

Abstract
myo-Inositol 1,3,4,5,6-pentakisphosphate (Ins(1,3,4,5,6)P5), an inositol polyphosphate of emerging significance in cellular signalling, and its C-2 epimer scyllo-inositol pentakisphosphate (scyllo-InsP5) were synthesised from the same myo-inositol-based precursor. Potentiometric and NMR titrations show that both pentakis-phosphates undergo a conformational ring-flip at higher pH, beginning at pH 8 for scyllo-InsP5 and pH 9 for Ins(1,3,4,5,6)P5. Over the physiological pH range, however, the conformation of the inositol rings and the microprotonation patterns of the phosphate groups in Ins(1,3,4,5,6)P5 and scyllo-InsP5 are similar. Thus, scyllo-InsP5 should be a useful tool for identifying biologically relevant actions of Ins(1,3,4,5,6)P5, mediated by specific binding sites, and distinguishing them from nonspecific electrostatic effects. We also demonstrate that, although scyllo-InsP5 and Ins(1,3,4,5,6)P5 are both hydrolysed by multiple inositol polyphosphate phosphatase (MINPP), scyllo-InsP5 is not dephosphorylated by PTEN or phosphorylated by Ins(1,3,4,5,6)P5 2-kinases. This finding both reinforces the value of scyllo-InsP5 as a biological control and shows that the axial 2-OH group of Ins(1,3,4,5,6)P5 plays a part in substrate recognition by PTEN and the Ins(1,3,4,5,6)P5 2-kinases.
Keywords: cyclitols, enzymes, inositol phosphates, protecting groups, structure—activity relationships
Introduction
myo-Inositol 1,3,4,5,6-pentakisphosphate (Ins(1,3,4,5,6)P5, 1; Scheme 1) which is present in nearly all eukaryotic cells at levels of 15–50 μm, has been called a signalling “hub” because it appears to serve several biological roles.[1] For example, Ins(1,3,4,5,6)P5 has been proposed to regulate the rate of cellular proliferation,[2, 3] apoptosis,[3] viral assembly,[4] chromatin remodelling[5] and the activity of L-type Ca2+ channels.[6] In addition, Ins(1,3,4,5,6)P5 inhibits angiogenesis and blocks growth of tumour cell xenografts.[7] Ins(1,3,4,5,6)P5 can also regulate signalling by 3-phosphorylated lipids—by binding to 3-phosphoinositide-dependent protein kinase 1 (PDK1) Ins(1,3,4,5,6)P5 can anchor the kinase in the cytosol and prevent it from being activated by phosphatidylinositol 3,4,5-trisphosphate (PtdIns- (3,4,5)P3).[8] Moreover, Ins(1,3,4,5,6)P5 can competitively inhibit PtdIns(3,4,5)P3 metabolism by phosphatase and tensin homologue deleted on chromosome ten (PTEN).[9]
Scheme 1.
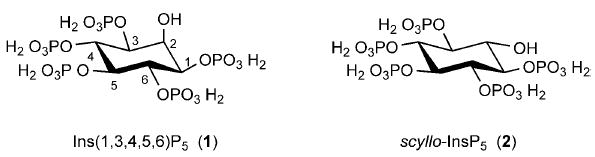
Structures of myo-inositol 1,3,4,5,6-pentakisphosphate (Ins(1,3,4,5,6)P5, 1) and scyllo-inositol pentakisphosphate (scyllo-InsP5, 2).
It is important to understand the structural determinants of the interactions between Ins(1,3,4,5,6)P5 and the proteins that it regulates. Such knowledge helps our understanding of basic biological processes at a molecular level, while also aiding the rational design of ligand agonists and antagonists that might have therapeutic value. The biological actions of inositol phosphates often rely on stereospecific ligand recognition by dedicated receptors. This applies, for example, to Ca2+ mobilization by Ins(1,4,5)P3[10] and Cl− channel inhibition by Ins (3,4,5,6)P4.[11] These ligand–protein interactions involve electrostatic interactions between the protein and the negatively charged phosphate groups on the ligand. Geometric constraints imposed by the bulky nature of the phosphate groups will also contribute to ligand specificity. However, spatial restrictions will be less important if the protein’s binding cleft is relatively spacious or if the ligand-binding site is on the surface of the protein. In such situations, electrostatic interactions can be delocalized, in which case ligand–protein association is driven more by the number of phosphate groups in a molecule, rather than their exact placement around the inositol ring.[12] One example of this phenomenon is the binding of inositol phosphates to pleckstrin homology (PH) domains.[12] This is a biologically relevant mechanism which could, in some cases, be exploited by a molecule such as Ins(1,3,4,5,6)P5.[8]
Unfortunately, delocalized electrostatic interactions between inositol polyphosphates and proteins, in vitro, can drive biologically irrelevant ligand binding, especially when the experimental milieu does not appropriately imitate the situation in vivo.[13] In such cases, the apparent functional data that might emerge from such assays can be misleading. Thus, it is useful when examining the biological activity of an inositol polyphosphate to have a closely related analogue that cannot imitate the actions of the “active” parent molecule. A good example in the literature is the demonstration that scyllo-inositol hexakis-phosphate (scyllo-InsP6) does not imitate the ability of myo-InsP6 to inhibit an inwardly rectifying K+ current in plant guard-cell protoplasts.[14] All of the phosphate groups in the naturally occurring scyllo-InsP6 are esters of equatorial hydroxyl groups (i.e., approximately in the plane of the inositol ring). In contrast, the 2-phosphate in myo-InsP6 is linked as an ester of an axial hydroxyl group, that is, it is perpendicular to the plane of the ring. The demonstration that myo-InsP6, but not scyllo-InsP6, is biologically active indicates that a receptor protein has a discriminating, stereospecific ligand-binding site, rather than merely reacting to high negative-charge density. Such specificity is often the hallmark of a biologically relevant process.
In this study, we have investigated the value of scyllo-inositol pentakisphosphate (scyllo-InsP5,[15] 2; Scheme 1), a totally synthetic molecule, as a control for studies into the biological activity of Ins(1,3,4,5,6)P5. We describe convenient synthetic routes to both pentakisphosphates from myo-inositol from an orthoester intermediate and compare the physicochemical properties of the two InsP5 epimers. The physicochemical part of the study was prompted by the consideration that the difference in the orientation of the hydroxyl group between the two polyphosphates might differentially affect the basicity of the adjacent phosphates through intramolecular hydrogen bonding, which in turn affects the hydration shell and association with cations.[16] Next, the metabolism of Ins(1,3,4,5,6)P5 and scyllo-InsP5 by two phosphatases that are known to hydrolyse Ins(1,3,4,5,6)P5 (multiple inositol polyphosphate phosphatase (MINPP) and PTEN) was compared. Finally, we examined whether scyllo-InsP5 can be phosphorylated by Ins(1,3,4,5,6)P5 2-kinases.[17,18] This highlights another reason for selecting scyllo-InsP5 as a control polyphosphate, because myo-InsP6 has itself drawn attention as an intracellular signal.[10,19] Thus, in experiments with cell-free systems, it would be useful when adding Ins(1,3,4,5,6)P5, to be able to exclude effects that might arise from its phosphorylation to InsP6. Moreover, an insight into the reactivity of 2-kinases towards scyllo-InsP5 could further our understanding of the origin of scyllo-InsP6—an enigmatic molecule of unknown metabolic origin that is nevertheless present in large quantities in soils.[20]
Results
Chemical synthesis of Ins(1,3,4,5,6)P5 and scyllo-InsP5
The versatile alcohol precursor 3[21] (Scheme 2) was chosen as starting material for the synthesis of both Ins(1,3,4,5,6)P5 and scyllo-InsP5. Oxidation of 3 at C-2,[21] followed by stereoselective reduction by using sodium borohydride[22] exclusively gave the axial alcohol 5. Interestingly, the fact that 5 has the scyllo-configuration is immediately apparent from the 1H NMR spectrum, because the signal corresponding to the methylidyne proton appears as a singlet, while in the myo-epimer 3 it appears as a narrow doublet due to a long range 5J spin–spin coupling to the axial H-2 proton. Benzylation of the free axial OH group in 5 gave 6 and treatment of 6 with HCl in refluxing ethanol cleaved both the orthoformate ester and the p-methoxybenzyl ethers. The pentaol product, 1-O-benzyl-scyllo-inositol (7) crystallised from the reaction mixture on cooling. Phosphitylation of 7, followed by oxidation of phosphite esters with mCPBA gave fully protected 8. Finally, hydrogenolytic deprotection of 8 gave scyllo-inositol pentakisphosphate (2),[15] which was purified by ion-exchange chromatography on Q-Sepharose fast-flow resin by using a gradient of aqueous triethylammonium bicarbonate. Ins(1,3,4,5,6)P5[23] (1) was synthesised in a similar way from 3 from the 2-O-benzyl ether 9 (Scheme 2).
