

Abstract
Many n-acylethanolamines utilize the anandamide membrane transporter (AMT) to gain facilitated access to the intracellular compartment, hence, we hypothesized that this mechanism might be important for anandamide (AEA)- and N-arachidonoyl-dopamine (NADA)-evoked CGRP release from cultured trigeminal ganglion (TG) neurons. Using [14C]AEA we demonstrated that TG neurons transported AEA in a FAAH- and AMT-inhibitable fashion. Although TRPV1-positive TG neurons were found to express fatty acid amide hydrolase, the application of FAAH inhibitors had no effect on AEA-evoked CGRP release. In contrast, application of the AMT inhibitors OMDM-2 or VDM-11 significantly reduced the potency and efficacy of AEA-, NADA- and capsaicin-evoked CGRP release. Moreover OMDM-2 (IC50 values ranging from 6.4–9.6 μM) and VDM-11 (IC50 values ranging from 5.3–11 μM) inhibited CGRP release evoked by EC80 concentrations of AEA, NADA and CAP and these values were consistent with IC50s obtained for inhibition of uptake. OMDM-2 had no effect on CGRP release per se while VDM-11 evoked CGRP release on its own (EC50 ~35 μM) in a CPZ-insensitive, but ruthenium red (RR)-sensitive fashion. This is the first demonstration that TG sensory neurons possess an AMT-like mechanism suggesting that this mechanism is important for the pharmacological action of AEA and NADA at native TRPV1 channels.
Keywords: Anandamide membrane transport, Cannabinoid, Vanilloid, Pain, Sensory neuron, Neuropeptide release
1. Introduction
The endogenous cannabinoid/vanilloid agonists anandamide (AEA) and N-arachidonoyl-dopamine (NADA) bind to both G-protein-coupled cannabinoid receptors (eg., CB1 and CB2 Bisogno et al., 2000; Devane et al., 1992) and the capsaicin (CAP)-activated ion channel vanilloid receptor type 1 (TRPV1 Huang et al., 2002; Zygmunt et al., 1999). AEA and NADA primarily exert their effects on trigeminal ganglion (TG) nociceptors through TRPV1 (Jennings et al., 2003; Price et al., 2004b; Roberts et al., 2002) and the cellular uptake of AEA (Beltramo et al., 1997; Di Marzo et al., 1994) and NADA (Bisogno et al., 2000; Huang et al., 2002) is mediated by the putative anandamide membrane transporter (AMT). Since the proposed binding site for TRPV1 agonists is on an intracellular domain of the receptor (Jordt and Julius, 2002; Jung et al., 1999, 2002; Welch et al., 2000), we hypothesized that the AMT might be utilized by AEA and NADA in TG neurons to gain access to this intracellular binding site.
Although the AMT has yet to be molecularly identified, multiple lines of evidence suggest its existence. The cellular uptake of AEA is a saturable process that is time- and temperature-dependent (Beltramo et al., 1997). Moreover, AEA uptake is inhibited by multiple proposed AMT inhibitors and there are strict molecular determinants for the pharmacological inhibition of uptake (Hillard and Jarrahian, 2000; Hillard and Jarrahian, 2003; Reggio and Traore, 2000) and uptake itself (Piomelli et al., 1999). AEA uptake has been demonstrated in many cell lines as well as in CNS neurons (Beltramo et al., 1997; Di Marzo et al., 1994; Hajos et al., 2004; Ortega-Gutierrez et al., 2004) and astrocytes (Beltramo et al., 1997). Additionally, AMT inhibitors augment extracellular AEA concentrations in vivo (Giuffrida et al., 2000) and increase the behavioral effects of exogenously administered AEA (de Lago et al., 2004). Hence, the primary role of the AMT in the CNS has been proposed as concentrating AEA within neurons and glia that contain the AEA-degrading enzyme fatty acid amide hydrolase (FAAH) (Giuffrida et al., 2001; Hillard and Jarrahian, 2003; Piomelli, 2003). FAAH also appears to be involved in concentrating AEA within cells (Day et al., 2001; Glaser et al., 2003; Ortega-Gutierrez et al., 2004), and so the relative contribution of FAAH and AMT to AEA uptake remains controversial (Hillard and Jarrahian, 2003). However, the persistence of AEA accumulation in FAAH knockout mice indicates that another mechanism, different from FAAH, and pharmacologically blocked by AMT inhibitors, plays an essential role in this process (Fegley et al., 2004; Ortega-Gutierrez et al., 2004).
The present study was undertaken to address the following hypotheses concerning AEA- and NADA-mediated actions on TG neurons. First, do TG neurons take up AEA by a process that is inhibited by AMT antagonists? Second, do AMT inhibitors inhibit TRPV1-mediated actions of AEA and NADA in TG neurons? To address these hypotheses we utilized two structurally distinct AMT inhibitors, OMDM-2 (Ortar et al., 2003) and VDM-11 (De Petrocellis et al., 2000) that have negligible activity at TRPV1 and do not inhibit FAAH. The findings presented here indicate that TG neurons do contain an AMT-like mechanism and that AMT antagonists limit the activation of TRPV1 by AEA, NADA and, somewhat surprisingly, CAP.
2. Materials and methods
2.1. Reagents
The cannabinoid/vanilloid agonists AEA, arachidonyl-2-chloroethylamide (ACEA) and NADA were all from Tocris (Ellisville, MO). The AMT inhibitors OMDM-2 and VDM-11 were from Tocris. The FAAH inhibitors included methyl-ester-phosphonofluoridic acid (MAFP, Tocris), URB-597 (Sigma) and CAY10400 (Cayman Chemical, Ann Arbor, MI). The TRPV1 agonists CAP and resiniferatoxin (RTX) were from Sigma and Tocris, respectively. The CB1 antagonist SR141716A was from the NIMH chemical synthesis and drug supply program. The TRPV1 antagonist CPZ was from Tocris. In-domethacin, thiorphan, meloxicam and nimesulide were from Sigma. Nerve growth factor (NGF; 7.0 S) was from Harlan (Indianapolis, IN). [14C]AEA 55 mCi/mmol was from American Radiolabeled Chemicals (St. Louis, MO).
2.2. Animals
Adult, male, Sprague–Dawley rats weighing 250–300 g were used in this study. All procedures utilizing animals were approved by the Institutional Animal Care and Use Committee of The University of Texas Health Science Center at San Antonio and were conducted in accordance with policies for the ethical treatment of animals established by the National Institutes of Health and the International Association for the Study of Pain.
2.3. TG culture
TGs were rapidly dissected (within ~30 s) and placed in ice-cold Ca+ +- and Mg+ +-free Hank’s balanced salt solution (HBSS, Gibco, Carlsbad, CA). TGs were enzymatically digested for 30 min with 1.5 mg/ml collagenase followed by 25 min with 0.1% trypsin Type IX supplemented for the last 10 min with 10 units of DNase I (Roche, Indianapolis, IN). TG homogenates were then centrifuged at 2000 RPM for 2 min, triturated briefly by vortexing and then recentrifuged. They were then resuspended in basal culture media, containing high glucose Dulbecco’s Modified Eagle’s Media (DMEM, Gibco), 1X pen-strep (Gibco), 1X glutamine (Gibco), 3 μg mL−1 5-FDU and 7 μg mL−1 uridine. TG homogenates were gently triturated with a Pasteur pipette followed by successive triturations through 19 and 23 gauge needles. TG homogenates were then transferred to a separate container, wherein the volume was adjusted such that a plating density of ~5000 neurons well−1 would be achieved.
2.4. [14C]AEA accumulation assays
TG cultures were prepared as described above except they were plated, at the same density, on 24-well poly-D-lysine coated plates. Neurons were maintained for 4 or 5 days at which time the accumulation assays were performed. Wells were washed twice with release buffer, without BSA, and FAAH or AMT inhibitors were added from 10 × stocks 10 min prior to [14C]AEA addition. For cold competitor assay neurons were preincubated for 10 min with CPZ and MAFP and 1 min prior to [14C]AEA addition the cold competitor was added. [14C]AEA was added at the indicated concentrations and incubated for 10 min at 37 °C, unless otherwise noted. Following the incubation time the plates were transferred to ice and the release buffer was aspirated and plates were washed twice with release buffer containing 0.1% BSA. We found that, for our assay conditions, 0.1% BSA was sufficient to release ~95% of the [14C]AEA bound to plastic from the wells. Following washing, neurons were incubated with 0.5% trypsin for 15 min at room temperature on a shaker. In initial experiments we compared removal of neurons with trypsin to lysis with 0.1 N NaOH for 15 min at 75 °C and obtained virtually identical results for both total (plates with neurons) and nonspecific (plates without neurons) uptake. The neurons were then pipetted from the plate with siliconized tips and transferred to vials and prepared for scintillation counting. For each experiment an identical plate was prepared and treated with drugs in exactly the same manner to account for nonspecific binding of [14C]AEA to plastic that could not be released by BSA. Moreover, the amount of [14C]AEA that was released by BSA was dependent on the compounds that were used to inhibit FAAH and AMT (as described by others Karlsson et al., 2004) hence, by subtracting this background absorption from the total [14C]AEA uptake measurement a more accurate measure of AEA uptake can be determined. Total [14C]AEA uptake in any given experiment never exceeded ~7.5% of the total added, hence, due to inhibition of [14C]AEA binding to plastic by OMDM-2 and VDM-11 we were never able to observe [14C]AEA uptake from the media as a measure of AEA accumulation that was sufficiently different from plates with neurons to warrant consideration. For assays with CHO cells, cells were grown to confluence in 24-well plates and experiments were conducted in the same manner (without modification) as those with TG neurons except that cells were lysed with 0.1 N NaOH at 75 °C for 15 min.
