hours after the first conditioning dose, mice were challenged by i.p. injection with 300 ug of LPS (LD90). Because CpG-preconditioned mice developed signs of severe toxicity at six hours after LPS challenge, mice were all euthanized before six hours post-challenge. Where indicated, mice were treated with 20 ug of recombinant mouse IFNγ daily for two days. Selected mice were injected with 0.5 mg of XMG1.2 anti-IFNγ IgG i.p. 4 hours or irrelevant rat IgG1 prior to initial pretreatment with CpG. All procedures were approved by the CWRU IACUC.
ELISA assays
Cytokine concentrations in serum and conditioned culture media were determined for IFN-γ using antibody pairs from BD-Pharmingen, as described [11] Serum IL-18 was measured using an ELISA from Medical and Biological Laboratories International (Woburn, MA) that detects mature mouse IL-18 with minimal cross-reactivity to precursor forms of IL-18. Mouse IFNγ and IL-1β were assayed using ELISA reagents from Pharmingen-BD (San Diego, CA).
Statistics
Tests for significance of difference were made using the Student parametric t-test.
Results
CpG pretreatment expands LPS-inducible IL-18 synthesis by an IFNγ-dependent mechanism
We confirmed that pretreatment of C3H/HeN mice with 50 ug of CpG for two days resulted in 10 to 20-fold increases in LPS-induced serum IL-18 [11], relative to saline-pretreated mice. These responses peaked between 3 and 5 hours after LPS challenge in both groups (Figure 1A). Because we previously observed circulating IFNγ after CpG pretreatment, we tested whether IFNγ exposure prior to LPS challenge regulated IL-18 synthetic capacity (Figure 1B). Compared to PBS-pretreated controls, CpG-pretreated mice produced 10-fold more LPS-inducible serum IL-18. However, CpG-pretreated IFNγ−/− mice generated IL-18 concentrations that were only one-third of that produced by pretreated IFNγ-competent mice (significant at p < 0.05). Similarly, combined pretreatment of wild-type C57Bl/6 mice with anti-IFNγ monoclonal antibody four hours prior to the first CpG pretreatment also reduced the LPS-induced serum IL-18 response to 17% of that of CpG-pretreated controls (p < 0.05), although these remained greater than those of PBS-pretreated control mice. In contrast, IFNγ was not necessary to support IL-18 production in PBS-pretreated C57BL/6 after LPS challenge, as shown by comparable increases in wild-type and IFNγ−/− mice (2.14 ± 0.53 and 2.59 ± 0.85 ng/ml, respectively) relative to normal serum controls (0.23 ± 0.03 ng/ml, p < 0.05). A repeat experiment in CpG-pretreated C3H/HeN mice confirmed that XMG1.2 co-pretreatment reduced maximal IL-18 responses by 84%, compared to pretreatment with CpG and irrelevant rat IgG1 antibody (Figure 1C).
Figure 1. Interferon-gamma is both necessary and sufficient to amplify LPS-inducible IL-18 production in vivo.
