

Abstract
Multiple lines of evidence suggest that calcium/calmodulin-dependent kinase II α (CaMKIIα) plays an important role in the spinal dorsal horn in nociceptive models of chemical, inflammatory and nerve injury. Moreover, CaMKIIα phosphorylates the vanilloid receptor type 1 (TRPV1), thereby regulating vanilloid agonist binding to the receptor. Herein, we have explored a possible interaction of CaMKIIα activity with the TRPV1 receptor in rat trigeminal ganglion (TG) neurons in vitro. Inhibition of CaMKIIα with KN-93 (5 μM) inhibited capsaicin (CAP)- and n-arachidonoyl-dopamine (NADA)-evoked calcitonin gene-related peptide (CGRP) release effectively decreasing the Emax for both compounds. This effect was not mimicked by the inactive compound KN-92 (5 μM), indicating that the effect was mediated by CaMKIIα inhibition. CAP also stimulated a significant ~50% increase in autophosphorylation of CaMKIIα at Thr286/287. Immunocytochemistry for phospho-CaMKIIα indicated that this effect specifically occurred in TRPV1-positive TG neurons. These findings indicate that phopho-CaMKIIα is likely to play a role in presynaptic primary afferents in animal models of nociceptive hypersensitivity and provide support for CaMKIIα modulation of TRPV1 activity in sensory neurons.
Keywords: Calcitonin-gene related peptide, Calcium/calmodulin-dependent kinase IIα, Vanilloid receptor type 1, Trigeminal ganglion, Pain
Calcium/calmodulin-dependent kinase II α (CaMKIIα) plays a well-established role in synaptic plasticity in CNS neurons. CaMKIIα is activated by NMDA receptors and is involved in phosporylation and trafficking of AMPA receptors, both of which are important steps in long term potentiation (LTP, recently reviewed in [7]). The importance of CaMKIIα is most firmly established postsynaptically; however, CaMKIIα is also involved in presynaptic facilitation of neurotransmitter release [6], likely through phosphorylation of vesicular release proteins [20].
Recently, a role for CaMKIIα has emerged in nociception, wherein CaMKIIα is believed to be involved in central sensitization [27]. CaMKIIα is expressed by PKC-gamma-expressing interneurons of lamina II in the dorsal horn [28]. CaMKIIα is also expressed by vanilloid receptor type 1 (TRPV1 [4])-immunoreactive dorsal root ganglion (DRG) neurons. Moreover, CaMKIIα is expressed in calcitonin gene-related peptide (CGRP)- and TRPV1-immunoreactive neurons of the trigeminal ganglion (TG [14]). CaMKIIα is also transported to central and peripheral terminals [3] of nociceptors, and it colocalizes to both substance P (SP)- immunoreactive and isolectin B4-binding afferent terminals in the dorsal horn [14]. Dorsal horn CaMKIIα protein levels increase following intraplantar formalin injection [18], and CaMKIIα protein in the medullary dorsal horn is like-wise increased following nerve injury to the inferior alveolar nerve wherein mechanical allodynia (as measured by escape behavior) is alleviated by CaMKIIα inhibition [21]. Peripheral CaMKIIα immunoreactivity also increases following complete Freund’s adjuvant (CFA)-induced inflammation [3]. CaMKIIα appears to be involved in spinal LTP, as inhibition of CaMKIIα in identified nociceptive dorsal horn neurons blocks the development of high-frequency stimulation-induced LTP [22]. Finally, autophosphorylation-deficient CaMKIIα mutant mice display deficiencies in ongoing nociceptive responses in the formalin model, leading to the hypothesis that CaMKIIα is primarily involved in spontaneous nociceptive responses [28].