2.5. Immunocytochemistry
TG cultures were generated as described above and plated at a reduced density (~500 neurons/cover-slip) onto poly-D-lysine and laminin coated coverslips (Becton Dickinson) placed in 24-well plates. After 5 days in culture the neurons were fixed in 2% formalin for 30 min and then permeabilized in 0.2% triton X-100 for 30 min. Neurons were then blocked in normal goat serum (10%) 3 × 10 min and incubated overnight at 4 °C with a rabbit anti-FAAH antibody at 1:1000 dilution (Cayman Chemical). Primary antibody was then washed off and exposed to alexa-fluor conjugated secondary antibody (1:600, goat anti-rabbit alexa-fluor 488, Molecular Probes) for 1 h. Following another set of PBS washes the neurons were then incubated overnight at 4 °C with a guinea pig anti-TRPV1 antibody at 1:3000 dilution (Neuromics). The TRPV1 antibody was washed off by successive PBS washes, and the neurons were then exposed to the appropriate alexa-fluor conjugated secondary antibody (1:600, goat anti-guinea pig alexa-fluor 568, Molecular Probes). Following washing, coverslips were mounted to slides, and labeling was analyzed by confocal microscopy.
2.6. CGRP release assays
Experiments were performed in 48-well, poly-D-lysine pre-coated plates (Becton Dickinson, Franklin Lakes, NJ). All cultures received 100 ng/ml NGF. Culture media was changed at 24 and 72 h and included fresh growth factor supplementation, and all CGRP assays were performed on day 5. TG cultures were washed free of culture media by two successive washes with release buffer (HBSS supplemented with 10.9 mM HEPES, 4.2 mM sodium bicarbonate, 10 mM dextrose and 0.1% bovine serum albumin (BSA), unless in cases where BSA was excluded, pH 7.4). Growth factors were not included in the release buffer. Following washing, TG cultures were exposed to the indicated concentrations of pretreatment FAAH, COX or AMT inhibitors for 10 min and followed by the stimulus drug for 10 min, after which the CGRP containing release buffer was removed and transferred to glass culture tubes. All drugs were diluted from their stock solutions to 10 × concentrations into siliconized glass culture tubes and then 1:10 into the culture plate wells with siliconized pipette tips. Drugs were made fresh, directly before each experiment from their respective stock solutions. For concentration–response curves concentrations of agonist were staggered throughout the plate and 24 of the 48 wells received pretreatment drug while the other 24 received vehicle. Due to small inter-experimental variations, all data for each plate were standardized to the maximal response to agonist with vehicle pretreatment (defined as 100%) and data are presented as % of the maximal effect for all concentrations of agonist in the presence or absence of the challenge drug. CGRP was measured by radioimmunoassay. Data shown are representative of at least two identically performed experiments with consistent results.
2.7. CGRP radioimmunoassay
Following culture release assays, individual aliquots of the superfusate (0.5 mL) were incubated with a C-terminally directed anti-CGRP antiserum kindly donated by Dr Michael Iadarola (NIDCR, NIH, Bethesda, MD, USA). After 24 h, 100 μL of [125I] CGRP28–37 (approximately 20,000–25,000 cpm.) and 50 μL of goat anti-rabbit antibody conjugated to ferric beads were added. Following another 24 h, bound peptide was separated from free peptide via immunomagnetic separation (PerSeptive Biosystems, Framingham, MA, USA). All incubations were carried out at 4 °C. The minimum detection limit for this assay is approximately 1–2 fmol per tube, with 50% displacement occurring at 20–40 fmol per tube. To account for the possibility of any nonspecific effects on the RIA, all drugs used in the release experiments were included in separate standard curves for the purposes of data analysis. No drugs significantly altered the standard curves.
2.8. Statistics
All graphical data are presented as mean ± SEM unless otherwise noted. Significant differences between groups were assessed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparison post hoc test, unless otherwise stated. Concentration–response curves were analyzed by variable slope non-linear regression, and differences between curves were assessed using global curve fitting, comparing variables (i.e., EC50 and Emax) by an F-test. Where EC50/80 or IC50 values are presented, the data are shown as pEC50/80 or pIC50 ± 95% confidence intervals. Emax data are also shown as Emax ± 95% confidence interval. For significant differences between given concentrations in a concentration–response function, differences were analyzed by two-way ANOVA with Bonferroni post hoc test. All data were analyzed with GraphPad Prism 4.0 for Mac OS X (GraphPad, San Diego, CA).
3. Results
To determine whether AMT inhibitors alter TG neuronal responses, we first evaluated the ability of OMDM-2 and VDM-11 to evoke CGRP release on their own. VDM-11 alone evoked CGRP release from TG neurons in a concentration-dependent fashion (Fig. 1A). The EC50 for VDM-11-evoked CGRP release was 18 μM (pEC50 = −4.73 ± 0.50) and reached an Emax of 28.4 ± 5.24 fmol CGRP per well. To examine the possibility that VDM-11-evoked CGRP release was due to TRPV1 agonism, we challenged VDM-11 (50 μM)-evoked CGRP release with CPZ (10 μM) or RR (10 μM). While CPZ did not inhibit VDM-11-evoked CGRP release, RR completely reversed VDM-11-evoked CGRP release (Fig. 1B). On the other hand, OMDM-2 did not evoke CGRP release on its own, (Fig. 1C). We next tested the calcium-dependence of VDM-11-evoked CGRP release. VDM-11-evoked CGRP release appeared to be only partially dependent on extracellular calcium and, in the absence of extracellular calcium, VDM-11-evoked CGRP release was only partially reversed by RR (10 μM, Fig. 1E) as opposed to a complete reversal in the presence of extracellular calcium (Fig. 1D).
Fig. 1.
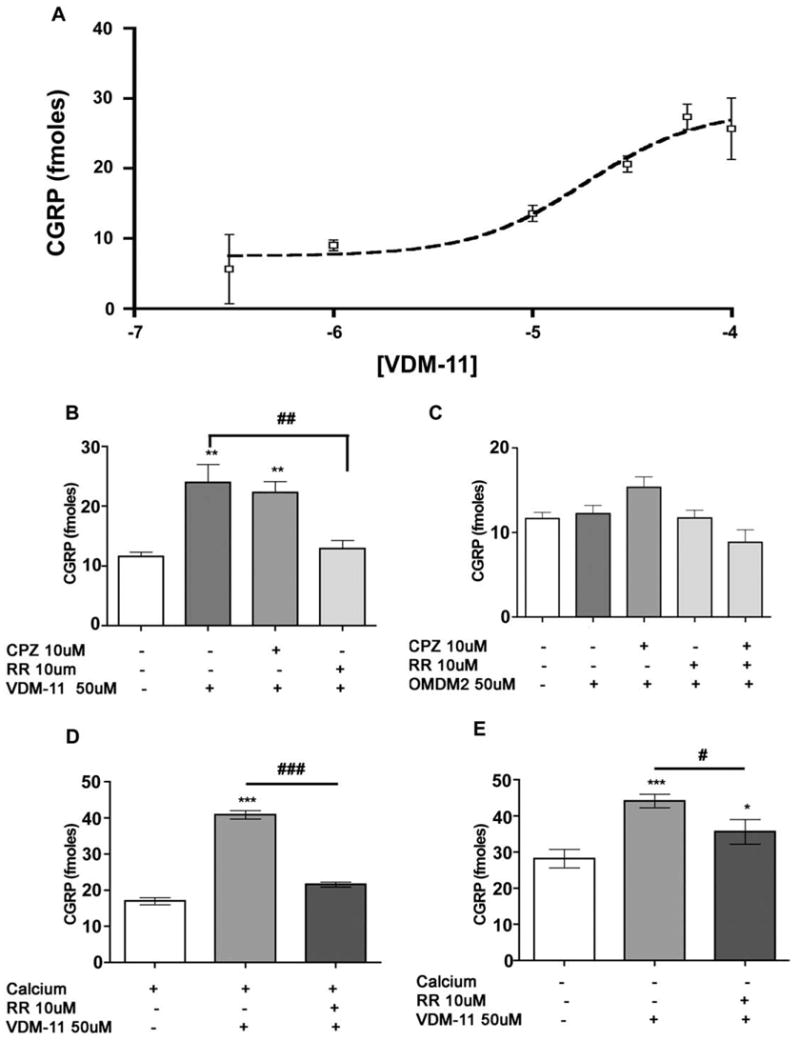
Effect of VDM-11 and OMDM-2 alone. TG neurons were exposed to the indicated concentrations of VDM-11 for 10 min to assess VDM-11-evoked CGRP release. VDM-11 (50 μM)-evoked CGRP release was unaffected by CPZ (10 μM) but was completely blocked by RR (10 μM; B). OMDM-2 (50 μM) did not evoke CGRP release, nor was an effect observed with CPZ (10 μM) or RR (10 μM; C). VDM-11 (50 μM)-evoked CGRP release was only partially dependent on extracellular calcium (E) and VDM-11 (50 μM)-evoked CGRP release was fully reversible in the presence of extracellular calcium (D) but only partially in the absence of extracellular calcium (E; *p < 0.05, **p < 0.01, ***p < 0.001 vs. baseline, #p < 0.05, ##p < 0.01, ###p < 0.001 vs. VDM-11 alone, one-way ANOVA Tukey’s post hoc test).