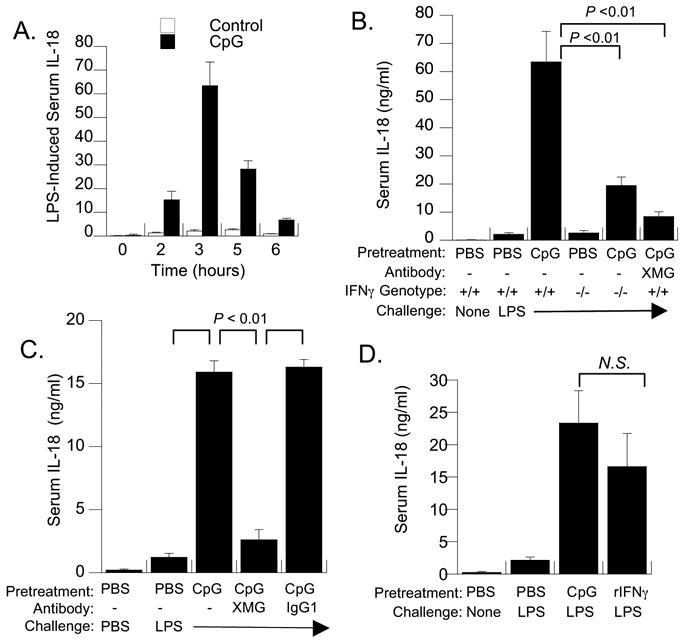
(A) Kinetics of IL-18 release into serum after endotoxin challenge. C3H/HeN mice were pretreated with PBS or CpG (50 ug) spaced 24 hours apart, followed by i.p. challenge with LPS (300 ug). Serum was obtained from groups of 4 to 6 mice at the indicated times after LPS challenge. Shown are the mean and standard error of the mean. CpG resulted in significantly greater IL-18 responses compared to PBS-pretreatment at 2, 3 and 5 hours (p < 0.05; Student T test). (B) Wild-type (+/+) and IFNγ (−/−) C57Bl/6 mice (n = 5) were pretreated with PBS or CpG as described above and mean concentrations of serum IL-18 ± SEM determined at 3 hours post-LPS challenge. A separate group of wild-type C57BL/6 mice were pretreated with neutralizing anti-IFNγ IgG XMG1.2 (XMG). PBS-preconditioned sera contained 155 ± 11 pg/ml of IL-18 in the absence of LPS challenge, which was significantly reduced relative to LPS-stimulated levels in all groups (p < 0.05). Brackets indicate significant decreases in serum IL-18 in CpG-pretreated IFNγ (−/−) or anti-IFNγ-pretreated mice compared to wild-type controls. (C) Pretreatment with anti-IFNγ antibody before CpG treatment blocks expansion of IL-18 synthesis. Groups of five C57BL/6 mice were treated with 0.5 mg of XMG1.2 (rat IgG1) or irrelevant rat IgG1 monoclonal antibody 2 hrs before the first CpG pretreatment. At 3 hrs post-LPS challenge, XMG1.2-pretreated mice produced 16% of the IL-18 levels observed in mice pretreated with CpG and isotype control IgG (p < 0.02). (D) Pretreatment with recombinant IFNγ was sufficient to amplify subsequent production of serum IL-18 in response to LPS challenge. Groups of (n =4) C3H/HeN mice were pretreated either with two doses of PBS, CpG (50 ug), or PBS containing recombinant IFNγ (10 ug) spaced 24 hours apart and challenged with PBS or LPS at 72 hours. LPS-induced IL-18 levels in rIFNγ-pretreated mice were not significantly different than those in CpG-pretreated mice; both were significantly greater (p < 0.01) than IL-18 levels induced by LPS in PBS-pretreated mice.
Recombinant IFNγ is sufficient to expand LPS-inducible IL-18 production in vivo
Since IFNγ was necessary for high-level IL-18 responses in CpG-pretreated mice, we tested whether IFNγ pretreatment alone could increase LPS-inducible IL-18 production. Groups of five C3H/HeN mice were pretreated with PBS, CpG (50 ug), or recombinant mouse IFNγ (20 ug) once daily for two days and then challenged with LPS as before. At four hours after challenge with LPS, IFNγ-pretreated mice produced serum IL-18 levels comparable to those of CpG-pretreated control mice (p =0.32), both of which were significantly increased (p < 0.05) relative to PBS-pretreated controls challenged with PBS or LPS (Figure 1D).
CpG-expanded IL-18 responses are optimally triggered by TLR4-dependent activation by LPS
We confirmed previous observations [11] that high-level IL-18 responses in CpG-pretreated C3H/HeN mice were elicited by LPS, but not by CpG challenge (Figure 2A). To determine if CpG-enhanced IL-18 responses were selectively elicited by specific TLR agonists, we broadened the comparison of TLR challenges to include bacterial lipopeptide (BLP; Pam3CysSerLys4), R848 and Poly(IC), using doses previously determined to be optimal for induction of signature cytokines in normal mice. We again observed that LPS was the most potent stimulus for generating high-level IL-18 in serum (20-fold increase relative to PBS-pretreated endotoxemic controls). Patterns of IL-1α production in CpG-pretreated mice were similar to IL-18, showing the greatest response to LPS challenge. Non-LPS challenges of CpG-pretreated mice significantly increased (p < 0.05) levels of cytokines other than IL-18. In particular, IFNγ was highly produced in response to CpG and R848 and IFNα was increased five-fold in response to poly(IC). This showed distinct and TLR-specific patterns of cytokine production in mice preconditioned by CpG.