CaMKIIα appears to be particularly involved in the capsaicin (CAP) pain model and has been implicated in the modulation of the capsaicin receptor, TRPV1. Thus, spinal CaMKIIα protein and phospho-CaMKIIα increase following intraplantar injection of capsaicin [9]. Moreover, spinal CaMKIIα inhibition abrogates decreased exploratory behavior, central sensitization of nociceptive dorsal horn neurons and GluR1 receptor phosphorylation [9] following intra-plantar CAP injection. Intracolonic CAP injection-stimulated delivery of dorsal horn GluR1 receptors to the membrane is also blocked by inhibition of CaMKIIα [10]. In DRG neurons, CaMKIIα has been implicated in the sensitization of TRPV1 responses induced by nerve growth factor (NGF [2]), and CaMKIIα directly phosphorylates the TRPV1 receptor at residues at which protein kinase A and C also phosphorylate TRPV1 [15]. Phosphorylation of TRPV1 by CaMKIIα regulates the binding of vanilloid compounds to the receptor, and CaMKIIα phosphorylation appears to be necessary for CAP-induced activation of TRPV1 [15]. Hence, CaMKIIα is important for CAP actions at TRPV1 and CAP-induced central sensitization. However, it is unclear whether presynaptically activated CaMKIIα might be involved in the effects of CAP injection. Here, we demonstrate that CAP stimulates CaMKIIα phosphorylation in TG neurons and that inhibition of CaMKIIα modulates CAP-evoked neuropeptide release from these neurons in vitro. These findings indicate that presynaptic effects of increased CaMKIIα activity might be involved in the effects of CAP injection in vivo.
To study CAMKIIα phosphorylation state and the effects of inhibition of CaMKIIα on neuropeptide release specifically in sensory neurons, we utilized cultured TG neurons from adult (250–300 g) rats (Sprague–Dawley, Charles River). All animal procedures were approved by The University of Texas Health Science Center at San Antonio Animal Care and Use Committee and were in accordance with NIH guidelines. TG neurons were cultured as previously described [23], at a density of ~5000 neurons/well in 48-well plates for CGRP release experiments and at a density of ~40,000 neurons/well in 6-well plates for phospho-CaMKIIα assessment. CGRP release assays and radioimmunoassays were performed as described previously [23]. For immunocytochemistry (ICC, method described in [24]), neurons were grown on glass coverslips in 12-well plates at a density of ~500 neurons/well. Before assaying, all TG neuronal cultures were maintained for 5 days in DMEM media supplemented with 10% fetal bovine serum and 100 ng/ml NGF. The anti-phospho-CaMKIIα (Thr286/287, 1:1000 for ICC and Western blotting) antibody and anti-CaMKIIα (1:1000 for Western blotting) antibody were from Chemicon. Both CaMKIIα antibodies recognize only a single band at 50 kDa corresponding to the molecular weight of CaMKIIα by Western blot. The anti-TRPV1 antibody (1:2000) was from Neuromics (see [12] for details on antibody specificity). Western blotting was performed by SDS–PAGE, and phospho-CaMKIIα levels were standardized to total CaMKIIα expression.
We first explored the effect of CaMKIIα inhibition on CAP-evoked CGRP release from TG neurons. TG neurons were preincubated for 10 min with the CaMKIIα inhibitor KN-93 (5 μM) or vehicle and then exposed to CAP (0.3 nM–30 μM) for 10 additional minutes in the continued presence of KN-93. CAP-evoked CGRP release concentration–response functions showed a U-shaped curve, wherein the downward phase of the curve is likely due to rapid desensitization of TRPV1 by higher concentrations of CAP. KN-93 had no effect on the upward EC50 for CAP-evoked CGRP release but did significantly reduce the Emax measured at 100 nM (Fig. 1). KN-93 also significantly inhibited 300 and 600 nM CAP-evoked CGRP release (Fig. 1). The CaMKIIα-inactive, but structurally related compound KN-92 (5 μM) had no effect on CAP-evoked release at concentrations of CAP inhibited by KN-93 (Fig. 1).
Fig. 1.
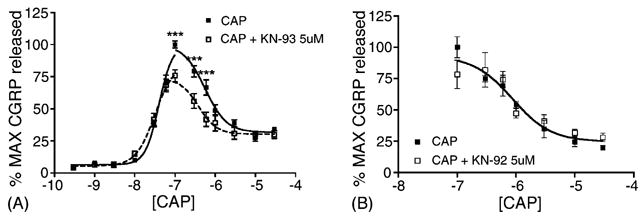
TG neurons were preincubated with VEH (closed squares), KN-93 (A, 5 μM open squares) or KN-92 (B, 5 μM, open squares) for 10 min and then exposed to the indicated concentrations of CAP in log units for an additional 10 min in the continued presence of the KN compound (***p < 0.001, two-way ANOVA, n = 6–9 for each concentration of CAP).