The postulated binding site for TRPV1 agonists is intracellular (Jordt and Julius, 2002; Jung et al., 1999); hence, we reasoned that AMT inhibitors might be capable of inhibiting TRPV1 agonist-evoked CGRP release by reducing the ability of agonists to gain access to the intracellular compartment in TG neurons. To examine this effect, concentration–response functions were constructed for AEA-, NADA- and CAP-evoked CGRP release in the presence and absence of OMDM-2 (10 μM) or VDM-11 (10 μM). At a concentration of 10 μM, OMDM-2 and VDM-11 each significantly reduced the Emax for AEA-evoked CGRP release, and OMDM-2 significantly right shifted the EC50 for AEA-evoked release (Fig. 2A and Table 1). AEA-evoked CGRP release was significantly inhibited by OMDM-2 at 6–60 μM and by VDM-11 at 30 and 60 μM. Likewise, OMDM-2 and VDM-11 each significantly right shifted the EC50 for NADA-evoked CGRP release (Fig. 2B and Table 1). NADA-evoked CGRP release was significantly inhibited by OMDM-2 at 0.6–3 μM and by VDM-11 at 0.6–10 μM. In addition, OMDM-2 and VDM-11 each significantly shifted the EC50 for CAP-evoked CGRP release to the right (Fig. 2C and Table 1). CAP-evoked CGRP release was significantly inhibited by OMDM-2 at 30–100 nM and by VDM-11 at 60 nM. To attempt to address whether this effect was due to CAP utilizing the AMT, or a possible competitive antagonist effect of these AMT inhibitors on TRPV1, we constructed CAP-evoked CGRP release curves in the presence of increasing concentrations of OMDM-2 (Fig. 2D). While 10, 30 or 60 μM OMDM-2 significantly right shifted the EC50 for CAP-evoked CGRP release, 30 and 60 μM OMDM-2 each significantly decreased the Emax for CAP-evoked release to 55.6 ± 8.92% and 49.9 ± 5.17%, respectively, while 10 μM OMDM-2 did not. We then tested the ability of OMDM-2 and VDM-11 to inhibit RTX-evoked CGRP release. While OMDM-2 at 10 μM did not influence RTX-evoked CGRP release (data not shown), 30 μM OMDM-2 significantly right shifted the EC50 for RTX from 10.60 ± 0.10 (25 pM) to 10.33 ± 0.15 (46 pM) without significantly reduced the Emax (Fig. 2E). OMDM-2 significantly inhibited RTX-evoked CGRP release at 30–300 pM. OMDM-2 at 60 μM also significantly right shifted the EC50 for RTX-evoked CGRP release without reducing the Emax (data not shown). VDM-11 (30 μM) significantly right shifted the EC50 for RTX-evoked CGRP release (pEC50 in the presence of VDM-11 10.03 ± 0.194 (94 pM)) but did not reduce the Emax (Fig. 2E). To the contrary, RTX-evoked release in the presence of VDM-11 was significantly augmented at 300 and 1000 pM RTX. Finally we evaluated whether OMDM-2 and VDM-11 were capable of inhibiting 2-aminoethoxydiphenyl borate (2-APB)-evoked CGRP release. 2-APB is an antagonist of internal calcium store channels, but is also a common activator of TRPV1-3 (Chung et al., 2004; Hu et al., 2004). Interestingly, 2-APB action at recombinant TRPV1 channels is reversed by RR, but not by CPZ, indicating that the binding site for 2-APB at TRPV1 might be extracellular (Hu et al., 2004). 2-APB (300 μM) significantly evoked CGRP release vs. baseline, and 2-APB-evoked release was not inhibited by CPZ (10 μM), OMDM-2 (10 or 30 μM) or VDM-11 (10 or 30 μM, Fig. 2F). On the other hand, 2-APB-evoked CGRP release was significantly inhibited by RR (10 μM, Fig. 2F).
Fig. 2.
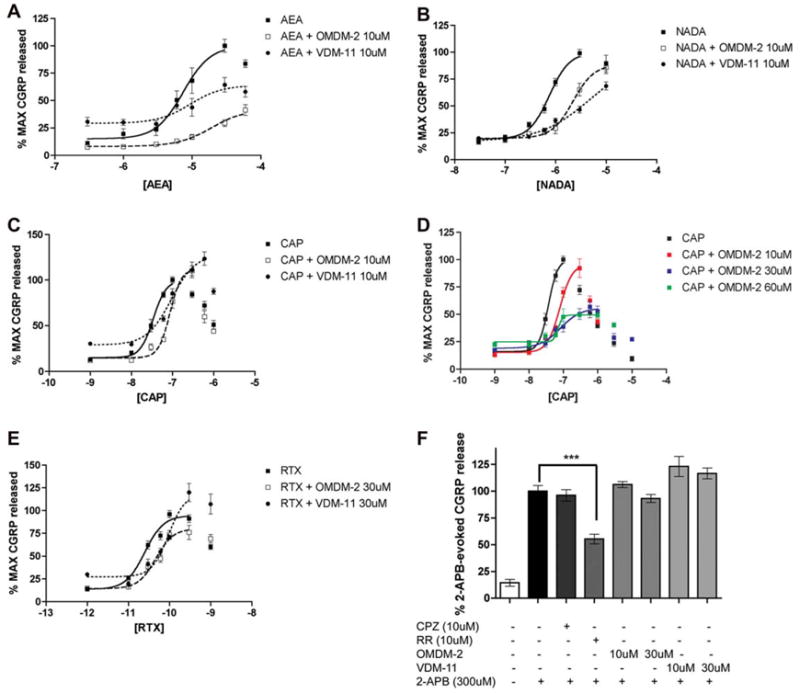
OMDM-2 and VDM-11 inhibit TRPV1-mediated evoked CGRP release. OMDM-2 and VDM-11 right shifted concentration–response curves for AEA (A), NADA (B) and CAP (C). (D) OMDM-2 shifted the concentration–response curve for CAP-evoked CGRP release to the right at 10 μM, 30 μM and 60 μM. Moreover, the Emax of CAP-evoked CGRP release was significantly reduced with both 30 μM and 60 μM OMDM-2. (E) OMDM-2 (30 μM) and VDM-11 (30 μM) right shifted the EC50 for RTX-evoked CGRP release. (F) 2-APB evoked CGRP release from TG neurons in a ruthenium red (RR)-inhibitable fashion (***p < 0.001, one-way ANOVA, Tukey’s post hoc test). Neither OMDM-2 nor VDM-11 inhibited 2-APB-evoked CGRP release.
Table 1.
pEC50 (molar units in parenthesis) ± 95% confidence intervals for AEA-, NADA- and CAP-evoked CGRP release in the presence or absence of OMDM-2 (10 μM) or VDM-11 (10 μM)
| Agonist | Alone | +OMDM-2 (10 μM) | CVDM-11 (10 μM) |
|---|---|---|---|
| AEA | 5.13 ± 0.12 (7.4 μM) | 4.72 ± 0.14 (19 μM)a | 5.03 ± 0.24 (9.4 μM) |
| NADA | 6.12 ± 0.05 (760 nM) | 5.65 ± 0.13 (2.2 μM)a | 5.49 ± 0.12 (3.3 μM)a |
| CAP | 7.47 ± 0.06 (34 nM) | 7.06 ± 0.07 (86 nM)a | 7.03 ± 0.17 (94 nM)a |
Non-overlapping 95% confidence intervals for pEC50 calculations vs. no AMT inhibitor.
Having established that OMDM-2 and VDM-11 inhibit AEA-, NADA- and CAP-evoked CGRP release, we sought to determine IC50s for each of these compounds for inhibition of EC80 concentrations of AEA, NADA and CAP. We also utilized the synthetic TRPV1 partial agonist ACEA in these experiments using a previously established EC80 concentration (Price et al., 2004b). OMDM-2 and VDM-11 each concentration-dependently inhibited AEA-, NADA-, CAP- and ACEA-evoked CGRP release, reaching a maximal inhibition of approximately 50% at 60 μM. Moreover, the pIC50s for this effect with each AMT inhibitor were roughly equivalent for each of the four TRPV1 agonists (Table 2).