Scheme 2.
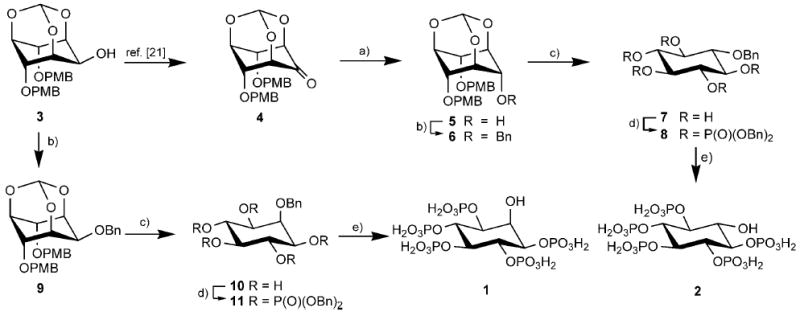
Synthesis of Ins(1,3,4,5,6)P5 (1) and scyllo-InsP5 (2). Reagents and conditions: a) NaBH4, THF, MeOH; b) BnBr, NaH, DMF; c) 1.0 m HCl/EtOH (1:2), reflux; d) i. (BnO)2PNPri2, 1H-tetrazole, CH3CN; ii. mCPBA, CH2Cl2, −78°C to room temperature; e) Pd-C, MeOH, H2O, H2, 3.5 bar; Bn = benzyl, PMB = 4-methoxybenzyl.
pH-dependent conformational changes of Ins(1,3,4,5,6)P5 and scyllo-InsP5
For inositol phosphates, the analysis of 31P and 1H NMR titration curves provides valuable information on both the charged species likely to be present at physiological pH and the conformational dependence of the inositol ring with respect to the ionisation states of the individual phosphate groups. NMR and potentiometric titrations of Ins(1,3,4,5,6)P5 and scyllo-InsP5 were performed in KCl (0.2 m) at 37 °C, a medium that mimics the ionic strength and temperature conditions encountered in the cell. To facilitate the discussion, the inositol ring in scyllo-InsP5 will here be numbered so that the attached phosphate groups correspond with those in Ins(1,3,4,5,6)P5, although this is a departure from the strict rules of inositol nomenclature.
The 31P NMR titration curves of Ins(1,3,4,5,6)P5 and scyllo-InsP5 are shown in Figure 1. Due to the symmetry of the molecules, only three curves are observed for each. Their general shape differs only slightly between the two molecules. In each pentakisphosphate, P4–P6 and P5 behave similarly, and show an intermediate plateau or even a slight deshielding upon protonation, whereas P1–P3 keeps a more attenuated biphasic shape. Interestingly, the chemical shifts of the phosphorus atoms in the protonated and deprotonated species differ by more than 1.30 ppm. This, along with the broadening of the phosphorus resonances that occur for Ins(1,3,4,5,6)P5 and scyllo-InsP5 in the pH ranges 9–11 and 8–10 respectively, suggest the presence of an interconverting mixture of conformers with either five equatorial (lower pH) or five axial phosphate groups (higher pH).
Figure 1.
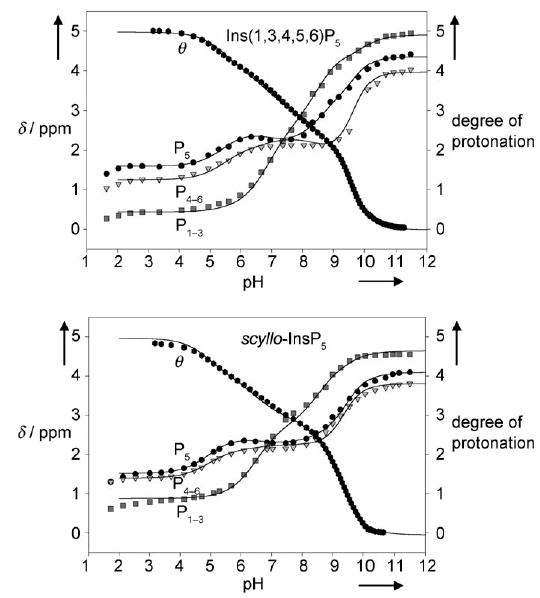
31P NMR titrations curves [δ = f(pH)] for Ins(1,3,4,5,6)P5 and scyllo-InsP5 along with the potentiometric site-specific degree of protonation [θ = f(pH)] determined in 0.2 m KCl at 37 °C (D2O). Least-squares fits are shown as solid lines; see text for details.
Examination of the 1H NMR titration curves for the two compounds confirmed that an inositol ring flip occurs at the earliest protonation steps. This is illustrated for scyllo-InsP5 in Figure 2, where a marked upfield shift for the signals that correspond to the protons of the inositol ring can be observed as the pH decreases from 10 to 8. This corresponds to the switching of the ring protons from an equatorial to an axial position. In line with this ring flip, the coupling constants between two vicinal protons in an equatorial–equatorial relationship (Jeq–eq, ca. 2–3 Hz) rapidly increase to 8–10 Hz; this corresponds to the couplings of two axial vicinal protons (Jax–ax). The same conclusions can be drawn from 1H NMR titration curves for Ins(1,3,4,5,6)P5 (not shown), with the only difference being that the conformational transition arises in the 11 to 9 pH range. Barrientos and Murthy,[24] who thoroughly investigated the conformational preferences of inositol mono- to hexakisphosphates by 1H NMR spectroscopy, also observed pH-dependent ring conformational changes for Ins(1,2,3,4,6)P5 and Ins(1,2,3,5,6)P5.
Figure 2.
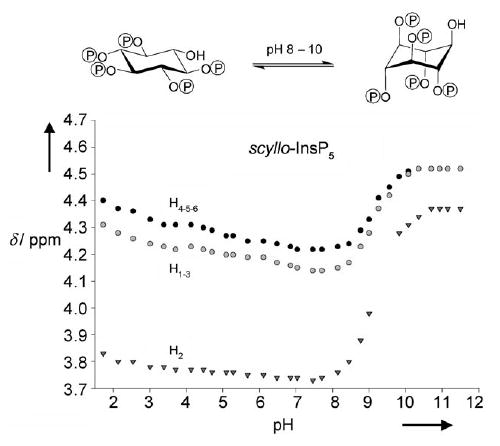
1H NMR titrations curves for scyllo-InsP5 (0.2 m KCl at 37 °C in D2O) show that pH-dependent inositol ring flipping occurs over the pH range 8–10.