Because the largest effect of KN-93 on CAP-evoked release was observed at 300 nM CAP, we tested whether this concentration of CAP was capable of stimulating CaMKIIα autophosphorylation at Thr286/287, a requisite step in CaMKIIα activation. When TG neurons were exposed to 300 nM CAP for 10 min, a significant, ~50% increase in phospho-CaMKIIα was observed (Fig. 2A and B). We next utilized the same concentration and time exposure of CAP in ICC studies to examine whether the phospho-CaMKIIα occurred specifically in TRPV1-immunoreactive neurons. As shown in Fig. 2C, phospho-CaMKIIα immunoreactivity was evident in TRPV1-positive TG neurons, while no above background immunoreactivity was noted in TRPV1-negative neurons. Due to the inherent difficulties in accurately quantifying immunoreactivity signals, no attempt was made to assess increases in phospho-CaMKIIα in these neurons; however, these data indicate that the increases in phospho-CaMKIIα observed by Western blot were likely to have occurred specifically in TRPV1-immunoreactive neurons.
Fig. 2.
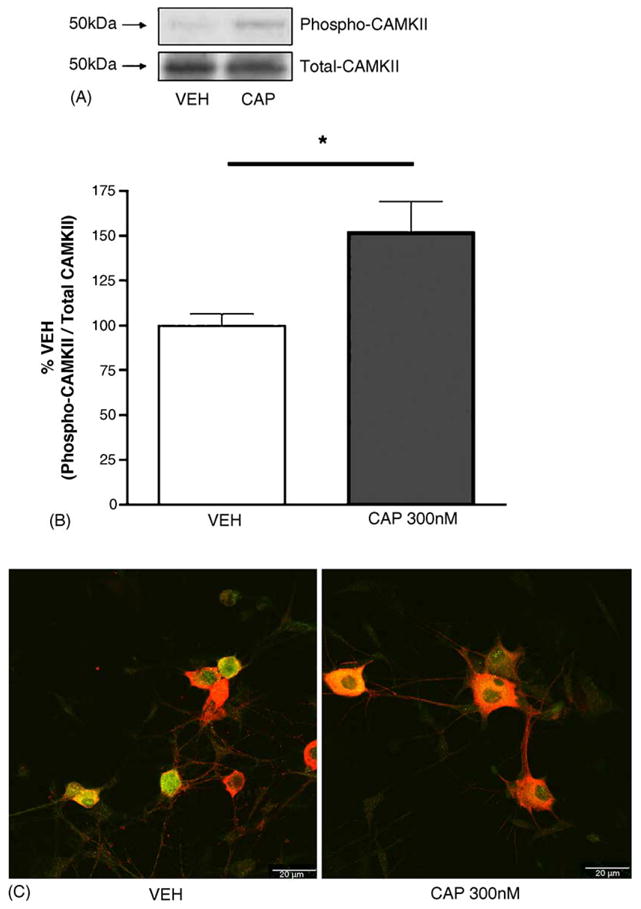
TG neurons were exposed to VEH or 300 nM CAP for 10 min, scraped into tubes on ice and prepared for protein analysis by SDS–PAGE. Panel A illustrates a representative Western blot for phopho-CaMKIIα (Thr286/287) and total CaMKIIα protein, which has been quantitated in panel B (*p < 0.05, Student’s t-test). Panel C: Confocal images show independent TG neuronal culture representative photomicrographs taken at 63× (scale bar = 20 μm) of phospho-CaMKIIα immunoreactivity (green) and TRPV1-immunoreactivity (red, colocalization in yellow) in TG neurons exposed to VEH or CAP for 10 min.
In addition to the prototypic TRPV1 agonist CAP, several endogenous TRPV1 agonists have been identified. Of particular interest is the endogenous cannabinoid/vanilloid ligand n-arachidonoyl dopamine (NADA), which exhibits greater potency than other endogenous cannabinoid/vanilloid agonists, such as anandamide. To evaluate whether KN-93-induced inhibition of CGRP release also occurs with other TRPV1 agonists, we examined the effect of KN-93 on NADA (10 nM–30 μM)-evoked CGRP release. KN-93 (5 μM) pre-treatment inhibited NADA-evoked CGRP release at 3 and 10 μM (Fig. 3), effectively decreasing the Emax for NADA-evoked CGRP release. Concentrations of NADA at and above 30 μM were not utilized because we have previously shown non-TRPV1 mediated effects of NADA on evoked CGRP release at these higher concentrations [23].