Table 2.
pIC50 (μM units in parenthesis) ± 95% confidence intervals for OMDM-2 and VDM-11 inhibition of EC80 concentrations of AEA-, ACEA-, NADA- and CAP-evoked CGRP release
| AMT INH | AEA (30 μM) | ACEA (30 μM) | NADA (1 μM) | CAP (60 nM) |
|---|---|---|---|---|
| OMDM-2 | 5.02 ± 0.45 (9.6) | 5.14 ± 0.39 (7.3) | 5.20 ± 0.79 (6.3) | 5.96 ± 0.34 (6.4) |
| VDM-11 | 5.28 (5.3) | 5.04 ± 0.18 (9.1) | 4.98 ± 0.64 (11) | 5.21 (6.2) |
95% Confidence intervals for VDM-11 with AEA and CAP could not be calculated due to the steepness of the curve.
AEA metabolism has also been implicated in terminating AEA actions at TRPV1 in TRPV1-expressing cell lines (De Petrocellis et al., 2001a). While we, and others, have shown that PMSF does not alter the concentration–response function for AEA-evoked responses in TG neurons (Jennings et al., 2003; Price et al., 2004b), this compound is nonselective; hence, we examined the effect of a number of more specific FAAH inhibitors. In addition, we examined the possible effect of a number of COX inhibitors since COX enzymes degrade AEA (Burstein et al., 2000; Kozak et al., 2003; Yu et al., 1997) and are known to be expressed by sensory neurons (Chopra et al., 2000). Because it is not known whether TG neurons express FAAH protein, we utilized a FAAH antibody to evaluate FAAH expression in TRPV1-positive TG neurons in culture. FAAH-like immunoreactivity was found in TRPV1-positive TG neurons in culture (Fig. 3 top panel). While the proportions of TRPV1-positive neurons that contained FAAH-like immunoreactivity were not determined, we did not observe TRPV1-positive neurons that did not contain FAAH-like immunoreactivity. While it appears that TG TRPV1-positive neurons express FAAH, pre-treatment with the FAAH inhibitor MAFP did not influence AEA-evoked CGRP release at either 100 nM (Fig. 3A) or 1 μM (data not shown). In addition, the FAAH inhibitor CAY10400 100 nM (Fig. 3B) or 1 μM (data not shown) also did not augment AEA-evoked CGRP release at any concentration and neither did URB-597 (data not shown). Similarly, the nonselective COX inhibitor indomethacin (10 μM, data not shown) and the selective COX-2 inhibitors meloxicam (30 μM, Fig. 3C) or nimesulide (30 μM, data not shown) also had no effect on AEA-evoked CGRP release. While no effects were observed with FAAH or COX inhibitors under normal conditions, we hypothesized that inhibition of AMT with OMDM-2 might unmask an effect of FAAH inhibitors. However, when TG neurons were pretreated with either OMDM-2 (10 μM) alone or OMDM-2 plus MAFP (100 nM) and then exposed to AEA, we did not observe any change in the AEA-evoked CGRP release (Fig. 3D).
Fig. 3.
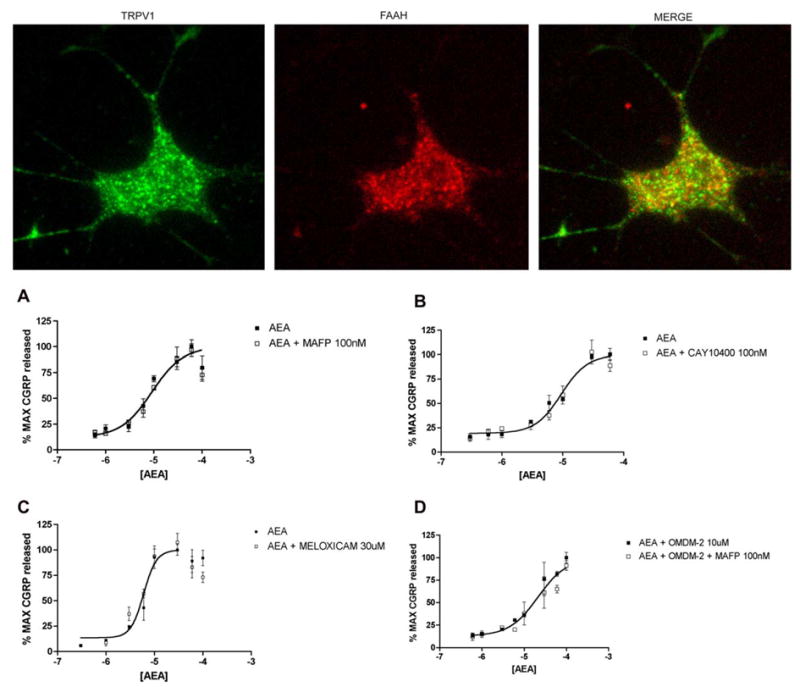
FAAH and COX-2 inhibitors do not alter AEA-evoked CGRP release. (TOP) Image showing FAAH-like immunoreactivity in a TRPV1-positive TG neuron in culture. Concentration–response curves for AEA were unaltered by the FAAH inhibitors MAFP (A, 100 nM) and CAY10400 (B, 100 nM) or the COX-2 inhibitor meloxicam (C, 30 μM). (D) Inclusion of MAFP (100 nM) did not reverse the effect of OMDM-2 (10 μM) on AEA-evoked CGRP release.
In TRPV1-expressing cell lines the presence of BSA in the extracellular media decreases the potency of AEA (De Petrocellis et al., 2001b), and this effect appears to be due to inhibition of the ability of cells to take up AEA in the presence of BSA (Ligresti et al., 2004). Since, to this point, all of our experiments with TRPV1 agonists had included BSA (0.1%), we examined the effect of the absence of BSA on TRPV1-mediated CGRP release. In the absence of BSA, the EC50 for AEA-evoked CGRP release was significantly left shifted (from −4.96 ±0.09 to −5.47 ± 0.14, Fig. 4A). A similar significant shift to the left was seen in the absence of BSA for NADA- (from −5.90 ± 0.08 to −6.19 ± 0.13, Fig. 4B) and CAP- (from −7.21 ± 0.09 to 7.39 ± 0.09, Fig. 4C) evoked CGRP release. The Emax was not altered for any agonist in the absence of BSA. We next examined the effect of OMDM-2 on EC80 concentrations of AEA, NADA and CAP in the absence of BSA. OMDM-2 concentration-dependently inhibited AEA-, NADA- and CAP-evoked CGRP release in the absence of BSA with roughly equivalent pIC50s for each agonist (AEA: 5.03 ± 0.83, NADA: 5.34 ± 0.24 and CAP: 6.02 ± 0.41, Fig. 4D). In the absence of BSA, the FAAH inhibitors URB-597 (1 μM) and MAFP (1 μM) were without effect (data not shown).
Fig. 4.
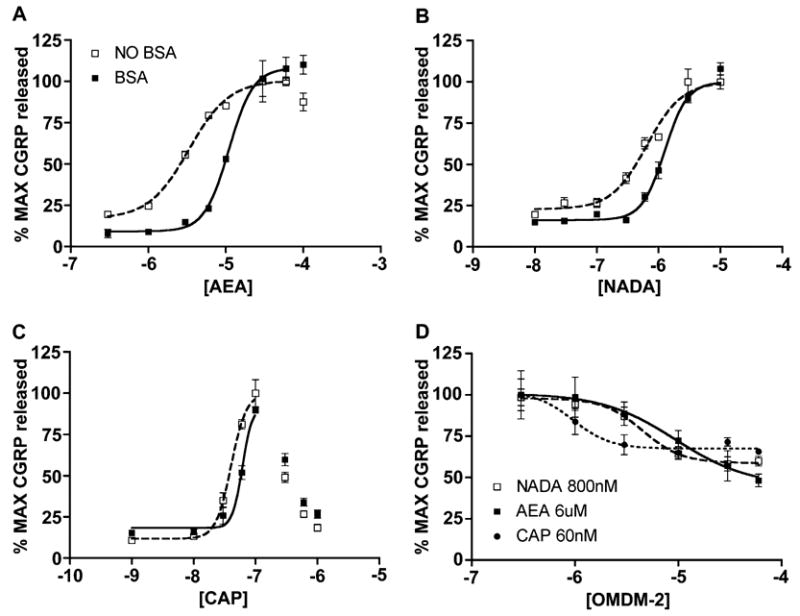
Effect of BSA on AEA-, NADA- and CAP-evoked CGRP release, effect of OMDM-2 in the absence of BSA. TG neurons were exposed to the indicated concentrations of AEA, NADA and CAP in the presence and absence of BSA (0.1%). In the absence of BSA the EC50 for AEA (A), NADA (B) and CAP (C) evoked CGRP release were significantly left shifted. (D) In the absence of BSA OMDM-2 concentration-dependently inhibited EC80 concentration evoked release with AEA (6 μM), NADA (800 nM) and CAP (60 nM).