Protonation patterns of Ins(1,3,4,5,6)P5 and scyllo-InsP5
Quantitative information about the intrinsic acid–base character of the phosphate groups in Ins(1,3,4,5,6)P5 and scyllo-InsP5 can be drawn from both the potentiometric and the NMR titration curves. Although each inositol pentakisphosphate carries ten protonatable sites (two on each phosphate group), only five of them will accept a proton for pHs ranging from 12 down to 3. Thus, for each pentakisphosphate, six possible macrostates exist over this pH range, with total electrostatic charges ranging from −10 at pH 12 (zero protons, n = 0) to −5 at pH 3 (five protons, n = 5). The probabilities of these macrostates, calculated from the potentiometric titration curves for Ins(1,3,4,5,6)P5 and scyllo-InsP5 are shown in Figure 3. It can be seen that the pH-dependent distributions of the macrostates for the two pentakisphosphates are similar, with the triprotonated species (n = 3) predominating at pH 7.5 in each case.
Figure 3.
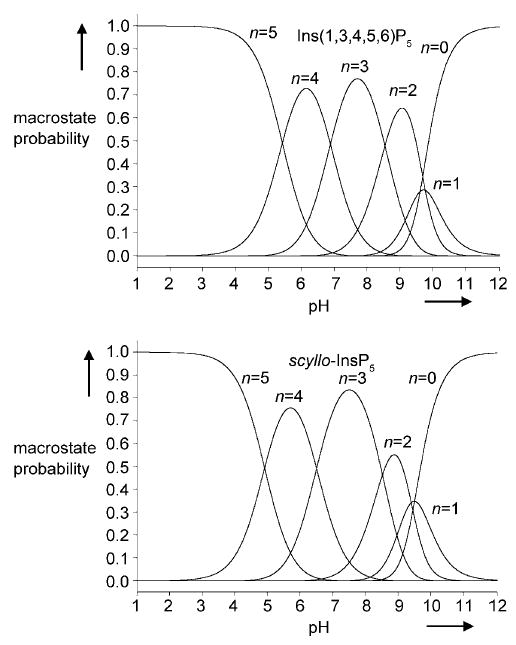
Macrostate probabilities for Ins(1,3,4,5,6)P5 and scyllo-InsP5 (0.2 m KCl at 37 °C in D2O) calculated from the potentiometric titration curves in Figure 1.
More complex microprotonation processes, considering each phosphate group individually, were treated by analysis of the 31P NMR curves by using the recently published “cluster-expansion method”.[25] This analysis enables the detailed microscopic protonation patterns over the studied pH range to be calculated for each pentakisphosphate. In the present study, we are primarily concerned with the properties of Ins(1,3,4,5,6)P5 and scyllo-InsP5 under physiological conditions and so only a part of this complex pattern will be considered in each case. The conditional probabilities calculated for Ins(1,3,4,5,6)P5 and scyllo-InsP5 at pH 7.5 (Figure 4) describe the proportions of the different microspecies of each pentakisphosphate that exist at a pH close to physiological conditions. It can be observed that for Ins(1,3,4,5,6)P5 and scyllo-InsP5, the same triprotonated and tetraprotonated species predominate at pH 7.5, with small differences in their populations. Thus, under these conditions, the two pentakisphosphates are expected to behave similarly with respect to overall electrostatic properties and charge distribution.
Figure 4.
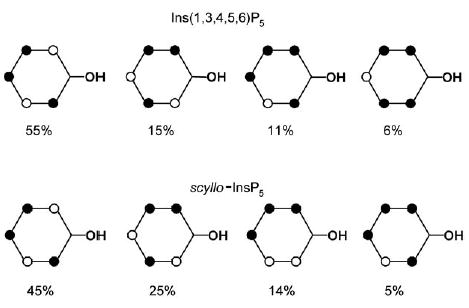
Conditional probabilities (%) for Ins(1,3,4,5,6)P5 and scyllo-InsP5 at pH 7.5; • : protonated sites.
Metabolism of Ins(1,3,4,5,6)P5 and scyllo-InsP5 by human MINPP and PTEN
One of the goals of the current study was to compare the metabolism of scyllo-InsP5 and Ins(1,3,4,5,6)P5. MINPP[26] and PTEN[9] are two inositol phosphate phosphatases that are known to be capable of actively hydrolysing Ins(1,3,4,5,6)P5 in mammals. We found that recombinant human MINPP dephosphorylated scyllo-InsP5 and Ins(1,3,4,5,6)P5 at similar rates (9.0 ± 0.1 and 6.2 ± 0.3 nmolmg −1min−1, respectively). Therefore, the active site of MINPP does not distinguish between these two epimers of InsP5.
In contrast, we found that recombinant human PTEN showed no detectable metabolism of scyllo-InsP5, even though Ins(1,3,4,5,6)P5 is a substrate (11.8 ± 1.3 nmolmg−1min−1). Furthermore, 250 μm scyllo-InsP5 did not inhibit the dephosphorylation of 25 μm Ins(1,3,4,5,6)P5 (12.1 vs. 11.6 nmol mg−1min−1 in the absence or presence of scyllo-InsP5, respectively). Thus, scyllo-InsP5 would be predicted to bind less effectively than Ins(1,3,4,5,6)P5 to the catalytic site of PTEN.
Metabolism of Ins(1,3,4,5,6)P5 and scyllo-InsP5 by Ins(1,3,4,5,6)P5 2-kinase
We next investigated the ability of Ins(1,3,4,5,6)P5 and scyllo-InsP5 to be phosphorylated by Ins(1,3,4,5,6)P5 2-kinase. Using recombinant enzymes from Arabidopsis thaliana, Sacromyces cerevisiae and Homo sapiens, we measured the incorporation of 32P into each pentakisphosphate from the phosphate donor γ-32P-ATP (Experimental Section). Ins(1,3,4,5,6)P5 was readily phosphorylated by each species of 2-kinase. However, there was no detectable phosphorylation of scyllo-InsP5. Furthermore, in a representative experiment with the Arabidopsis 2-kinase, phosphorylation of 50 μm Ins(1,3,4,5,6)P5 was not significantly affected by the addition of 50 μm scyllo-InsP5 (3621 vs. 3336 c.p.m. 32P-InsP6 formed, respectively) but 500 μm scyllo-InsP5 inhibited the reaction by approximately 80 % (766 c.p.m. 32P-InsP6 formed).
Discussion
Nonspecific electrostatic interactions can introduce a difficult practical problem when exploring the mechanism of action of a highly charged molecule such as Ins(1,3,4,5,6)P5. Some delocalized electrostatic interactions of inositol phosphates with proteins might be biologically relevant.[10,11] However, other delocalized electrostatic interactions, which can be permitted in a particular set of experimental conditions, might not be relevant to the intracellular milieu.[13]
Based on earlier studies of other inositol phosphates, a larger difference between the acid–base properties of Ins(1,3,4,5,6)P5 and scyllo-InsP5 might have been expected. For example, marked differences were previously observed between Ins(1,4,5)P3 and epi-Ins(1,4,5)P3, which has an axially orientated 6-OH group,[27] or Ins(1,4,6)P3, which has its 2-OH and 3-OH groups in inverted configurations with regard to Ins-(1,4,5)P3.[28] However, the physicochemical studies described above show that the protonation pattern of scyllo-InsP5 parallels that of Ins(1,3,4,5,6)P5 over the physiological pH range. Detailed analysis by using cluster expansion (not shown) suggests that this similarity between the two epimers could arise from a strong P1–P3 interaction for scyllo-InsP5, which is compensated by large P1–P6 and P3–P4 interactions for Ins(1,3,4,5,6)P5. The differences in the strengths of these interactions are presumably a sign of different hydrogen-bonding patterns for the two epimers due to the difference in orientation of the hydroxyl group. While these effects might have biological consequences if the 1,2,3 moiety of the inositol ring is important in binding to a selective binding site, their overall effect is to make the charge distribution of Ins(1,3,4,5,6)P5 and scyllo-InsP5 similar at physiological pH.