Fig. 3.
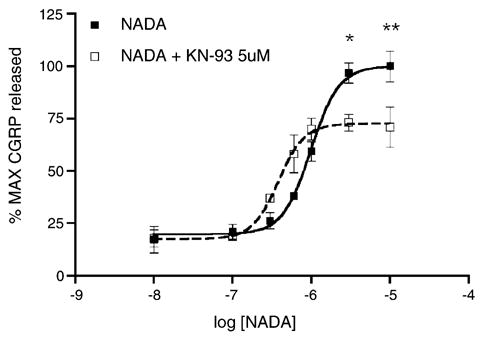
TG neurons were preincubated with KN-93 (5 μM, open squares) or VEH (closed squares) for 10 min and then exposed to the indicated concentrations of NADA in log units for an additional 10 min in the continued presence of KN-93 (*p < 0.05, **p < 0.01, two-way ANOVA, n = 6 per concentration of NADA).
We have demonstrated that CAP stimulates CaMKIIα phosphorylation at Thr286/287 in TRPV1-immunoreactive neurons. This is likely relevant to the in vivo CAP model, because previous studies have indicated that CaMKIIα kinase activity plays an important role in the activation of downstream effectors, such as AMPA receptors, as well as in behavioral manifestations of the model [9]. Many of these effects have been attributed to postsynaptic actions of CaMKIIα [9,10]; however, the present findings indicate that presynaptic actions might also be involved. In that regard, it has been shown that CaMKIIα [28] and phospho-CaMKIIα [17] are present in normal animals in presynaptic C-fiber terminals of the dorsal horn. CaMKIIα plays a role in augmenting synaptic vesicle release through phosphorylation of proteins involved in vesicle fusion [20]. Moreover, the release of neuropeptides, especially SP [1] and CGRP [25], is believed to play an important role in central sensitization, and is increased following peripheral inflammation [11]. To this end, we have also illustrated that CaMKIIα inhibition is capable of inhibiting CAP-evoked CGRP release, particularly at higher CAP concentrations, and that CAP stimulates CaMKIIα autophosphorylation in sensory neurons themselves. Our findings thus indicate that these presynaptic effects might be important with regard to dorsal horn CaMKIIα inhibition in the CAP model.
CaMKIIα has been implicated in regulating vanilloid binding to the TRPV1 receptor [15]. In this regard, there appears to be an interplay between calcineurin and CaMKIIα, wherein calcineurin is involved in regulating calcium-sensitive desensitization through direct phosphatase activity [8]. CaMKIIα, on the other hand, appears to be involved in the resensitization of the TRPV1 receptor through re-phosphorylation of residues required for vanilloid binding to the receptor [15]. There are numerous examples of opposing actions of CaMKIIα and calcineurin on calcium-permeable ion channels (such as the α 7 nicotinic receptor [19]) and synaptic plasticity [26] and the balance of kinase to phosphatase activity appears to depend on local calcium concentrations [26]. We have demonstrated that inhibition of CaMKIIα diminishes CAP-evoked CGRP release, especially at concentrations at which the CAP-evoked CGRP release concentration–response function illustrates a downward inflection. Insofar as CaMKIIα inhibition shifts the CAP-evoked CGRP release concentration–response function toward greater desensitization (as measured by decreased CGRP release), the present finding supports the notion that CaMKIIα is involved in resensitization of TRPV1, at least when CAP is the agonist utilized. Because we also observed an increase in phospho-CaMKIIα with CAP treatment, calcium entry through TRPV1 itself might be involved in an autoregulatory loop through which CaMKIIα becomes autophosphorylated and resensitizes TRPV1. Since both desensitization and resensitization of TRPV1 appear to involve calcium-dependent mechanisms, gaining a more complete understanding of how calcium signals mediate these distinct mechanisms might yield novel insight into how TRPV1 activity can be therapeutically modulated.