AEA is a lipophilic compound, and although a specific transport mechanism has been proposed for its uptake, AEA should still be capable of entering cells through passive diffusion. To test whether OMDM-2 inhibition of AEA-evoked CGRP release might be overcome with longer incubation periods of AEA due to passive diffusion, we exposed TG neurons to AEA in the presence or absence of OMDM-2 for time periods ranging from 2.5 to 60 min. OMDM-2 (30 μM) reduced the amount of AEA (30 μM)-evoked CGRP released at all time points with a significant effect observed at 5–60 min (Fig. 5A). Interestingly, AEA (30 μM)-evoked CGRP release in the presence of OMDM-2 was static from 5 to 60 min. MAFP (1 μM), included with OMDM-2 during the preincubation, did not reverse the inhibition observed with OMDM-2. In the absence of BSA (Fig. 5B), simple diffusion appeared to have a greater role in AEA (10 μM)-evoked CGRP release, as AEA-evoked release was not static over the measured time points through the full 60 min in the presence of OMDM-2 (10 μM). AEA-evoked release was decreased at all time points in the presence of OMDM-2, with significant effects observed at 15–60 min. The effect of OMDM-2 was not altered by the inclusion of MAFP (1 μM) in the absence of BSA (data not shown).
Fig. 5.
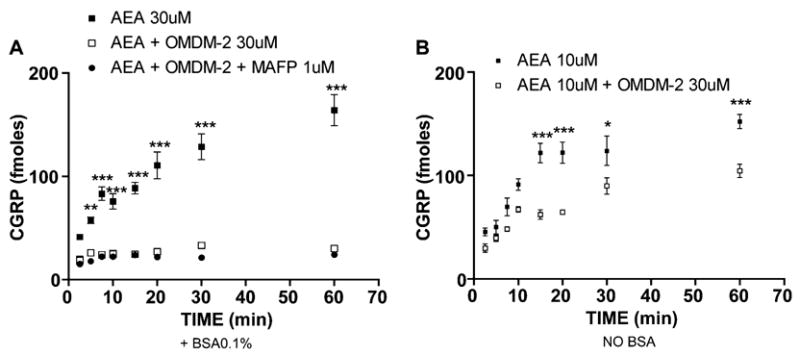
Time Course of OMDM-2 inhibition of AEA-evoked CGRP release. TG neurons were pretreated with OMDM-2 (30 μM) or OMDM-2 and MAFP (1 μM) and then treated with AEA for the indicated time points in the presence (A) or absence (B) of BSA. In the presence of BSA, AEA (30 μM)-evoked CGRP release was significantly inhibited by OMDM-2 or OMDM-2 and MAFP from 5 to 60 min. In the absence of BSA, AEA (10 μM)-evoked CGRP release was significantly inhibited by OMDM-2 from 15 to 60 min (*p <0.05, ***p <0.001, two-way ANOVA, Bonferroni post hoc test).
Since n-acylethanolamines are thought to modulate TRPV1 by neuronal uptake followed by binding to an intracellular domain on the receptor, we evaluated whether TG neurons take up AEA in a manner that is inhibitable by either FAAH or AMT inhibitors. To address this question we pretreated TG neurons with antagonists of FAAH, CB1, TRPV1 or AMT, either singly or in combination, to examine the relative contributions of each of these proteins (or putative proteins in the case of the AMT) on AEA uptake. First, we utilized a concentration of [14C]AEA relevant to TRPV1 agonism (10 μM). Pretreatment of TG neurons with the FAAH inhibitor MAFP (1 μM) led to a significant, approximately 15% reduction in [14C]AEA accumulation over a 10-min period (Fig. 6A). This effect was not altered by the inclusion of either SR141716A (1 μM) or SR141716A plus CPZ (10 μM). On the other hand, inclusion of OMDM-2 (60 μM) significantly reduced [14C]AEA uptake, such that it was cumulatively inhibited by approximately 60% (Fig. 6A). We performed the same experiment with 100 nM [14C]AEA, although in this case, because 100 nM AEA has no effect on TRPV1, CPZ was not evaluated (Fig. 6B). Using 100 nM [14C]AEA, we observed an approximately 40% inhibition with MAFP (1 μM) alone, and again, this effect was not altered by SR141716A (1 μM). The addition of OMDM-2 (60 μM) led to a nearly 80% cumulative inhibition of [14C]AEA uptake. Similar results were obtained for both concentrations of [14C]AEA when MAFP was replaced with the FAAH inhibitor URB-597 (1 μM, data not shown). Since there appears to be both a FAAH- and an AMT-dependent mechanism for AEA uptake in TG neurons, we examined the concentration-dependent inhibition of [14C]AEA uptake by AMT inhibitors in the presence of MAFP (1 μM). Both OMDM-2 and VDM-11 concentration-dependently inhibited 100 nM (Fig. 6C) and 1 μM (Fig. 6D) [14C]AEA uptake. The pIC50 values for OMDM-2 inhibition of 100 nM and 1 μM [14C]AEA uptake were 5.50 ± 0.43 (3.0 μM) and 5.91 ± 0.24 (1.2 μM), respectively, and for VDM-11 5.21 ± 0.64 (6.1 μM) and 5.53 ± 0.58 (2.9 μM), respectively. [14C]AEA uptake by TG neurons in the presence of MAFP (1 μM) was time-dependent with a t1/2 of 6.16 ± 0.36 min (Fig. 6E). Temperature-dependence of uptake was not measured due to the presence of multiple temperature-sensitive channels in TG neurons.
Fig. 6.
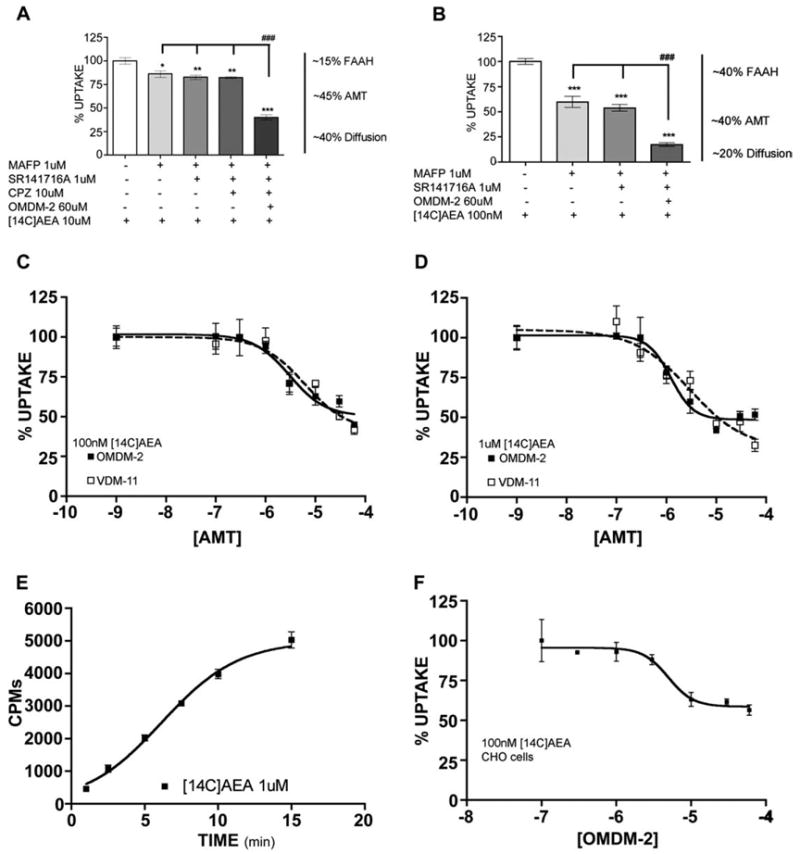
[14C]AEA uptake by TG neurons, effects of FAAH and AMT inhibitors. TG neurons were pretreated with the indicated concentrations of MAFP, SR141716A, CPZ and OMDM-2 and then the indicated concentration of [14C]AEA was added and accumulation was measured as described in Section 2. (A) MAFP (1 μM) inhibited ~15% of 10 μM [14C]AEA accumulation. No further effect was observed with either SR141716A (1 μM) or CPZ (10 μM). Addition of OMDM-2 (60 μM) further inhibited [14C]AEA accumulation by a total of ~60%. (B) 100 nM [14C]AEA accumulation was inhibited by ~40% by MAFP (1 μM) and the inclusion of OMDM-2 (60 μM) further augmented this inhibition to ~80%. No effect was observed with SR141716A (1 μM *p < 0.05, **p < 0.01, ***p < 0.001 vs. control, ###p <.001 between groups, one-way ANOVA Tukey’s post hoc test). Both OMDM-2 and VDM-11, in the presence of MAFP (1 μM) concentration-dependently inhibited 100 nM (C) and 1 μM (D) [14C]AEA accumulation. (E) Time course of [14C]AEA (1 μM) accumulation by TG neurons (V50 =6.16 ± 0.36 min). (F) OMDM-2, in the presence of MAFP (1 μM) concentration-dependently inhibited [14C]AEA (100 nM) accumulation in CHO cells.