Figure 2. CpG-expanded production of serum IL-18 is elicited by lipopolysaccharide, but not by biologically active challenges with CpG, bacterial lipopeptide (BLP), R848 or poly(IC).
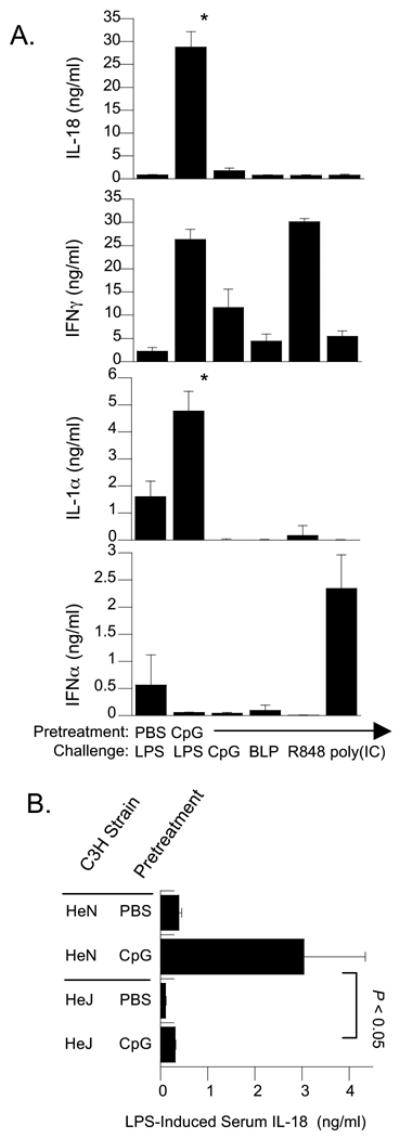
(A) Groups of 5 C3H/HeN mice were pretreated with two i.p. injections of 50 ug of CpG at 0 and 24 hrs and challenged by i.p. injection at 72 hours with either Salmonella enteriditis LPS (300 ug), CpG-containing oligonucleotide 1826 (150 ug), bacterial lipopeptide Pam3Cys (100ug), R848 (100 ug), or Poly(IC) (100 ug). Sera were collected at 4 hours after stimulation and analyzed for circulating IL-18, IFNγ, IL-1α, and IFNα by ELISA. The increased levels of IL-18 induced by LPS in CpG-pretreated mice were significantly greater (p < 0.01) than those stimulated by challenge with each of the other toll agonists or induced by LPS in PBS-pretreated mice. (B) LPS-induced production of CpG-expanded IL-18 is TLR4-dependent. Groups of five TLR4-dysfunctional C3H/HeJ and TLR4-competent C3H/HeN mice were pretreated with PBS or CpG (50 ug) twice 24 hours apart, followed by LPS stimulation at 72 hours. Shown are mean serum IL-18 concentrations present at 3 hours after LPS injection. The bracket indicates significant increases in LPS-induced serum IL-18 by TLR4-competent mice compared to C3H/HeJ mice.
We also confirmed that LPS induced IL-18 by a TLR4-dependent mechanism (Figure 2B). After CpG-pretreatment, TLR4-competent C3H/HeN mice produced 6.5-fold more IL-18 than saline pretreated mice, but TLR4-dysfunctional C3H/HeJ mice produced only 11% of the maximal response observed in the C3H/HeN strain (significant decrease at p < 0.05). These findings suggest that the CpG-mediated increases in IL-18 synthetic potential are optimally triggered by a TLR4-restricted mechanism.