We have also demonstrated that CaMKIIα inhibition attenuates NADA-evoked CGRP release. Since concentrations of NADA above 30 μM have non-specific effects on evoked CGRP release [23] we were unable to examine desensitization effects in a manner analogous to that performed for CAP. However, CaMKIIαinhibition reduced the Emax for NADA-evoked CGRP release much as it did for CAP-evoked CGRP release. Other methods will be required, such as patch-clamp electrophysiology, to determine the effects of CaMKIIα on NADA-induced TRPV1 desensitization/resensitization. The TRPV1 receptor plays an important role in inflammation-induced nociception [5], and multiple lines of evidence suggest that endogenous agonists are involved in TRPV1-mediated nociception. Intrathecal and local peripheral administration of the TRPV1 antagonist A-425619 attenuated thermal hyperalgesia in the inflammatory CFA model and mechanical allodynia in the neuropathic sciatic nerve ligation model [13]. Moreover, spinal application of capsazepine inhibits A∂ and C-fiber-driven responses of spinal dorsal horn neurons [16]. NADA is an attractive candidate for this action due to its potency and efficacy at TRPV1 receptors. Additionally, central [9] and peripheral [3] CaMKIIα appears to be involved in inflammation-induced nociception. Our observation that inhibition of CaMKII attenuates endogenous mediator activation of TRPV1-mediated sensory neuron activity (as measured by CGRP release) indicates that targeting CaMKIIα interactions with TRPV1 might be an effective manner through which TRPV1-mediated nociception can be modulated.
In summary, we have provided evidence that CAP stimulates autophosphorylation of CaMKIIα in sensory neurons and that pharmacological inhibition of CaMKIIα reduces TRPV1-mediated CGRP release. These results suggest that presynaptic CaMKIIα autophosphorylation is likely to contribute to increases in phospho-CaMKIIα observed in the dorsal horn in the CAP inflammation model and support the proposed role of CaMKIIα in regulation of TRPV1-mediated afferent activity.
Acknowledgments
This work was supported by NIDA grant DA11959.
References
- 1.Afrah AW, Fiska A, Gjerstad J, Gustafsson H, Tjolsen A, Olgart L, Stiller CO, Hole K, Brodin E. Spinal substance P release in vivo during the induction of long-term potentiation in dorsal horn neurons. Pain. 2002;96:49–55. doi: 10.1016/s0304-3959(01)00414-6. [DOI] [PubMed] [Google Scholar]
- 2.Bonnington JK, McNaughton PA. Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J Physiol. 2003;551:433–446. doi: 10.1113/jphysiol.2003.039990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlton SM. Localization of CaMKIIalpha in rat primary sensory neurons: increase in inflammation. Brain Res. 2002;947:252–259. doi: 10.1016/s0006-8993(02)02932-3. [DOI] [PubMed] [Google Scholar]
- 4.Carlton SM, Hargett GL. Stereological analysis of Ca(2+)/calmodulin-dependent protein kinase II alpha-containing dorsal root ganglion neurons in the rat: colocalization with isolectin Griffonia simplicifolia, calcitonin gene-related peptide, or vanilloid receptor 1. J Comp Neurol. 2002;448:102–110. doi: 10.1002/cne.10250. [DOI] [PubMed] [Google Scholar]
- 5.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 6.Chapman PF, Frenguelli BG, Smith A, Chen CM, Silva AJ. The alpha-Ca2+/calmodulin kinase II: a bidirectional modulator of presynaptic plasticity. Neuron. 1995;14:591–597. doi: 10.1016/0896-6273(95)90315-1. [DOI] [PubMed] [Google Scholar]
- 7.Colbran RJ, Brown AM. Calcium/calmodulin-dependent protein kinase II and synaptic plasticity. Curr Opin Neurobiol. 2004;14:318–327. doi: 10.1016/j.conb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Docherty RJ, Yeats JC, Bevan S, Boddeke HW. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch. 1996;431:828–837. doi: 10.1007/s004240050074. [DOI] [PubMed] [Google Scholar]
- 9.Fang L, Wu J, Lin Q, Willis WD. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 2002;22:4196–4204. doi: 10.1523/JNEUROSCI.22-10-04196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Garry MG, Hargreaves KM. Enhanced release of immunoreactive CGRP and substance P from spinal dorsal horn slices occurs during carrageenan inflammation. Brain Res. 1992;582:139–142. doi: 10.1016/0006-8993(92)90328-7. [DOI] [PubMed] [Google Scholar]