To determine whether AEA, NADA or CAP are capable of competing for AEA uptake in TG neurons, we examined the effect of 1, 10 and 30 μM AEA, NADA and CAP on 100 nM [14C]AEA uptake in the presence of MAFP (1 μM). CPZ (10 μM) was also included in these experiments to exclude any possible influence of TRPV1-mediated effects of excess unlabelled ligand on TG neurons. AEA, at 10 or 30 μM effectively reduced [14C]AEA accumulation (Fig. 7A) as did NADA at 10 or 30 μM (Fig. 7B). On the other hand, CAP had no effect on [14C]AEA accumulation in TG neurons (Fig. 7C). We have shown previously that AEA, NADA and CAP are all capable of overcoming CPZ antagonism to evoke CGRP release in TG neurons (Price et al., 2004b). Due to this we were concerned that high concentrations of AEA, NADA and CAP could all cause excitation of TG neurons even in the presence of CPZ and that this effect would confound uptake results in TG neurons in the presence of excess concentrations of these agonists. To overcome this problem, we utilized CHO cells, which do not express TRPV1 channels. We first tested the ability of CHO cells to take up [14C]AEA in an OMDM-2-inhibitable fashion. CHO cells did take up 100 nM [14C]AEA in a similar fashion to TG neurons, and this uptake was inhibited by either MAFP (data not shown) or OMDM-2 (Fig. 6F). In competition assays in CHO cells, AEA and NADA each effectively competed for [14C]AEA (100 nM) uptake (Fig. 7D and E). In contrast to our finding with TG neurons, CAP also competed for 100 nM [14C]AEA uptake in CHO cells, albeit at very high concentrations of 100 and 1000 μM (Fig. 7F).
Fig. 7.
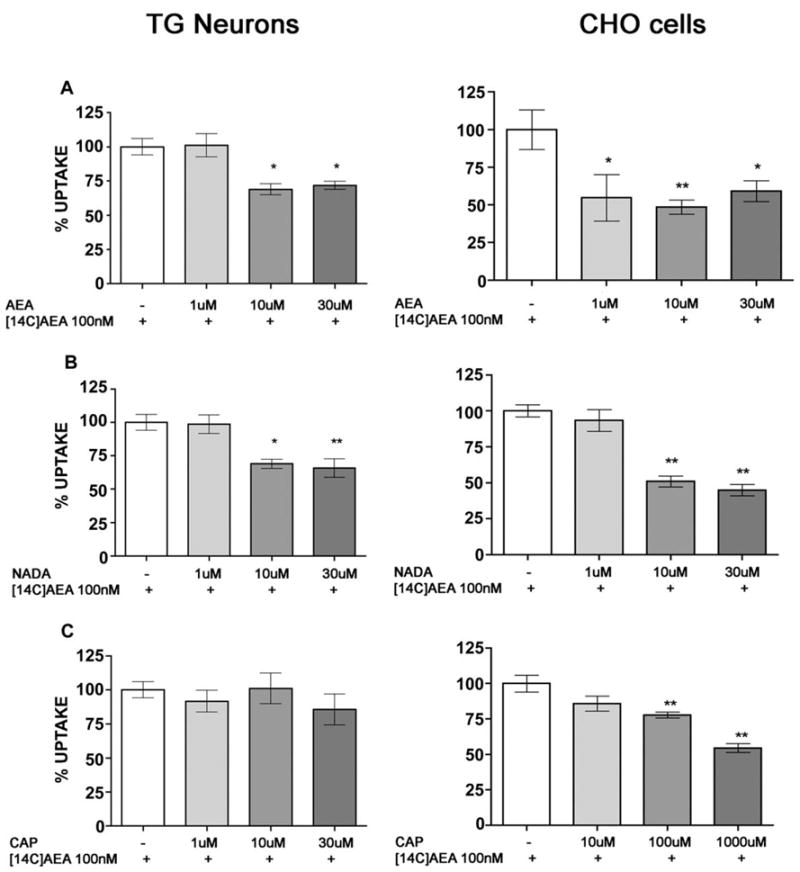
Completion for [14C]AEA accumulation by excess unlabelled TRPV1 agonists. TG neurons (left panels) or CHO cells (right panels) were exposed to AEA, NADA or CAP at the indicated concentrations in the presence of MAFP (1 μM) and CPZ (10 μM), except CPZ was excluded with CHO cells. (A) AEA at (1 μM with CHO cells) 10 μM and 30 μM significantly inhibited [14C]AEA (100 nM) accumulation. Likewise 10 μM and 30 μM NADA (B) inhibited [14C]AEA accumulation. On the other hand, no significant effect of [14C]AEA accumulation was observed with CAP in TG neurons, whereas 100 and 1000 μM CAP significantly inhibited [14C]AEA accumulation in CHO cells (C, *p < 0.05, **p < 0.01, one-way ANOVA, Dunnett’s post hoc test).
4. Discussion
The present study indicates that TG neurons appear to express an AMT-like mechanism for uptake of AEA, which is inhibitable by OMDM-2 and VDM-11 and can be displaced by excess AEA and NADA. AEA uptake in TG neurons is also partially dependent on FAAH; however, FAAH does not appear to be involved in AEA-dependent TRPV1-mediated CGRP release. Moreover, the AMT inhibitors OMDM-2 and VDM-11 limit the neurosecretory actions of AEA, ACEA, NADA and CAP at TRPV1 in TG neurons. Taken together, these findings demonstrate that the AMT, or an AMT-like mechanism, is integrally involved in TRPV1-mediated responses to AEA and NADA in sensory neurons.
In this study, we have demonstrated that TG neurons take up AEA and that this uptake mechanism can be inhibited by antagonists of FAAH and AMT. Interestingly, the relative contribution of FAAH and AMT was dependent on the concentration of [14C]AEA. At 100 nM [14C]AEA, the contribution of FAAH and AMT to uptake of AEA was roughly equal, whereas at 10 μM [14C]AEA, the AMT played a more dominant role in uptake. The concentration at which [14C]AEA uptake was primarily dependent on an AMT-like mechanism occurs within the concentration range that is observed for TRPV1 activation. It should be noted that, at this concentration, no effect on evoked CGRP release was seen with FAAH inhibitors while an inhibitory effect was evident with AMT inhibitors. This is consistent with a dominant role of an AMT-like mechanism in [14C]AEA uptake at concentrations relevant to TRPV1 agonism. It has recently been shown that CB1 appears to also play a role in AEA uptake (Ortega-Gutierrez et al., 2004). We did not observe such an effect in our study using SR141716A; however, TG neuronal cultures grown in the presence of NGF are greatly enriched for CGRP-positive neurons (Price et al., 2005), and these neurons rarely express CB1 (Bridges et al., 2003; Hohmann and Herkenham, 1999; Price et al., 2003). Hence, this discrepancy may not represent a lack of contribution of CB1 to AEA uptake in TG neurons but only a lack of CB1 in the CGRP-expressing subclass of TG neurons. Both OMDM-2 and VDM-11 concentration-dependently inhibited [14C]AEA uptake in TG neurons, and the IC50 values for this inhibition were consistent with those obtained for inhibition of evoked CGRP release by AEA, NADA, CAP and ACEA as well as for inhibition of uptake in other cell lines (De Petrocellis et al., 2000; Fowler et al., 2004; Ortar et al., 2003).
Although FAAH appears to be involved in AEA uptake in TG neurons and FAAH-immunoreactivity is present in TRPV1-positive TG neurons, FAAH inhibitors did not influence exogenously applied AEA-evoked CGRP release. We and others have shown previously that the amide hydrolase inhibitor PMSF does not augment exogenously applied AEA-mediated TRPV1 effects in TG neurons (Jennings et al., 2003; Price et al., 2004b). In the present study, we addressed this issue more rigorously using more potent and selective inhibitors of FAAH. Neither MAFP, URB-597 nor CAY10400 exerted any effect on exogenously applied AEA-evoked CGRP release in TG neurons. FAAH does not appear to be associated with the cell membrane in most cells, and in fact, its intracellular localization appears to be mostly on, or very near, intracellular organelles known to store calcium (Gulyas et al., 2004). The AMT, however, should be found on the plasma membrane where TRPV1 is also localized to permit cation influx. n-Acylethanolamines, including AEA, can exert genomic effects indirectly via intracellular proteins (Derocq et al., 1998; Ramer et al., 2001; Sancho et al., 2003). One such intracellular target is the peroxisome proliferator-activated receptor (PPAR) alpha receptor that binds oleoylethanolamide (OEA) (Fu et al., 2003; Guzman et al., 2004) and palmitoylethanolamide (PEA) (Lo Verme et al., 2004). While it is not known whether TG neurons express these receptors, FAAH might be involved in limiting n-acylethanolamine actions in TG neurons at intracellular targets, whereas its cellular localization renders the protein incapable of limiting AEA actions at TRPV1. Likewise, since AEA has been reported to be produced by sensory neurons themselves (Ahluwalia et al., 2003), the effects of FAAH inhibitors on AEA-evoked actions at TRPV1 might be quite pronounced if the source of AEA is intracellular, which was not tested here. Hence, we interpret these findings to indicate that the AMT is primarily involved in providing extracellularly derived/applied n-acylethanolamines access to intracellular TRPV1 agonist recognition sites. Even over a 60-min time period, wherein simple diffusion of AEA across biological membranes should occur, OMDM-2 nearly completely blocked the secretory effects of AEA on TG neurons.