The conformation of organic molecules influences their biological activity by impacting their binding interactions with enzymes and receptors. Barrientos and Murthy[24] have shown that Ins(1,2,3,4,6)P5 and Ins(1,2,3,5,6)P5 undergo a pH-dependent ring flip, whereupon the substituents around the inositol ring switch from a 5-equatorial/1-axial conformation to a 1-equatorial/5-axial arrangement. The actual pH at which this conformational change occurs is strongly dependent upon the spatial placement of phosphate groups around the inositol ring. We therefore compared the influence of pH on the conformations of Ins(1,3,4,5,6)P5 and scyllo-InsP5, because these were factors that could potentially establish different biological activities for these compounds. Our results show that pH-dependent conformational changes occur for both Ins(1,3,4,5,6)P5 and scyllo-InsP5 and that the conformational change for scyllo-InsP5 occurs at about one pH unit lower than that of Ins(1,3,4,5,6)P5, with the inositol ring flip beginning at pH 8 and 9, respectively. However, the NMR titrations show that, in solution, under normal physiological conditions, Ins(1,3,4,5,6)P5 and scyllo-InsP5 will both exist in the conformation that has five equatorial phosphate groups.
The physiological relevance of the differences in conformational behaviour at higher pH for the interaction of Ins(1,3,4,5,6)P5 and scyllo-InsP5 with enzymes and receptors is more difficult to assess, because the pH and the dielectric constant in the vicinity of a protein-binding site are difficult to measure. In theory, both parameters can change the acid–base properties of the ligands with respect to the aqueous medium that was used in this study. However, with this reservation in mind, we can conclude that scyllo-InsP5 will likely mimic interactions between Ins(1,3,4,5,6)P5 and proteins that are largely based on delocalized electrostatics. Such a determination would be an important step forward from a mechanistic perspective, but additional experiments would still be necessary to determine if such interactions were to be biologically relevant. It can also be anticipated that Ins(1,3,4,5,6)P5 will interact with certain proteins in a manner in which the precise spatial geometry of the ligand helps to dictate specificity. Such specific interactions have evolved in order to distinguish a particular inositol phosphate from the many others that cells contain. Indeed, we show in the current study that scyllo-InsP5 cannot be metabolized by PTEN or by Ins(1,3,4,5,6)P5 2-kinases.
An X-ray structure of PTEN in complex with l-(+)-tartrate is available.[29] Furthermore, the catalytic mechanism of the phosphatase activity of PTEN can be inferred[29] from studies on related protein-tyrosine phosphatases (PTPs), whose catalytic domains have sequence and structural homology with that of PTEN. We docked a flexible model of Ins(1,3,4,5,6)P5 into a model of the PTEN binding site (see refs. [9, 29] and Experimental Section) to examine which features of Ins(1,3,4,5,6)P5 might be important for its interaction with PTEN (Figure 5). Detailed structural and mechanistic studies of PTPs[30] have shown that binding of substrates (phosphotyrosine) to their active sites induces a large conformational change which brings a flexible loop of residues (the “general acid loop” [30a]) down into the binding pocket. This generates additional stabilising interactions and allows an aspartate residue to protonate the scissile phenolic oxygen of phosphotyrosine. By applying this mechanism to the hydrolysis of Ins(1,3,4,5,6)P5 by PTEN, our model suggests that, on binding of Ins(1,3,4,5,6)P5 to the active site, the general acid loop would move towards the area of the binding site above the axial 2-OH group so as to approach the O-3 atom of Ins(1,3,4,5,6)P5. Thus, one possibility is that the axial 2-OH group of Ins(1,3,4,5,6)P5 interacts with the general acid loop—an interaction that would be impossible for the equatorial hydroxyl group of scyllo-InsP5. Another possibility is that the conformation of the 1-phosphate group is influenced by intramolecular interactions with the adjacent hydroxyl group so that, in scyllo-InsP5, this phosphate is not optimally positioned for binding. Our model predicts that the 1-phosphate group of Ins(1,3,4,5,6)P5, which is equivalent to the phosphate diester in myo-inositol phospholipids, could interact with Lys125 of the PTEN active site (Figure 5).
Figure 5.
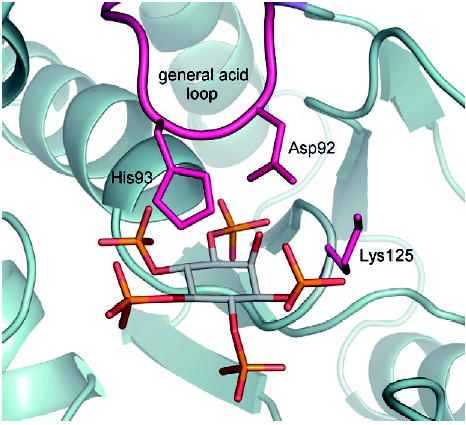
Model of Ins(1,3,4,5,6)P5 docked into the catalytic site of PTEN (PDB ID: 1D5R). The axial 2-OH group of Ins(1,3,4,5,6)P5 is predicted to be close to Asp92 of the general acid loop, which is thought to be involved in the mechanism of catalysis.[9,29] The 2-OH group might also influence the conformation of the 1-phosphate group, which is predicted to have stabilising interactions with Lys125. For details, see Experimental Section and Discussion. For clarity, hydrogen atoms are not shown.
In the case of Ins(1,3,4,5,6)P5 2-kinases no X-ray structure is currently available. The human and plant Ins(1,3,4,5,6)P5 2-kinases show some limited (27 %) sequence identity, but the human and yeast homologues are even more divergent (only 14% sequence identity). Nevertheless, in each case the active site discriminates between scyllo-InsP5 and Ins(1,3,4,5,6)P5, this suggests important similarities in the structure of this region. Our results imply that, as for PTEN, the axial 2-OH group is important for substrate recognition by 2-kinases. Considerable quantities of scyllo-InsP6, are found in soils[20] and our results reinforce the enigma of the metabolic origin of this material.
Because Ins(1,3,4,5,6)P5 and scyllo-InsP5 are physicochemically similar at physiological pH, we argue that, in those experimental situations in which scyllo-InsP5 does not imitate a biological action of Ins(1,3,4,5,6)P5, then a strong case can be made that Ins(1,3,4,5,6)P5 is acting specifically, and therefore, in a physiologically relevant manner. Moreover, the inability of scyllo-InsP5 to be phosphorylated by 2-kinases could exclude the possibility that the synthesis of InsP6 contributes to any of the effects detected in the experiment. scyllo-InsP5 will not be metabolically inert in all experiments with cell extracts; in the current study we did find it to be a substrate of MINPP. This finding is not unexpected, as MINPP is also capable of binding and hydrolysing InsP6. This suggests that the active site of MINPP can tolerate modifications to the 2-hydroxyl group of Ins(1,3,4,5,6)P5. However, in most mammalian cells MINPP is restricted to the interior of the endoplasmic reticulum and only has very limited access to cytosolic inositol phosphates such as Ins(1,3,4,5,6)P5. Thus, there are several experimental situations, such as during the microinjection of inositol phosphates into cell cytosol or during patch-clamp experiments, in which MINPP-dependent InsP5 metabolism will not be a complicating factor. In such circumstances, scyllo-InsP5 will be metabolically inert. Overall, there are a number of experimental paradigms in which scyllo-InsP5 could be a useful practical tool.