Disparate priming for high-level IL-18 responses by distinct TLR agonists
In contrast to the IL-18-enhancing effects of CpG pretreatment, we confirmed that LPS pretreatment could not amplify serum IL-18 responses to later LPS challenge [11], although LPS-pretreatment generated low levels of IL-18 present at 72 hrs. A similar constitutive IL-18 response was not seen at 72 hrs after CpG-pretreatment, but these mice generated high-level IL-18 in response to LPS challenge (Figure 3A). To determine if other TLR agonists might similarly mediate divergent regulatory effects on LPS-inducible serum IL-18, C3H/Hen mice were pretreated with PBS or with toll receptor agonists Pam3Cys (BLP), CpG or R848 (Figure 3B). CpG pretreatment resulted in the greatest increases in LPS-triggered serum IL-18 concentrations, with levels that were 5.5-fold greater than those in R848-pretreated mice, and at least 8-fold greater than the other groups tested (p < 0.005).
Figure 3. LPS, R848, BLP and CpG pretreatments differentially regulate LPS-inducible IL-18 responses that correlate with serum and splenic IFNγ levels prior to challenge.
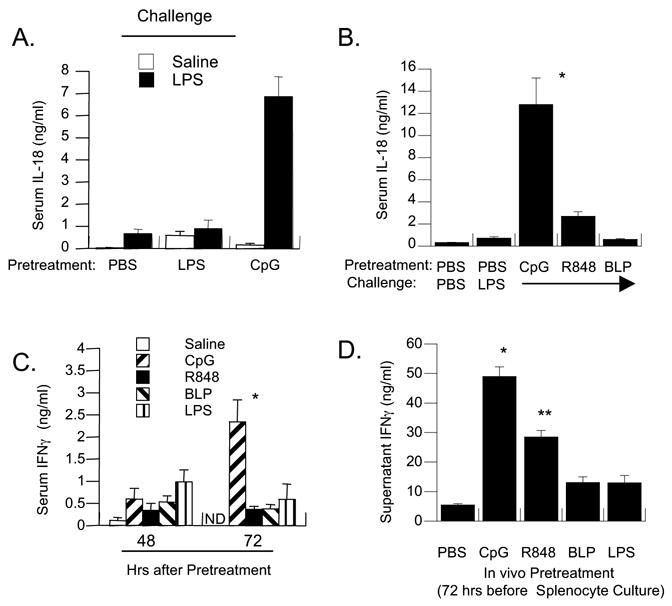
(A) C3H/HeN mice (n=5 per group) were pretreated twice with PBS, LPS (50 ug) or CpG (50 ug) 24 hours apart. Shown are mean concentrations of serum IL-18 present 3 hrs after challenge with PBS or LPS (300 ug) given at 72 hours. The asterisk indicates IL-18 levels significantly increased relative to all other values in the panel (p < 0.005). Findings are representative of five experiments. (B) C3H/HeN mice were pretreated with PBS, CpG (50 ug), R848 (20 ug), or Pam3Cys bacterial lipopeptide (BLP; 50 ug) 24 hours apart. Serum IL-18 was measured 3 hrs after LPS challenge. A single asterisk indicates a significant increase over all other values (p < 0.005). The double asterisk indicates a significant increase in LPS-induced IL-18 in R848-pretreated, compared to PBS, LPS and BLP-pretreated mice (p < 0.05). These findings are representative of two experiments. (C) C3H/HeN mice (n=5 per group) were pretreated with PBS, LPS (50 ug), R848 (20 ug), BLP (50 ug) or CpG (50 ug) at 0 and 24 hrs. The amount of IFNγ spontaneously released into serum was assayed at 48 and 72 hours after pretreatment. Shown are the mean and SEM of IFNγ in ng/ml (N.D = not done). The single asterisk indicates levels significantly higher than all other groups (p < 0.05) (D) Spontaneous production of IFNγ by splenocytes obtained from LPS, R848, BLP and CpG-pre-treated mice. Splenocytes were obtained at 72 hours from the mice shown in Figure 3C and cultured in endotoxin-free media for 18 hrs without in vitro stimulation. Shown are IFNγ levels and standard errors in ng/ml. The single asterisk indicates significant increases for CpG pretreated cultures compared to all other cultures (p < 0.05). R848-pretreated cultures (dual asterisks) produced significantly more IL-18 than media controls, but not compared to BLP or LPS-pretreated cultures.