The first potent AMT inhibitor, AM404 (Beltramo et al., 1997), was subsequently found to be a potent TRPV1 agonist (Zygmunt et al., 2000), thereby confounding the interpretation of the effects of this compound in TRPV1-expressing cells. The discovery of potent AMT inhibitors, such as OMDM-2 (Ortar et al., 2003) and VDM-11 (De Petrocellis et al., 2000), that were either not TRPV1 agonists or had only weak effects on this channel, made the study of the contribution of the AMT to TRPV1-mediated responses possible in TRPV1-expressing cells. On the other hand, compounds that influence the endogenous cannabinoid system, such as cannabinol (Jordt et al., 2004; Zygmunt et al., 2002) and WIN 55,212-2 (Price et al., 2004a), exert effects through mechanisms other than CB1, CB2 or TRPV1. Hence, we examined the effects of VDM-11 and OMDM-2 on TG neurons. OMDM-2 per se did not evoke CGRP release from TG neurons, whereas VDM-11 did. VDM-11-evoked CGRP release does not appear to be dependent on TRPV1, as it was not blocked by CPZ. VDM-11-evoked release was blocked by RR, suggesting that VDM-11 might be an agonist of other TRP channels that are also antagonized by RR. Interestingly, the VDM-11-evoked CGRP release was only partially dependent on extracellular calcium, suggesting that VDM-11 might also release calcium from intracellular stores. While we have not identified the molecular target of VDM-11 in TG neurons, these findings emphasize the importance of assessing the effects of AMT inhibitors at other channels and on sensory neurons themselves, as the repertoire of molecular targets for cannabimemetics is becoming increasingly diverse.
OMDM-2 and VDM-11 each inhibited AEA-, NA-DA- and CAP-evoked CGRP release from TG neurons. These effects are consistent with previous findings of shifts in potency for AEA in the presence of AMT inhibitors, either in cell lines ectopically expressing TRPV1 (De Petrocellis et al., 2001a) or at certain visceral terminals of capsaicin-sensitive primary sensory neurons (Andersson et al., 2002), and suggest that the AMT is involved in AEA actions at TRPV1 in TG neurons. The inhibition of NADA-evoked CGRP release was expected, since NADA is also a substrate for the AMT. CAP has also been shown to interact with the AMT, albeit at mid-μM concentrations (Di Marzo et al., 1998). On the other hand, in HEK cells expressing human TRPV1, VDM-11 had no effect on CAP-mediated alterations in cytosolic calcium concentration (De Petrocellis et al., 2001a), and in mesenteric arteries, VDM-13 had no effects on CAP-induced vasodilation (Andersson et al., 2002). These discrepancies may reflect differences between either AMT properties in HEK cells, native vasculature and TG neurons or sensitivities of the assays (i.e., calcium imaging and vasodilation vs. CGRP release and [14C]AEA uptake). The proposition that CAP might be a substrate for the AMT is supported by the demonstration here that CAP competes for [14C]AEA in CHO cells, although CAP was without effects in TG neurons. Additionally, the presence of BSA, which inhibits uptake of AEA (Karlsson et al., 2004; Ligresti et al., 2004), right shifted the concentration–response function for CAP-evoked CGRP release, as it did for AEA and NADA. Since the shifts observed in the potency and efficacy of CAP-evoked CGRP release with OMDM-2 and VDM-11 were less than those observed with either AEA or NADA, it is reasonable to propose that CAP is a weaker substrate for the AMT than either AEA or NADA. Hence, higher concentrations of CAP would be needed to compete with AEA. However, such concentrations would be capable of overcoming competitive antagonism with CPZ, which would confound the experiment (in TG neurons) due to excitation of the neurons by higher concentrations of CAP (Price et al., 2004b). While our findings in CHO cells indicate that CAP might be a substrate for the AMT, the concentrations of CAP needed to achieve this effects were very high (in agreement with Di Marzo et al., 1998).
The effects of AMT inhibitors on CAP-evoked CGRP release raises an important question in relation to the pharmacological actions of OMDM-2 and VDM-11. Do OMDM-2 and VDM-11 exert their primary action through AMT inhibition or are these compounds also TRPV1 antagonists? The efficacy and potency of AEA-, NADA- and CAP-evoked CGRP release was decreased with increasing concentrations of OMDM-2. On the other hand, the same concentrations of OMDM-2 and VDM-11 did not decrease the efficacy of RTX-evoked CGRP release but rather right shifted its potency. The structure of RTX is not consistent with the requirements for recognition by the AMT; hence, it is unlikely that RTX is an AMT substrate. Moreover, neither OMDM-2 nor VDM-11 inhibited 2-APB-evoked CGRP release. 2-APB is a common agonist of TRPV1-3, but its action at TRPV1 is not reversible by CPZ (Hu et al., 2004). Based on these findings, it is likely that OMDM-2 and VDM-11 possess antagonist activity at the CPZ/RTX-binding site of TRPV1 (as evidenced by the right shift in potency for RTX and lack of effects against 2-APB). On the other hand, AMT inhibition accounts for the decrease in efficacy observed with AEA, NADA and CAP but not RTX. It is also likely that AMT inhibition accounts for a portion of the shift in potency with AEA, NADA and CAP, but this possibility was not directly assessed in these experiments. Proof of this pharmacological model will have to await competition radioligand binding experiments at TRPV1, which were beyond the scope of the present experiments, and the molecular identification of the AMT.
The data presented here provide evidence that TG neurons possess an AMT-like mechanism and that this mechanism is important for the pharmacological action of AEA and NADA at TRPV1. TG neurons also contain FAAH, although FAAH does not appear to be directly linked to exogenously applied AEA actions on TRPV1-mediated CGRP release. Moreover, our findings indicate that the prototypical vanilloid, CAP, might also be a substrate for the AMT. These experiments provide the first demonstration of AMT-like activity in TG neurons and provide functional evidence that these compounds might be clinically utilized in pathological states where AEA and/or NADA are produced by extraneuronal sources to drive TRPV1-mediated neurogenic inflammation and/or pain.
Acknowledgments
This work was supported by NIDA grant DA11959. The authors wish to thank Andrea Giuffrida for invaluable technical advice and data discussion.
References
- Ahluwalia J, Yaqoob M, Urban L, Bevan S, Nagy I. Activation of capsaicin-sensitive primary sensory neurones induces anandamide production and release. J Neurochem. 2003;84:585–591. doi: 10.1046/j.1471-4159.2003.01550.x. [DOI] [PubMed] [Google Scholar]
- Andersson DA, Adner M, Hogestatt ED, Zygmunt PM. Mechanisms underlying tissue selectivity of anandamide and other vanilloid receptor agonists. Mol Pharmacol. 2002;62:705–713. doi: 10.1124/mol.62.3.705. [DOI] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Melck D, Bobrov M, Gretskaya NM, Bezuglov VV, De Petrocellis L, Di Marzo V. N-acyldopamines: novel synthetic CB(1) cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem J. 2000;351 (Pt 3):817–824. [PMC free article] [PubMed] [Google Scholar]
- Bridges D, Rice AS, Egertova M, Elphick MR, Winter J, Michael GJ. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience. 2003;119:803–812. doi: 10.1016/s0306-4522(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Burstein SH, Rossetti RG, Yagen B, Zurier RB. Oxidative metabolism of anandamide. Prostaglandins Other Lipid Mediat. 2000;61:29–41. doi: 10.1016/s0090-6980(00)00053-8. [DOI] [PubMed] [Google Scholar]
- Chopra B, Giblett S, Little JG, Donaldson LF, Tate S, Evans RJ, Grubb BD. Cyclooxygenase-1 is a marker for a subpopulation of putative nociceptive neurons in rat dorsal root ganglia. Eur J Neurosci. 2000;12:911–920. doi: 10.1046/j.1460-9568.2000.00979.x. [DOI] [PubMed] [Google Scholar]
- Chung MK, Lee H, Mizuno A, Suzuki M, Caterina MJ. 2-Aminoethoxydiphenyl borate activates and sensitizes the heat-gated ion channel TRPV3. J Neurosci. 2004;24:5177–5182. doi: 10.1523/JNEUROSCI.0934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day TA, Rakhshan F, Deutsch DG, Barker EL. Role of fatty acid amide hydrolase in the transport of the endogenous cannabinoid anandamide. Mol Pharmacol. 2001;59:1369–1375. doi: 10.1124/mol.59.6.1369. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agro A, Di Marzo V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J Biol Chem. 2001a;276:12856–12863. doi: 10.1074/jbc.M008555200. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Davis JB, Di Marzo V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001b;506:253–256. doi: 10.1016/s0014-5793(01)02934-9. [DOI] [PubMed] [Google Scholar]