Conclusion
Ins(1,3,4,5,6)P5 and its C-2 epimer scyllo-InsP5 were synthesised from the same myo-inositol-based precursor and their conformational and protonation behaviours were compared by using potentiometric and NMR titrations. The results show that, over the physiological pH range, the acid–base properties and inositol-ring conformation of Ins(1,3,4,5,6)P5 and scyllo-InsP5 are similar, and that at pH 7.5, the same triprotonated and tetraprotonated species of each compound predominate with only small differences in their populations. These results suggest that scyllo-InsP5 can be a useful tool for biological studies of Ins(1,3,4,5,6)P5, because under experimental conditions where delocalised electrostatic interactions result in nonspecific effects, the two pentakisphosphates should behave in a similar way. In contrast, when Ins(1,3,4,5,6)P5 and scyllo-InsP5 are demonstrated to have different biological effects, then stereospecific binding sites that require the characteristic axial 2-OH group of Ins(1,3,4,5,6)P5 are likely to be involved. This concept is illustrated by our demonstration that scyllo-InsP5, unlike Ins(1,3,4,5,6)P5, is neither dephosphorylated by PTEN nor phosphorylated by several Ins(1,3,4,5,6)P5 2-kinases; this points to the importance of the axial 2-OH group in substrate recognition by these enzymes. In contrast, MINPP was able to dephophosphorylate both pentakisphosphates; this shows that, in this case, substrate recognition does not require the axial 2-OH group of Ins(1,3,4,5,6)P5.
Experimental Section
Synthesis
General synthetic methods were carried out as previously reported.[21]
2,4-Di-O-(p-methoxybenzyl)-1,3,5-O-methylidyne-scyllo-inositol (5)
Sodium borohydride (430 mg, 11.6 mmol) was added in portions to a stirred solution of ketone 4[21] (1.98 g, 4.63 mmol) in a mixture of THF (20 mL) and methanol (80 mL) at room temperature. After 30 min, TLC (ethyl acetate/hexane, 1:1) showed complete conversion of 4 (streak on TLC plate) into a single product (Rf 0.44). Water (100 mL) was added and the product was extracted with dichloromethane (3 0 100 mL). The combined organic extracts were washed with brine, dried (MgSO4) and concentrated to give a white solid, which was recrystallised from ethyl acetate/hexane, to give alcohol 5 (1.77 g, 4.12 mmol, 89%); m.p. 125–126 °C (from ethyl acetate/hexane). 1H NMR (270 MHz, CDCl3): δ = 3.79 (s, 6H; 2 OCH3), 4.10 (d, D2O exch., 3J(H,H) = 12.45 Hz, 1 H; OH), 4.34–4.42 (m, 3 H; H-6; 2 inositol H), 4.43–4.48 (m, 2 H; 2 inositol H), 4.56 (s, 4 H; OCH2Ar), 4.56 (m, buried, 1 H; H-3), 5.49 (s, 1 H; O3CH), 6.77–6.84 (m, 4 H; C6H4OMe), 7.08–7.16 (m, 4H; C6H4OMe); elemental analysis calcd (%) for C23H26O8 (430.45): C 64.18, H 6.09; found: C 63.9, H 6.08.
6-O-Benzyl-2,4-di-O-(p-methoxybenzyl)-1,3,5-O-methylidyne-scyllo-inositol (6)
Sodium hydride (400 mg of a 60% suspension in oil, 10 mmol) was added to a solution of alcohol 5 (2.15 g, 5.00 mmol) in dry DMF at room temperature. After 30 min, the suspension was cooled to 0 °C and benzyl bromide (0.7 mL, 6 mmol) was added dropwise over 5 min. The mixture was allowed to reach room temperature and stirred overnight. Excess NaH was destroyed by careful addition of water and the solvents were removed by evaporation under reduced pressure. The residue was partitioned between dichloromethane and water (100 mL of each), the organic layer was separated, dried (MgSO4) and concentrated to give an oily residue. Purification by flash chromatography (ethyl acetate/hexane, 1:4 then 1:3) gave 6 (2.31 g, 4.44 mmol, 89%) as a white solid; m.p. 106–107 °C (from ethyl acetate/hexane). 1H NMR (400 MHz, CDCl3): δ = 3.76 (s, 6 H; 2 OCH3), 4.27–4.32 (m, 3 H; 3 inositol H), 4.47–4.52 (m, 3 H; 3 inositol H), 4.53 (s, 4 H; OCH2Ar), 4.59 (s, 2H; OCH2Ph), 5.49 (s, 1 H; O3CH), 6.66–6.69 (m, 4 H; C6H4OMe), 7.10–7.13 (m, 4 H; C6H4OMe), 7.15–7.24 (m, 5H; Ph); elemental analysis calcd (%) for C30H32O8 (520.57): C 69.22, H 6.20; found: C 69.5, H 6.23.
1-O-Benzyl-scyllo-inositol (7)
A suspension of 6 (1.56 g, 3.00 mmol) in ethanol (30 mL) and HCl (1.0 m, 15 mL) was heated at reflux for 4 h. When the clear solution was allowed to cool to room temperature, crystals of pentaol 7 appeared. The crystals were removed by filtration, washed well with ethanol and dried in vacuo at 60°C to give pure 7 (667 mg, 2.47 mmol, 82%). The crystals sublimed above 220 °C to give new crystals on the cover slip, m.p. 270–272°C. 1H NMR (270 MHz, [D6]DMSO): δ = 2.92–3.08 (m, 4H; 4 inositol H), 3.16 (ddd, 3J(H,H) = 8.4, 8.2, 4.5 Hz, 2H; 2 inositol H), 4.73 (d, 3J(H,H) = 3.2 Hz, 1H; OH-4), 4.77 (s, 2H; OCH2Ph), 4.78 (d, 3J(H,H) = 4.2 Hz, 2H; 2 OH), 4.84 (d, 3J(H,H) = 4.7 Hz, 2 H; 2OH), 7.20–7.34 (m, 3H, Ph), 7.38–7.45 (m, 2 H, Ph); elemental analysis calcd (%) for C13H18O6 (270.3): C 57.77, H 6.71; found: C 57.8, H 6.70.
1-O-Benzyl-scyllo-inositol 2,3,4,5,6-pentakis(dibenzylphosphate) (8)
A solution of 1H-tetrazole (10 mL of a 0.45m solution in acetonitrile, 4.5 mmol) was added to finely powdered pentaol 7 (135 mg, 0.500 mmol) under N2, followed by bis(benzyloxy)diisopropylaminophosphine (1.3 mL, 3.75 mmol). The mixture was stirred at room temperature for 1.5 h and the solvents were then removed by evaporation under reduced pressure. The residue was taken up in dichloromethane (10 mL) and cooled to −78°C before addition of 3-chloroperoxybenzoic acid (60 %, 1.4 g, 5.0 mmol). The mixture was allowed to warm to room temperature, then diluted with dichloromethane (50 mL), washed with aqueous Na2SO3 (10 %) and dried over MgSO4. Evaporation, followed by flash chromatography on silica (ethyl acetate/hexane (1:2) then ethyl acetate) gave 8 as an oil (543 mg, 0.346 mmole, 69%). 1H NMR (270 MHz, CDCl3): δ = 4.16 (t, 3J(H,H) = 4.7 Hz, 1 H; H-1), 4.61 (s, 2H; OCH2Ph), 4.84–5.16 (m, 25H; H-2, H-3, H-4, H-5, H-6, 10OCH2Ph), 7.12–7.32 (m, 55H; 11Ph); 31P NMR (100 MHz, CDCl3, 1H-decoupled): δ = −1.34 (2 P), −1.31 (2 P), −1.20 (1 P). High resolution MS-FAB (positive ion): calcd C83H84O21P5 +: 1571.4193; found: 1571.4172.