- 12.Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- 13.Honore P, Wismer CT, Mikusa JP, Zhu CZ, Zhong C, Gauvin DM, Gomtsyan A, El Kouhen R, Lee CH, Marsh K, Sullivan JP, Faltynek CR, Jarvis MF. A-425619, a novel TRPV1 receptor antagonist, relieves pathophysiological pain associated with inflammation and tissue injury in rats. J Pharmacol Exp Ther. 2005 doi: 10.1124/jpet.105.083915. [DOI] [PubMed] [Google Scholar]
- 14.Ichikawa H, Gouty S, Regalia J, Helke CJ, Sugimoto T. Ca2+/calmodulin-dependent protein kinase II in the rat cranial sensory ganglia. Brain Res. 2004;1005:36–43. doi: 10.1016/j.brainres.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 15.Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U. Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem. 2004;279:7048–7054. doi: 10.1074/jbc.M311448200. [DOI] [PubMed] [Google Scholar]
- 16.Kelly S, Chapman V. Spinal administration of capsazepine inhibits noxious evoked responses of dorsal horn neurons in non-inflamed and carrageenan inflamed rats. Brain Res. 2002;935:103–108. doi: 10.1016/s0006-8993(02)02552-0. [DOI] [PubMed] [Google Scholar]
- 17.Larsson M, Broman J. Different basal levels of CaMKII phosphorylated at Thr 286/287 at nociceptive and low-threshold primary afferent synapses. Eur J Neurosci. 2005;21:2445–2458. doi: 10.1111/j.1460-9568.2005.04081.x. [DOI] [PubMed] [Google Scholar]
- 18.Liang DY, Li X, Clark JD. Formalin-induced spinal cord calcium/calmodulin-dependent protein kinase II alpha expression is modulated by heme oxygenase in mice. Neurosci Lett. 2004;360:61–64. doi: 10.1016/j.neulet.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 19.Liu Q, Berg DK. Actin filaments and the opposing actions of CaM kinase II and calcineurin in regulating alpha7-containing nicotinic receptors on chick ciliary ganglion neurons. J Neurosci. 1999;19:10280–10288. doi: 10.1523/JNEUROSCI.19-23-10280.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Llinas R, McGuinness TL, Leonard CS, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci USA. 1985;82:3035–3059. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogawa A, Dai Y, Yamanaka H, Iwata K, Niwa H, Noguchi K. Ca(2+)/calmodulin-protein kinase IIalpha in the trigeminal subnucleus caudalis contributes to neuropathic pain following inferior alveolar nerve transection. Exp Neurol. 2005;192:310–319. doi: 10.1016/j.expneurol.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Pedersen LM, Lien GF, Bollerud I, Gjerstad J. Induction of long-term potentiation in single nociceptive dorsal horn neurons is blocked by the CaMKII inhibitor AIP. Brain Res. 2005;1041:66–71. doi: 10.1016/j.brainres.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM. Modulation of trigeminal sensory neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-arachidonoyl-dopamine and arachidonyl-2-chloroethylamide. Br J Pharmacol. 2004;141:1118–1130. doi: 10.1038/sj.bjp.0705711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Price TJ, Patwardhan AM, Flores CM, Hargreaves KM. A role for the anandamide membrane transporter in TRPV1-mediated neurosecretion from trigeminal sensory neurons. Neuropharmacology. 2005;49:25–39. doi: 10.1016/j.neuropharm.2005.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun RQ, Tu YJ, Lawand NB, Yan JY, Lin Q, Willis WD. Calcitonin gene-related peptide receptor activation produces PKA- and PKC-dependent mechanical hyperalgesia and central sensitization. J Neurophysiol. 2004;92:2859–2866. doi: 10.1152/jn.00339.2004. [DOI] [PubMed] [Google Scholar]
- 26.Wang HX, Gerkin RC, Nauen DW, Bi GQ. Coactivation and timing-dependent integration of synaptic potentiation and depression. Nat Neurosci. 2005;8:187–193. doi: 10.1038/nn1387. [DOI] [PubMed] [Google Scholar]
- 27.Willis WD. Long-term potentiation in spinothalamic neurons. Brain Res Brain Res Rev. 2002;40:202–214. doi: 10.1016/s0165-0173(02)00202-3. [DOI] [PubMed] [Google Scholar]
- 28.Zeitz KP, Giese KP, Silva AJ, Basbaum AI. The contribution of autophosphorylated alpha-calcium-calmodulin kinase II to injury-induced persistent pain. Neuroscience. 2004;128:889–898. doi: 10.1016/j.neuroscience.2004.07.029. [DOI] [PubMed] [Google Scholar]