Because CpG has been shown to maintain elevated serum IFNγ in mice, we tested whether differences in serum IFNγ levels correlated with development of high-level IL-18 responses. C3H/HeN mice were pretreated with the same panel of TLR agonists used previously and serum levels of IFNγ compared at 48 and 72 hrs after pretreatment (Figure 3C). Splenocytes obtained at 72 hrs were cultured under non-stimulatory conditions and spontaneous IFNγ production in vitro determined (Figure 3D). In sera obtained at 72 hrs and in spleen culture, spontaneous IFNγ levels were highest in CpG-preconditioned tissues, compared to those derived from saline, LPS and BLP-pretreated mice (p < 0.01) and relative to R848-pretreated mice (p = 0.04). R848-pretreated spleen produced significantly more IFNγ than media controls, but this trend was non-significant relative to BLP- and LPS-pretreated spleen cultures.
CpG-enhanced IL-18 responses in vivo are caspase-1-dependent and P2X7 receptor-independent
Bioactive IL-18 and IL-1 are produced from peptide precursors through proteolytic cleavage by caspase-1. Release of activated cytokine is subsequently mediated by vesicular exocytosis. Using C57BL/6 mice that were genetically deficient for this enzyme, we confirmed that caspase-1 expression was necessary for both low- and high-level serum IL-18 responses mice pretreated with PBS and CpG, respectively (Figure 4A). In comparison to IFNγ−/− mice, in which CpG maintained residual increases in LPS-inducible IL-18, serum IL-18 after LPS challenge was not significantly increased in CpG-pretreated caspase-1−/− mice.
Figure 4. Normal and CpG-expanded IL-18 responses to LPS are Caspase-1 dependent, but P2X7 receptor-independent.
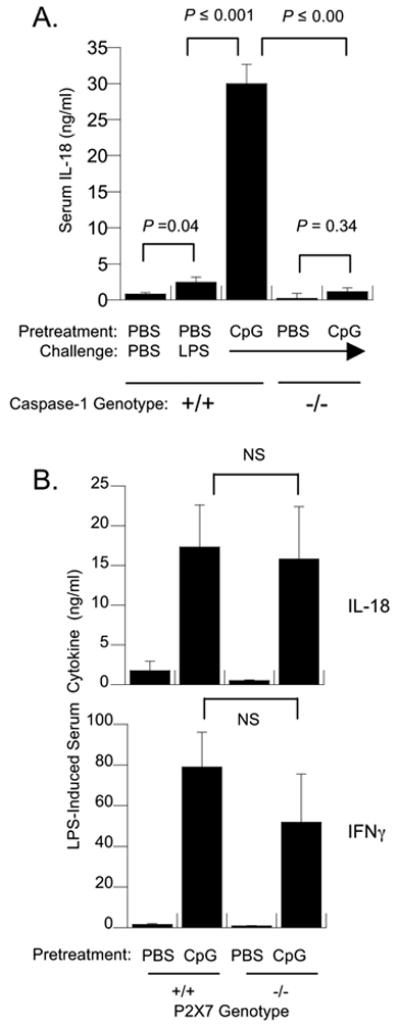
(A) Wild-type and Caspase-1 deficient C57BL/6 mice (n=5 per group) were pretreated with either PBS or CpG and challenged with saline or LPS (300 ug) at 72 hours. Shown are the mean serum IL-18 and IFNγ concentrations at three hours after LPS challenge. The brackets indicate pair-wise tests of significance. (B) Wild-type and P2X7R-deficient C57BL/6 mice were pretreated with either PBS or CpG (50 ug) as described above and then challenged i.p. with LPS at 72 hours. As indicated by brackets, the presence or absence of P2X7R did not significantly alter cytokine levels (NS, p > 0.05). These data are representative of two experiments using either 100 or 300 ug LPS challenges and five mice per group.
In the presence of its ATP ligand, the P2X7 receptor dramatically increases caspase-1-mediated processing of both IL-1 and IL-18 in LPS-primed macrophages and PBMC [15, 16]. To determine if CpG priming and/or LPS triggering of high-level IL-18 also required P2X7 receptor function, wild-type and P2X7 receptor deficient (P2X7R−/−) C57BL/6 mice were pretreated with PBS or CPG and challenged with LPS. In two experiments using either 100 ug or 300 ug LPS challenges, both P2X7R−/− and P2X7R+/+ mice showed large increases in serum IL-18 production after CpG pretreatment (Figure 4B). IFNγ production was also preserved in CpG-pretreated P2X7\ mice relative to wild-type controls.