scyllo-Inositol 1,2,3,4,5-pentakisphosphate (2) [15]
Palladium on activated charcoal (Aldrich; 10%, 50% water, 1.0 g) was added to a solution of 8 (500 mg, 0.318 mmol) in methanol (60 mL) and deionised water (10 mL). The mixture was shaken in a Parr hydrogenator under an atmosphere of hydrogen (3.5 bar) for 24 h. The catalyst was removed by filtration through a PTFE syringe filter and the solution was neutralised by the addition of triethylammonium bicarbonate (TEAB) buffer (1 m, 2 mL). The solvents were removed by evaporation under reduced pressure. The residue was purified by ion-exchange chromatography on Q-Sepharose fast-flow resin and eluted with a gradient of TEAB buffer (0 to 1.5 m over 950 mL, collecting 10 mL fractions). Fractions were tested for phosphate by using a modification of the Briggs phosphate assay.[31] The target compound (2) eluted at a concentration of 80% buffer. Fractions containing 2 were combined and concentrated by evaporation under reduced pressure. Methanol was repeatedly added and evaporated to decompose excess TEAB until a clear colourless glass remained. Lyophilisation of this residue from deionised water gave the pure triethylammonium salt of 2 as a colourless glassy solid (0.233 mmol as determined by total phosphate assay, 73%). 1H NMR (270 MHz, CD3OD): δ = 3.56 (t, 3J(H,H) = 9.2 Hz, 1 H; H-6), (ddd, 3J(H,H) = 9.4, 9.2 Hz, 3J(H,P) = 9.1 Hz, 2H; 2 inositol H); 4.18–4.30 (m, 3H; 3 inositol H); 31P NMR (109 MHz, CD3OD, 1H-decoupled): δ = 1.44 (2 P), 1.67 (2 P), 1.96 (1P); high-resolution mass MS-FAB (negative ion) calcd for C6H16O21P5−: 578.8872; found: 578.8871.
2-O-Benzyl-4,6-di-O-(p-methoxybenzyl)-1,3,5-O-methylidyne-myo-inositol (9)
Benzylation of alcohol 3[21] (as described for 5) and purification by flash chromatography (ethyl acetate/hexane, 1:3) gave 9 as an oil, which slowly crystallised; m.p. 57–59 °C (from ether). 1H NMR (270 MHz, CDCl3): δ = 3.80 (s, 6 H; 2OCH3), 3.98–4.02 (m, 1 H; H-2), 4.23–4.27 (m, 2H; H-1/3), 4.29 (dd, 3J(H,H) = 3.9, 3.5 Hz, 2 H; H-4/6), 4.37 (m, buried, 1 H; H-5), 4.39, 4.52 (AB system, 2J(H,H) = 11.1 Hz, 4 H; OCH2Ar), 4.63 (s, 2H; OCH2Ph), 5.52 (d, 5J-(H,H) = 1.3 Hz, 1 H; O3CH), 6.78–6.84 (m, 4 H; C6H4OMe), 7.08–7.15 (m, 4H; C6H4OMe), 7.27–7.40 (m, 5H; Ph); elemental analysis: calcd for C30H32O8 (520.57): C 69.22, H 6.20; found: C 69.3, H 6.22.
2-O-Benzyl-myo-inositol (10)
A suspension of 9 (1.56 g, 3.00 mmol) in ethanol (30 mL) and HCl (1.0 m, 15 mL) was heated at reflux for 4 h and then allowed to cool. Crystals of pentaol were isolated in the same way as for 7, to give 10 (547 mg, 2.07 mmol, 67%); the crystals sublimed above 200°C to give new crystals on the cover slip, m.p. 248–251 °C; ref. [23a]: m.p. 248–250°C (dec); ref. [32]: m.p. 250–251 °C. 1H NMR (270 MHz, [D6]DMSO): δ = 2.93 (td, 3J(H,H) = 8.9, 4.5 Hz, 1H; H-5), 3.26 (ddd, 3J(H,H) = 9.6, 4.5, 2.5 Hz, 2 H; H-1/3), 3.39 (ddd, 3J(H,H) = 9.4, 9.1, 4.5 Hz, 2 H; H-4/6), 3.71 (t, 3J(H,H) = 2.5 Hz, 1 H; H-2), 4.64–4.67 (m, 5 H; 5OH), 4.76 (s, 2 H; OCH2Ph), 7.21–7.34 (m, 3H; Ph), 7.37–7.43 (m, 2H; Ph); elemental analysis calcd (%) for C13H18O6 (270.3): C 57.77, H 6.71; found: C 57.5, H 6.65.
2-O-Benzyl-myo-inositol 1,3,4,5,6-pentakis(dibenzylphosphate) (11):[23a]
Phosphitylation of 10 (203 mg, 0.75 mmol) followed by oxidation and purification (as described in the synthesis of 8) gave 11 as a colourless oil (932 mg, 0.593 mmol, 79%). 1H NMR (400 MHz, CDCl3): δ = 4.27 (ddd, 3J(H,H) = 9.8, 2.3 Hz, 3J(H,P) = 9.8 Hz, 1 H; H-1/ 3), 4.38 (dt, 3J(H,P) = 9.8 Hz, 3J(H,H) = 9.4 Hz, 1 H; H-5), 4.73 (t, 3J-(H,H) = 2.3 Hz, 1H; H-2), 4.75 (s, 2 H; OCH2Ph), 4.87–5.07 (m, 22H; 10POCH2Ph, H-4/6), 7.12–7.26 (m, 55 H; 11 Ph); 31P NMR (162 MHz, CDCl3, 1H-decoupled) δ = −0.87 (2 P), −0.18 (2 P), 0.15 (1P); elemental analysis calcd (%) for C83H83O21P5 (1571.4): C 63.44, H 5.32; found: C 63.5, H 5.31.
myo-Inositol 1,3,4,5,6-pentakisphosphate (1):[23]
Hydrogenolytic deprotection of 11 (540 mg, 0.344 mmol) and purification (as described for 2) gave the pure triethylammonium salt of 1 as a colourless glassy solid (0.220 mmol as determined by total phosphate assay, 64 %). 1H NMR (400 MHz, CD3OD) δ = 4.13 (ddd, 3J(H,H) = 9.7, 2.3 Hz, 3J(H,P) = 9.8 Hz, 2 H; H-1/3), 4.18 (dt, 3J(H,P) = 9.8 Hz, 3J-(H,H) = 9.4 Hz, 1 H; H-5), 4.29 (t, 3J(H,H) = 2.3 Hz, 2H; H-2), 4.62 (ddd, 3J(H,P) = 9.7 Hz, 3J(H,H) = 9.7, 9.4 Hz, 2 H; H-4/6); 31P NMR (162 MHz, CD3OD, 1H-decoupled): δ = 1.18 (2 P), 2.00 (2 P), 2.25 (1 P).
Potentiometric studies and NMR determinations
Potentiometric and NMR experiments were performed in two steps, in which the same initial solution of the studied compounds (about 2 mm) was successively subjected to potentiometric and 31P or 1H NMR titrations. The processing of the pH measurements allowed the total concentration of the ligand and the acid as well as the macroscopic protonation constants (by using HYPERQUAD) to be determined. The NMR titrations were performed on 0.50 mL of solution in D2O on a Bruker DPX-300 Fourier transform spectrometer. One-dimensional 31P NMR spectra were recorded at 121.50 MHz and 31P chemical-shifts values were referenced to an external 85 % H3PO4 signal at 0.00 ppm with downfield shifts represented by positive values. Spectra were acquired over a spectral width of 10 ppm by using a 0.1 s relaxation delay and a π/2 pulse. Typically 1 K data points were sampled with a corresponding 0.4 s acquisition time. The spectra had a digital resolution of 1.19 Hz per point. 1H NMR spectra were acquired with water presaturation over a spectral width of 6 ppm by using a 3 s relaxation delay and a π/2 pulse. 4 K data points were sampled with a corresponding 1.14 s acquisition time. The spectra had a digital resolution of 0.44 Hz per point. Data were zero filled and a 1 Hz exponential line broadening function was applied prior to Fourier transformation. The temperature in both cases was controlled at 37 ± 0.5 °C. The proton and phosphorus resonances of Ins(1,3,4,5,6)P5 and scyllo-InsP5 were assigned by performing proton–proton and phosphorus–proton 2D correlation experiments with at least two suitable pH values, this allowed the titration curves to be unambiguously characterized. Cluster and chemical-shift parameters, which were used to calculate the conditional probabilities (Figure 4), were obtained by a nonlinear least-squares fitting procedure to the chemical-shift data. More details of the fitting procedure are provided elsewhere.[25, 33,34]
Cloning of human Ins(1,3,4,5,6)P5 2-kinase
The gene encoding human Ins(1,3,4,5,6)P5 2-kinase was amplified by PCR by using a TrueClone human full-length cDNA clone (OriGene) as template. The upstream primer was 5′-GGGGACAAGTAGTTTGTACAAAAAAGCAGGCACCATGGAAGAGGGGAAGATGGACG-3′, and the downstream primer was 5′-GGGGACCACTTTGTACAAGAAAGCTGGTTAGACCTTGTGGAGAACTAATG-3′. The PCR product was cloned into pDONR vector (Invitrogen) which was subsequently used to construct pDEST605-hIP2kinase by using the manufacturer’s instructions.