- Derocq JM, Bouaboula M, Marchand J, Rinaldi-Carmona M, Segui M, Casellas P. The endogenous cannabinoid anandamide is a lipid messenger activating cell growth via a cannabinoid receptor-independent pathway in hematopoietic cell lines. FEBS Lett. 1998;425:419–425. doi: 10.1016/s0014-5793(98)00275-0. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffn G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, Melck D, Ross R, Brockie H, Stevenson L, Pertwee R, De Petrocellis L. Interactions between synthetic vanilloids and the endogenous cannabinoid system. FEBS Lett. 1998;436:449–454. doi: 10.1016/s0014-5793(98)01175-2. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, Piomelli D. Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci USA. 2004;101:8756–8761. doi: 10.1073/pnas.0400997101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Tiger G, Ligresti A, Lopez-Rodriguez ML, Di Marzo V. Selective inhibition of anandamide cellular uptake versus enzymatic hydrolysis –a difficult issue to handle. Eur J Pharmacol. 2004;492:1–11. doi: 10.1016/j.ejphar.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Beltramo M, Piomelli D. Mechanisms of endocannabinoid inactivation: biochemistry and pharmacology. J Pharmacol Exp Ther. 2001;298:7–14. [PubMed] [Google Scholar]
- Giuffrida A, Rodriguez de Fonseca F, Nava F, Loubet-Lescoulie P, Piomelli D. Elevated circulating levels of anandamide after administration of the transport inhibitor, AM404. Eur J Pharmacol. 2000;408:161–168. doi: 10.1016/s0014-2999(00)00786-x. [DOI] [PubMed] [Google Scholar]
- Glaser ST, Abumrad NA, Fatade F, Kaczocha M, Studholme KM, Deutsch DG. Evidence against the presence of an anandamide transporter. Proc Natl Acad Sci USA. 2003;100:4269–4274. doi: 10.1073/pnas.0730816100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- Guzman M, Lo Verme J, Fu J, Oveisi F, Blazquez C, Piomelli D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor alpha (PPAR-alpha) J Biol Chem. 2004;279:27849–27854. doi: 10.1074/jbc.M404087200. [DOI] [PubMed] [Google Scholar]
- Hajos N, Kathuria S, Dinh T, Piomelli D, Freund TF. Endocannabinoid transport tightly controls 2-arachidonoyl glycerol actions in the hippocampus: effects of low temperature and the transport inhibitor AM404. Eur J Neurosci. 2004;19:2991–2996. doi: 10.1111/j.0953-816X.2004.03433.x. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A. The movement of N-arachidonoylethanolamine (anandamide) across cellular membranes. Chem Phys Lipids. 2000;108:123–134. doi: 10.1016/s0009-3084(00)00191-2. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A. Cellular accumulation of anandamide: consensus and controversy. Br J Pharmacol. 2003;140:802–808. doi: 10.1038/sj.bjp.0705468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience. 1999;90:923–931. doi: 10.1016/s0306-4522(98)00524-7. [DOI] [PubMed] [Google Scholar]
- Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, Lee LY, Wood JD, Zhu MX. 2-Aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J Biol Chem. 2004;279:35741–35748. doi: 10.1074/jbc.M404164200. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci USA. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings EA, Vaughan CW, Roberts LA, Christie MJ. The actions of anandamide on rat superficial medullary dorsal horn neurons in vitro. J Physiol. 2003;548:121–129. doi: 10.1113/jphysiol.2002.035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Julius D. Molecular basis for species-specific sensitivity to ‘‘hot’’ chili peppers. Cell. 2002;108:421–430. doi: 10.1016/s0092-8674(02)00637-2. [DOI] [PubMed] [Google Scholar]
- Jung J, Hwang SW, Kwak J, Lee SY, Kang CJ, Kim WB, Kim D, Oh U. Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. J Neurosci. 1999;19:529–538. doi: 10.1523/JNEUROSCI.19-02-00529.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Lee SY, Hwang SW, Cho H, Shin J, Kang YS, Kim S, Oh U. Agonist recognition sites in the cytosolic tails of vanilloid receptor 1. J Biol Chem. 2002;277:44448–44454. doi: 10.1074/jbc.M207103200. [DOI] [PubMed] [Google Scholar]
- Karlsson M, Pahlsson C, Fowler CJ. Reversible, temperature-dependent, and AM404-inhibitable adsorption of anandamide to cell culture wells as a confounding factor in release experiments. Eur J Pharm Sci. 2004;22:181–189. doi: 10.1016/j.ejps.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Prusakiewicz JJ, Rowlinson SW, Prudhomme DR, Marnett LJ. Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid anandamide. Biochemistry. 2003;42:9041–9049. doi: 10.1021/bi034471k. [DOI] [PubMed] [Google Scholar]
- de Lago E, Ligresti A, Ortar G, Morera E, Cabranes A, Pryce G, Bifulco M, Baker D, Fernandez-Ruiz J, Di Marzo V. In vivo pharmacological actions of two novel inhibitors of anandamide cellular uptake. Eur J Pharmacol. 2004;484:249–257. doi: 10.1016/j.ejphar.2003.11.027. [DOI] [PubMed] [Google Scholar]
- Ligresti A, Morera E, Van Der Stelt M, Monory K, Lutz B, Ortar G, Di Marzo V. Further evidence for the existence of a specific process for the membrane transport of anandamide. Biochem J. 2004;380:265–272. doi: 10.1042/BJ20031812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor PPAR-{alpha} mediates the antiinflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2004 doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- Ortar G, Ligresti A, De Petrocellis L, Morera E, Di Marzo V. Novel selective and metabolically stable inhibitors of anandamide cellular uptake. Biochem Pharmacol. 2003;65:1473–1481. doi: 10.1016/s0006-2952(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Ortega-Gutierrez S, Hawkins EG, Viso A, Lopez-Rodriguez ML, Cravatt BF. Comparison of anandamide transport in FAAH wild-type and knockout neurons: evidence for contributions by both FAAH and the CB1 receptor to anandamide uptake. Biochemistry. 2004;43:8184–8190. doi: 10.1021/bi049395f. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Beltramo M, Glasnapp S, Lin SY, Goutopoulos A, Xie XQ, Makriyannis A. Structural determinants for recognition and translocation by the anandamide transporter. Proc Natl Acad Sci USA. 1999;96:5802–5807. doi: 10.1073/pnas.96.10.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Helesic G, Parghi D, Hargreaves KM, Flores CM. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience. 2003;120:155–162. doi: 10.1016/S0306-4522(03)00333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM. Cannabinoid receptor-independent actions of the aminoalkylindole WIN 55,212-2 on trigeminal sensory neurons. Br J Pharmacol. 2004a;142:257–266. doi: 10.1038/sj.bjp.0705778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM. Modulation of trigeminal sensory neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-arachidonoyldopamine and arachidonyl-2-chloroethylamide. Br J Pharmacol. 2004b;141:1118–1130. doi: 10.1038/sj.bjp.0705711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Louria MO, Candelario-Soto D, Dussor GO, Jeske NA, Patwardhan AM, Diogenes A, Trott AA, Hargreaves KM, Flores CM. Treatment of trigeminal ganglion neurons in vitro with NGF, GDNF or BDNF: effects on neuronal survival, Neurochemical properties and TRPV1-mediated neuropeptide secretion. BMC Neurosci. 2005;6 (1):4. doi: 10.1186/1471-2202-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramer R, Brune K, Pahl A, Hinz B. R(–)-methanandamide induces cyclooxygenase-2 expression in human neuroglioma cells via a non-cannabinoid receptor-mediated mechanism. Biochem Biophys Res Commun. 2001;286:1144–1152. doi: 10.1006/bbrc.2001.5518. [DOI] [PubMed] [Google Scholar]
- Reggio PH, Traore H. Conformational requirements for endocannabinoid interaction with the cannabinoid receptors, the anandamide transporter and fatty acid amidohydrolase. Chem Phys Lipids. 2000;108:15–35. doi: 10.1016/s0009-3084(00)00185-7. [DOI] [PubMed] [Google Scholar]
- Roberts LA, Christie MJ, Connor M. Anandamide is a partial agonist at native vanilloid receptors in acutely isolated mouse trigeminal sensory neurons. Br J Pharmacol. 2002;137:421–428. doi: 10.1038/sj.bjp.0704904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho R, Calzado MA, Di Marzo V, Appendino G, Munoz E. Anandamide inhibits nuclear factor-kappaB activation through a cannabinoid receptor-independent pathway. Mol Pharmacol. 2003;63:429–438. doi: 10.1124/mol.63.2.429. [DOI] [PubMed] [Google Scholar]
- Welch JM, Simon SA, Reinhart PH. The activation mechanism of rat vanilloid receptor 1 by capsaicin involves the pore domain and differs from the activation by either acid or heat. Proc Natl Acad Sci USA. 2000;97:13889–13894. doi: 10.1073/pnas.230146497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Ives D, Ramesha CS. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem. 1997;272:21181–21186. doi: 10.1074/jbc.272.34.21181. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Andersson DA, Hogestatt ED. Delta 9-tetrahydrocannabinol and cannabinol activate capsaicin-sensitive sensory nerves via a CB1 and CB2 cannabinoid receptor-independent mechanism. J Neurosci. 2002;22:4720–4727. doi: 10.1523/JNEUROSCI.22-11-04720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Chuang H, Movahed P, Julius D, Hogestatt ED. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur J Pharmacol. 2000;396:39–42. doi: 10.1016/s0014-2999(00)00207-7. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]