Discussion
In these studies, we show that IFNγ is both necessary and sufficient to markedly increase the serum IL-18 synthetic response to LPS in vivo. This identifies a mechanism by which prior inflammation can regulate the extent of IL-18 production. The co-administration of anti-IFNγ antibody with CpG effectively localizes the action of IFNγ to a time after CpG pretreatment. This is consistent with the presence of significantly elevated serum levels of IFNγ in mice three days after CpG-pretreatment. Since recombinant IFNγ alone reproduces the IL-18-expanding effects of CpG pretreatment, CpG-induced IFNγ is likely the critical intermediate linking CpG pretreatment and modulated serum IL-18 response. However, residual increases in the IL-18 response of IFNγ-deficient mice after CpG pretreatment were observed, suggesting there may be additional factors elicited by CpG that can amplify IL-18 synthesis. These findings are not limited to IL-18, as CpG also expanded LPS-inducible serum IL-1α and IL-1β responses in vivo (data not shown), consistent with common regulatory effects on a family of cytokines that are processed and exported by nonclassical cellular mechanisms. These findings significantly increase our understanding of how IL-18 release is regulated in systemic and local inflammation and may be especially informative with regards to the anti-infective properties of CpG immunomodulatory therapy. The successful control of microbial infection by CpG oligonucleotide treatment and CpG-associated IFNγ was previously ascribed to IL-12-dependent mechanisms [12, 17–19]. We can now postulate that IFNγ-regulated IL-18 and/or IL-1 production may be equally important in the mediation of CpG immunoprophylactic and adjuvant effects in vivo.
Our findings show that IFNγ-expanded IL-18 responses maintain their canonical dependence on caspase-1 processing, ruling out recruitment of nonclassical mechanisms for processing pro-IL-18. Instead, this suggests IFNγ may regulate caspase-1 activity, synthesis of pro-IL-18 substrate and/or the efficiency of the exocytosis pathway for mature IL-18. Preliminary studies show increased pro-caspase-1 in liver tissue after IFNγ pretreatment (data not shown), consistent with a previous report of IFNγ-dependent increases in caspase-1 activity [20]. In contrast, we failed to show an IFNγ-mediated role for the P2X7 ATP-receptor shown to be necessary for high-level processing and release of bioactive IL-1 and IL-18 in the presence of exogenous ATP [14, 16]. Because both normal and CpG-expanded IL-18 responses to LPS challenge were P2X7-independent, these findings suggest that the mouse model of acute endotoxemia does not provide sufficient quantities of circulating ATP to support P2X7 activation. We cannot exclude a significant regulatory function for P2X7 in other disease processes that might trigger the release of abundant ATP.
Although low-level production of IL-18 by normal tissues or cells is known to be induced by a range of toll receptor agonists [6, 7], the IFNγ-expanded IL-18 response of CpG-pretreated mice was optimally triggered by LPS challenge acting through TLR4. Lesser responses were triggered by other TLR agonists. This was not due to inadequate dosing, as the challenges used were sufficient to increase production of other cytokines. The relative specificity for TLR4-dependent activation of IL-18 in CpG-pretreated mice may indicate IFNγ-mediated changes in the TLR repertoire, choice of signal pathway involved or identity of the IL-18-synthesizing cell. Future studies will need to distinguish between these mechanistic possibilities.
We also observed in these initial studies that preconditioning with CpG, compared to other TLR agonists, was optimal for expanding IL-18 responses in vivo. Using in vivo doses of toll agonist that acutely stimulated cytokine production in normal mice, Pam3Cys, LPS and R848 promoted LPS-triggered IL-18 responses that were significantly less than those generated in CpG-pretreated mice. Further escalation of LPS and Pam3Cys dosing was limited by toxicity. Consistent with our finding that IFNγ alone is sufficient to regulate the amplitude of the IL-18 response, CpG promoted significantly greater levels of splenocyte or serum IFNγ in mice at 72 hours after pretreatment. R848, which promoted lesser increases of LPS-inducible IL-18 also generated increased levels of IFNγ in splenocyte culture. These observations suggest that other inflammatory responses productive of IFNγ, such as seen after repeated dosing with IL-12 [10], may similarly expand IL-18 capacity in vivo.