Purification of human Ins(1,3,4,5,6)P5 2-kinase from Sf9 cells
The pDEST605-hIP2kinase vector containing the full length human Ins(1,3,4,5,6)P5 2-kinase was used to generate bacmid insect viral DNA (Invitrogen). The His-tagged fusion protein was expressed in Sf9 cells by using the baculovirus expression system (Invitrogen). Cells were infected for 3 days and then lysed in lysis buffer (Promega). The lysate was allowed to bind to Ni Sepharose high performance beads (Amersham Biosciences), washed with HEPES pH 7.3 buffer and eluted with imidazole (200 mm) gradient buffer. The peak elute was run on an SDS-PAGE gel and stained with Simply Blue stain (Invitrogen) and a single protein band of ~60000 kDa was identified.
Expression and purification of recombinant Arabidopsis Ins(1,3,4,5,6)P5 2-kinase
The 2-kinase plasmid (kindly provided by Drs. John York and Stevenson-Paulik, Duke University, NC, USA) was transformed into DH5 E. coli competent cells. DH5 E. coli cells (1 L) were grown overnight at 28°C to an absorbance of ~0.6 at 595 nm. Gene expression was induced by addition of IPTG (1.0 mm). The cells were continuously cultured for 4 h, harvested and frozen at −80 °C. Frozen cells were thawed at 4 °C and added to lysis buffer (PBS pH 7.4, NP40 0.05 %, PMSF 1 mm). The lysate was cleaned by centrifugation at 15000g for 15 min at 4°C and the supernatant was incubated with glutathione–Sepharose resin (1 mL, 50%) for 15 min at room temperature. The Sepharose was washed three times with PBS buffer and the GST fusion protein was eluted with reduced glutathione (1 mL, 10 mm) three times. Each eluate was kept at −80°C in aliquots.
Enzyme-assay conditions
Ins(1,3,4,5,6)P5 2-kinase assays were performed by a slight modification of the procedure described in ref. [35]. Either Ins(1,3,4,5,6)P5 or scyllo-InsP5 (50 μm) was incubated with purified recombinant 2-kinase (1.0 μg) from either Arabidopsis, H. sapiens or S. cerevisiae in buffer (10 μL) containing HEPES (20 mm, pH 7.3), KCl (100 mm), MgSO4 (15 mm) and γ-32P-ATP (0.5 μL, 300 000 cpm) for 5 h at 37°C. Assays were quenched, neutralized and analyzed on a 12.50 4.6 mm Partisphere SAX HPLC column eluted with an ammonium phosphate gradient as previously described.[36] The radioactivity was assessed “online” by using a Flo-1 counter (Packard Instruments) with a 1:3 mixture of HPLC eluate/scintillation fluid (MonoFlo4, National Diagnostics). Data were acquired by using Flo-1 for Windows (v3.61) and then exported as an ASCII file into SigmaPlot (v8.0).
Recombinant human PTEN and MINPP were obtained as previously described (see refs. [9] and [37], respectively). These enzymes were incubated in buffer (100 μL) containing Tris–HCl (50 mm, pH 7.2). Enzyme activity was determined against Ins(1,3,4,5,6)P5 (25 μm) at 37°C by using a phosphate-release assay.[38]
Molecular docking of Ins(1,3,4,5,6)P5 into the active site of PTEN
Molecular-docking experiments were carried out according to general methods previously described.[39] By using the SYBYL 7.0 program (Tripos Associates) Ins(1,3,4,5,6)P5 was extracted from the crystal structure of the PH domain of GRP1 (PDB ID: 1FHW; ligand identifier I5P1001). The crystal structure of PTEN was obtained from the Brookhaven Protein Data Bank (PDB ID: 1D5R). The GOLD docking program (v2.2)[40] was used to dock the flexible model of Ins(1,3,4,5,6)P5 into the PTEN active site, which was defined as a 10 & sphere around the Sγ atom of Cys124. The ligand was docked to the enzyme a total of 40 times and the docking modes that achieved the highest fitness score were retained and compared. The predicted position of Ins(1,3,4,5,6)P5 in the PTEN binding site (Figure 5) resembles earlier predictions[9] made by superimposing the 3- and 4-phosphate groups of a rigid model of Ins(1,3,4,5,6)P5 on the bound l-(+)-tartrate molecule of the X-ray structure; although, in the GOLD-docked model some interactions of phosphate groups with conserved active-site residues, predicted by mutagenesis studies,[29] are better reproduced. The scissile 3-phosphate group of Ins(1,3,4,5,6)P5 is well positioned for nucleophilic attack by Cys124 (not shown) and for stabilising interactions with the electrostatic dipole of a nearby α-helix. Figure 5 was produced by using PyMOL (http://www.pymol.org)
Acknowledgments
We thank the Wellcome Trust for Programme Grant support (060554 to B.V.L.P.). Recombinant Ins(1,3,4,5,6)P5 2-kinase from S. cerevisiae was kindly provided by Dr. John York and Dr. Andrew Seeds. We also thank the Commissariat 3 l’Energie Atomique (CEA) and the Conseil Régional d’Alsace for a Ph.D fellowship (P.K.).
References
- 1.S. B. Shears in The Handbook of Cell Signalling, Vol. 2 (Eds.: R. Bradshaw, E. Dennis), Academic Press, San Diego, 2003, pp. 233–236.