In summary, we conclude that IFNγ centrally promotes high-level production of IL-18 in CpG-pretreated mice and that, compared to other toll agonists, LPS best stimulates CpG-expanded IL-18 release. Because CpG expands IL-18 production in T- and B-cell deficient mice [11], cells of the innate immune system are the likely targets of these immunomodulatory effects. One limitation of this study is that we have not identified a cell source for IFNγ-augmented IL-18 synthesis, despite examination of IFNγ-preincubated macrophage and dendritic cell cultures or splenocytes and peritoneal macrophages derived from CpG-pretreated mice (data not shown). Future studies need to determine if IL-18 is produced by a non-traditional cell source --perhaps of endothelial or epithelial origin--that is uniquely recruited for IL-18 synthesis in response to circulating IFNγ. However, we can conclude that the synthetic pathway involved remains caspase-1 dependent. Recently, synchronous stimulation of multiple toll receptors has been shown to mediate synergistic increases in cytokine production [21]. The model of innate immune modulation described here differs in that the outcome is both sequence and TLR-specific and occurs over a period of days. We speculate that the innate immune system may possess additional differentiative pathways distinct to specific temporal patterns of exposure to microbial PAMP during infection.
Acknowledgments
Fred Heinzel is supported by the VA Medical Research Service and by NIH grant AI45602, Jeff Auletta by AI57801 and Sameer Gupta by T32 HL07889. We thank Drs. Eric Pearlman, George Dubyak and Michelle Lin for discussion, encouragement and sharing of reagents in these studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, Okura T, Nukada Y, Hattoria K, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 2.Joshi VD, Kalvakolanu DV, Hasday JD, Hebela RJ, Cross AS. IL-18 levels and the outcome of innate immune response to lipopolysaccharide: importance of a positive feedback loop with caspase-1 in IL-18 expression. J Immunol. 2002;169:2536–2544. doi: 10.4049/jimmunol.169.5.2536. [DOI] [PubMed] [Google Scholar]
- 3.Netea MG, Vonk AG, van den Hoven M, Verschueren I, Joosten LA, van Krieken JH, van den Berg WB, Van der Meera JW, Kullberg BJ. Differential role of IL-18 and IL-12 in the host defense against disseminated Candida albicans infection. Eur J Immunol. 2003;33:3409–3417. doi: 10.1002/eji.200323737. [DOI] [PubMed] [Google Scholar]
- 4.Pien GC, Satoskar AR, Takeda K, Akiraa S, Biron CA. Cutting edge: selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J Immunol. 2000;165:4787–4791. doi: 10.4049/jimmunol.165.9.4787. [DOI] [PubMed] [Google Scholar]
- 5.Stuyt RJ, Netea MG, van Krieken JH, van der Meera JW, Kullberg BJ. Recombinant interleukin-18 protects against disseminated Candida albicans infection in mice. J Infect Dis. 2004;189:1524–1527. doi: 10.1086/382955. [DOI] [PubMed] [Google Scholar]
- 6.Seki E, Tsutsui H, Nakano H, Tsuji N, Hoshino K, Adachi O, Adachi K, Futatsugi S, Kuida K, Takeuchi O, Okamura H, Fujimoto J, Akiraa S, Nakanishi K. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J Immunol. 2001;166:2651–2657. doi: 10.4049/jimmunol.166.4.2651. [DOI] [PubMed] [Google Scholar]
- 7.Netea MG, Kullberg BJ, Jacobs LE, Verver-Jansen TJ, van der Ven-Jongekrijg J, Galama JM, Stalenhoef AF, Dinarelloa CA, Van der Meer JW. Chlamydia pneumoniae stimulates IFN-gamma synthesis through MyD88-dependent, TLR2- and TLR4-independent induction of IL-18 release. J Immunol. 2004;173:1477–1482. doi: 10.4049/jimmunol.173.2.1477. [DOI] [PubMed] [Google Scholar]