- 2.Orchiston EA, Bennett D, Leslie NR, Clarke RG, Winward L, Downes CP, Safrany ST. J Biol Chem. 2004;279:1116–1122. doi: 10.1074/jbc.M310933200. [DOI] [PubMed] [Google Scholar]
- 3.Piccolo E, Vignati S, Maffucci T, Innominato PF, Riley AM, Potter BVL, Pandolfi PP, Broggini M, Iacobelli S, Innocenti P, Falasca M. Oncogene. 2004;23:1754–1765. doi: 10.1038/sj.onc.1207296. [DOI] [PubMed] [Google Scholar]
- 4.Campbell S, Fisher RJ, Towler EM, Fox S, Issaq HJ, Wolfe T, Phillips LR, Rein A. Proc Natl Acad Sci USA. 2001;98:10 875–10 879. doi: 10.1073/pnas.191224698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steger DJ, Haswell ES, Miller AL, Wente SR, O’Shea EK. Science. 2003;299:114–116. doi: 10.1126/science.1078062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quignard JF, Rakotoarisoa L, Mironneau J, Mironneau C. J Physiol. 2003;549:729–737. doi: 10.1113/jphysiol.2002.037473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maffucci T, Piccolo E, Cumashi A, Iezzi M, Riley AM, Saiardi A, Godage HY, Rossi C, Broggini M, Iacobelli S, Potter BVL, Innocenti P, Falasca M. Cancer Res. 2005;65:8339–8349. doi: 10.1158/0008-5472.CAN-05-0121. [DOI] [PubMed] [Google Scholar]
- 8.Komander D, Fairservice A, Deak M, Kular GS, Prescott AR, Downes CP, Safrany ST, Alessi DR, van Aalten DMF. EMBO J. 2004;23:3918–3928. doi: 10.1038/sj.emboj.7600379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caffrey JJ, Darden T, Wenk MR, Shears SB. FEBS Lett. 2001;499:6–10. doi: 10.1016/s0014-5793(01)02500-5. [DOI] [PubMed] [Google Scholar]
- 10.Irvine RF, Schell MJ. Nat Rev Mol Cell Biol. 2001;2:327–338. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- 11.Ho MWY, Shears SB. Curr Top Membr. 2002;53:345–363. [Google Scholar]
- 12.Lemmon MA, Ferguson KM, Abrams CS. FEBS Lett. 2002;513:71–76. doi: 10.1016/s0014-5793(01)03243-4. [DOI] [PubMed] [Google Scholar]
- 13.Shears SB. Cell Signal. 2001;13:151–158. doi: 10.1016/s0898-6568(01)00129-2. [DOI] [PubMed] [Google Scholar]
- 14.Lemtiri-Chlieh F, MacRobbie EAC, Brearley CA. Proc Natl Acad Sci USA. 2000;97:8687–8692. doi: 10.1073/pnas.140217497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung SK, Kwon YU, Chang YT, Sohn KH, Shin JH, Park KH, Hong BJ, Chung IH. Bioorg Med Chem. 1999;7:2577–2589. doi: 10.1016/s0968-0896(99)00183-2. [DOI] [PubMed] [Google Scholar]
- 16.Horne G, Maechling C, Fleig A, Hirata M, Penner R, Spiess B, Potter BVL. Biochem Biophys Res Commun. 2004;320:1262–1270. doi: 10.1016/j.bbrc.2004.06.079. [DOI] [PubMed] [Google Scholar]
- 17.Verbsky JW, Wilson MP, Kisseleva MV, Majerus PW, Wente SR. J Biol Chem. 2002;277:31 857–31 862. doi: 10.1074/jbc.M205682200. [DOI] [PubMed] [Google Scholar]
- 18.Stevenson-Paulik J, Bastidas RJ, Chiou ST, Frye RA, York JD. Proc Natl Acad Sci USA. 2005;102:12 612–12 617. doi: 10.1073/pnas.0504172102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shears SB. Biochem J. 2004;377:265–280. doi: 10.1042/BJ20031428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turner BL, Paphazy MJ, Haygarth PM, McKelvie ID. Philos Trans R Soc London Ser B. 2002;357:449–469. doi: 10.1098/rstb.2001.0837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riley AM, Guedat P, Schlewer G, Spiess B, Potter BVL. J Org Chem. 1998;63:295–305. [Google Scholar]
- 22.Lee HW, Kishi Y. J Org Chem. 1985;50:4402–4404. [Google Scholar]
- 23.Lu PJ, Gou DM, Shieh WR, Chen CS. Biochemistry. 1994;33:11586–11 597. doi: 10.1021/bi00204a021. a) [DOI] [PubMed] [Google Scholar]; Ozaki S, Koga Y, Ling L, Watanabe Y, Kimura Y, Hirata M. Bull Chem Soc Jpn. 1994;67:1058–1063. b) [Google Scholar]; Chung SK, Chang YT. Bioorg Med Chem Lett. 1996;6:2039–2042. c) [Google Scholar]; Rudolf MT, Kaiser T, Guse AH, Mayr GW, Schultz C. Liebigs Ann/Recl. 1997:1861–1869. doi: 10.1006/bbrc.1997.7629. d) [DOI] [PubMed] [Google Scholar]; Podeschwa MAL, Plettenburg O, Altenbach HJ. Eur J Org Chem. 2005:3101–3115. e) [Google Scholar]
- 24.Barrientos LG, Murthy PPN. Carbohydr Res. 1996;296:39–54. doi: 10.1016/s0008-6215(96)00250-9. [DOI] [PubMed] [Google Scholar]
- 25.Borkovec M, Koper GJM. Anal Chem. 2000;72:3272–3279. doi: 10.1021/ac991494p. [DOI] [PubMed] [Google Scholar]
- 26.Nogimori K, Hughes PJ, Glennon MC, Hodgson ME, Putney JW, Shears SB. J Biol Chem. 1991;266:16 499–16 506. [PubMed] [Google Scholar]
- 27.Ballereau S, Guédat P, Poirier SN, Guillemette G, Spiess B, Schlewer G. J Med Chem. 1999;42:4824–4835. doi: 10.1021/jm991084t. [DOI] [PubMed] [Google Scholar]
- 28.Felemez M, Bernard P, Schlewer G, Spiess B. J Am Chem Soc. 2000;122:3156–3165. [Google Scholar]
- 29.Lee JO, Yang HJ, Georgescu MM, Di Cristofano A, Maehama T, Shi YG, Dixon JE, Pandolfi P, Pavletich NP. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 30.Jackson MD, Denu JM. Chem Rev. 2001;101:2313–2340. doi: 10.1021/cr000247e. Reviewed in; a) [DOI] [PubMed] [Google Scholar]; Bialy L, Waldmann H. Angew Chem. 2005;117:3880–3906. b) Angew. Chem. Int. Ed. 2005, 44, 3814–3839; [Google Scholar]; Dewang PM, Hsu NM, Peng SZ, Li WR. Curr Med Chem. 2005;12:1–22. doi: 10.2174/0929867053363504. c) [DOI] [PubMed] [Google Scholar]
- 31.Lampe D, Liu CS, Potter BVL. J Med Chem. 1994;37:907–912. doi: 10.1021/jm00033a007. [DOI] [PubMed] [Google Scholar]
- 32.Sureshan KM, Shashidhar MS, Praveen T, Gonnade RG, Bhadbhade MM. Carbohydr Res. 2002;337:2399–2410. doi: 10.1016/s0008-6215(02)00298-7. [DOI] [PubMed] [Google Scholar]
- 33.Borkovec M, Spiess B. Phys Chem Chem Phys. 2004;6:1144–1151. [Google Scholar]
- 34.Kuad P, Borkovec M, Murr MDE, Le Gall T, Mioskowski C, Spiess B. J Am Chem Soc. 2005;127:1323–1333. doi: 10.1021/ja0483185. [DOI] [PubMed] [Google Scholar]
- 35.Stevenson-Paulik J, Odom AR, York JD. J Biol Chem. 2002;277:42 711–42 718. doi: 10.1074/jbc.M209112200. [DOI] [PubMed] [Google Scholar]
- 36.Saiardi A, Caffrey JJ, Snyder SH, Shears SB. J Biol Chem. 2000;275:24 686–24 692. doi: 10.1074/jbc.M002750200. [DOI] [PubMed] [Google Scholar]
- 37.Deleu S, Choi K, Pesesse X, Cho J, Sulis ML, Parsons R, Shears SB. Cell Signal. 2006;18:488–498. doi: 10.1016/j.cellsig.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 38.Ullah AHJ, Sethumadhavan K, Mullaney EJ, Ziegelhoffer T, Austin-Phillips S. Biochem Biophys Res Commun. 1999;264:201–206. doi: 10.1006/bbrc.1999.1501. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg HJ, Riley AM, Laude AJ, Taylor CW, Potter BVL. J Med Chem. 2003;46:4860–4871. doi: 10.1021/jm030883f. [DOI] [PubMed] [Google Scholar]
- 40.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]