- 8.Mehta VB, Harta J, Wewers MD. ATP-stimulated release of interleukin (IL)-1beta and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem. 2001;276:3820–3826. doi: 10.1074/jbc.M006814200. [DOI] [PubMed] [Google Scholar]
- 9.Sluyter R, Dalitza JG, Wiley JS. P2X7 receptor polymorphism impairs extracellular adenosine 5′-triphosphate-induced interleukin-18 release from human monocytes. Genes Immun. 2004;5:588–591. doi: 10.1038/sj.gene.6364127. [DOI] [PubMed] [Google Scholar]
- 10.Fantuzzi G, Reeda DA, Dinarello CA. IL-12-induced IFN-gamma is dependent on caspase-1 processing of the IL-18 precursor. J Clin Invest. 1999;104:761–767. doi: 10.1172/JCI7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gould MP, Greene JA, Bhoj V, DeVecchioa JL, Heinzel FP. Distinct modulatory effects of LPS and CpG on IL-18-dependent IFN-gamma synthesis. J Immunol. 2004;172:1754–1762. doi: 10.4049/jimmunol.172.3.1754. [DOI] [PubMed] [Google Scholar]
- 12.Cowdery JS, Chace JH, Yia AK, Krieg AM. Bacterial DNA induces NK cells to produce IFN-gamma in vivo and increases the toxicity of lipopolysaccharides. J Immunol. 1996;156:4570–4575. [PubMed] [Google Scholar]
- 13.Tsuji H, Mukaida N, Harada A, Kaneko S, Matsushita E, Nakanuma Y, Tsutsui H, Okamura H, Nakanishi K, Tagawa Y, Iwakura Y, Kobayashia K, Matsushima K. Alleviation of lipopolysaccharide-induced acute liver injury in Propionibacterium acnes-primed IFN-gamma-deficient mice by a concomitant reduction of TNF-alpha, IL-12, and IL-18 production. J Immunol. 1999;162:1049–1055. [PubMed] [Google Scholar]
- 14.Labasi JM, Petrushova N, Donovan C, McCurdy S, Lira P, Payette MM, Brissette W, Wicks JR, Audolya L, Gabel CA. Absence of the P2X7 receptor alters leukocyte function and attenuates an inflammatory response. J Immunol. 2002;168:6436–6445. doi: 10.4049/jimmunol.168.12.6436. [DOI] [PubMed] [Google Scholar]
- 15.Perregaux DG, McNiff P, Laliberte R, Conklyna M, Gabel CA. ATP acts as an agonist to promote stimulus-induced secretion of IL-1 beta and IL-18 in human blood. J Immunol. 2000;165:4615–4623. doi: 10.4049/jimmunol.165.8.4615. [DOI] [PubMed] [Google Scholar]
- 16.Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffithsa RJ, Gabel CA. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–132. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- 17.Deng JC, Moore TA, Newstead MW, Zeng X, Kriega AM, Standiford TJ. CpG oligodeoxynucleotides stimulate protective innate immunity against pulmonary Klebsiella infection. J Immunol. 2004;173:5148–5155. doi: 10.4049/jimmunol.173.8.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krieg AM, Love-Homan L, Yia AK, Harty JT. CpG DNA induces sustained IL-12 expression in vivo and resistance to Listeria monocytogenes challenge. J Immunol. 1998;161:2428–2434. [PubMed] [Google Scholar]
- 19.Zimmermann S, Egeter O, Hausmann S, Lipford GB, Rocken M, Wagnera H, Heeg K. CpG oligodeoxynucleotides trigger protective and curative Th1 responses in lethal murine leishmaniasis. J Immunol. 1998;160:3627–3630. [PubMed] [Google Scholar]
- 20.Cross AS. Endotoxin tolerance-current concepts in historical perspective. J Endotoxin Res. 2002;8:83–98. doi: 10.1179/096805102125000227. [DOI] [PubMed] [Google Scholar]
- 21.Napolitani G, Rinaldi A, Bertoni F, Sallustoa F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]