

Abstract
The stress-inducible gene heme oxygenase (HO-1) has previously been shown to provide cytoprotection against oxidative stress. The mechanism(s) by which HO-1 provides this cytoprotection is poorly understood. We demonstrate here that carbon monoxide (CO), a byproduct released during the degradation of heme by HO, plays a major role in mediating the cytoprotection against oxidant-induced lung injury. We show in vitro that CO protects cultured epithelial cells from hyperoxic damage. By using dominant negative mutants and mice deficient in the genes for the various MAP kinases, we demonstrate that the cytoprotective effects of CO are mediated by selective activation of the MKK3/p38β protein MAP kinase pathway. In vivo, our experiments demonstrate that CO at a low concentration protects the lungs, extends the survival of the animals, and exerts potent anti-inflammatory effects with reduced inflammatory cell influx into the lungs and marked attenuation in the expression of pro-inflammatory cytokines.
Heme oxygenase (HO) degrades heme to biliverdin, which is then reduced enzymatically to bilirubin by bilirubin reductase. 1 Three isoforms of HO exist, HO-1, HO-2, and HO-3, and are the products of different genes. HO-2 and -3 are constitutively expressed, while HO-1 is the inducible isoform. 1,2 HO-1 can be induced not only by the substrate heme but also by non-heme cellular stressors including endotoxin, heavy metals, cytokines, and agents causing oxidative stress including hyperoxia. 3 Studies by our laboratory and others have demonstrated that induction of endogenous HO-1 expression provides potent cytoprotection in various models of oxidative stress including rhabdomyolysis, endotoxic shock, hyperoxic lung injury, or xenotransplantation tissue rejection. 3-6 Recent analyses of HO-1(−/−) null mice as well as a HO-1-deficient human 7,8 have also strengthened the emerging paradigm that HO-1 serves as a critical molecule in the host defense against oxidant stress. Furthermore, our laboratory has demonstrated that administration of exogenous HO-1 by gene transfer provides potent cytoprotection in hyperoxia-induced lung injury. 9
Although the functional importance of HO-1 in the host’s defense against oxidative stress is amply supported by the aforementioned studies, the precise mechanism(s) by which HO-1 provides cytoprotection is unclear. Our laboratory has also demonstrated that exogenous administration of low concentrations of carbon monoxide (CO), a catalytic byproduct of HO degradation of heme, protects rats against hyperoxia-induced lung injury. 10 Most recently, Fujita et al 11 have shown that CO at similar concentrations to those used here and in previous work protects the lung against ischemia reperfusion injury. They suggest that the mechanism of protection observed in their model involves guanylate cyclase and cGMP, which is involved in the modulation of plasminogen activator inhibitor-1 (PAI-1). Sato et al 12 have also shown that CO can protect against xenotransplantation rejection in rodents. The ability of CO to provide cytoprotection in rats was very similar, if not identical, to the protective effects observed in studies using gene transfer of HO-1 in rats. 9 These studies strongly suggested to us that CO could mediate the cytoprotective effects of HO-1. To begin to understand the mechanism by which CO can confer cytoprotection against oxidant-induced lung injury, we used a mouse model for two major reasons. First, we believe that observing the cytoprotection of CO in mice (most studies have been done in rats) will further strengthen the emerging paradigm that CO-induced cytoprotection is a universal response. Second, establishment of cytoprotection in a mouse model enables us to perform mechanistic studies, because mice are amenable to genetic alteration or manipulation of specific gene products.
Exposure to high concentrations of O2 results in an increased oxidative burden, leading to the damage of lipids, proteins and DNA. In the lung this is followed by inflammation, fibrin deposition, and thickening of alveolar membranes. 13 Early evidence of injury consists of endothelial leakage, edema formation, inflammatory cell influx into the airways, and cytokine production, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, with subsequent alveolar hemorrhage and diffuse alveolar damage.
We demonstrate here that CO-induced cytoprotection is indeed observed in the mouse, with similar protective effects to that observed in the rat. 10 A low concentration of CO provides potent cytoprotective effects including attenuation of lung lipid peroxidation, edema as measured by wet/dry ratios and protein leakage into the airways. CO also exerted potent anti-inflammatory effects in hyperoxia-induced lung injury including inhibiting the expression of the pro-inflammatory cytokines TNFα, IL-1β, and IL-6. Since the mitogen-activated protein (MAP) kinase pathway plays a major role in regulating the expression of many of these pro-inflammatory cytokines, we investigated whether the anti-inflammatory properties of CO might involve the MAP kinase pathway. Using mice deficient for the various MAP kinases (gene knockout mice) and dominant negative mutants in an A549 epithelial cell culture model, we demonstrate that CO-induced cytoprotection and anti-inflammatory effects involve the MAP kinase cascade, specifically the MKK3/p38β pathway.
Experimental Procedures
Animals
Male C57BL6 mice (6 to 8 weeks of age) were purchased from Jackson Laboratory (Bar Harbor, ME) and allowed to acclimate for 1 week with rodent chow and water ad libitum. The mkk3(−/−) and jnk2(−/−) mice were generated as previously described. 14,15 Wild-type littermates were used as controls. All animals were housed in accordance with guidelines from the American Association for Laboratory Animal Care and Research Protocols and were approved by the Animal Care and Use Committee at Yale University.
Exposures and Survival Studies
The animals were exposed to >98% O2 or >98% O2 plus CO mixture at a flow rate of 12 L/minute in a 3.7-ft 3 glass exposure chamber. The animals were supplied food and water throughout exposure. CO at a concentration of 1% (10,000 ppm) in compressed air was mixed with >98% O2 in a stainless steel mixing cylinder to achieve a final concentration of 250 ppm (0.025%) before delivery into the exposure chamber. Mice in the CO/O2 group were pretreated 1 hour prior with CO (250 ppm) in air. A CO analyzer (Interscan, Chatsworth, CA) was used to measure CO levels continuously in the chambers. Gas samples were taken by the analyzer through a port in the top of the chambers at a rate of 1 L/minute and analyzed by electrochemical detection, with a sensitivity of 10 to 600 ppm. Concentration levels were measured hourly and there were no fluctuations in the CO concentrations once the chamber had equilibrated (approximately 5 minutes). O2 concentrations in the chamber were determined with a gas spectrometer and were maintained at >98% O2. Mice were placed into either the chamber receiving hyperoxia or an identical chamber receiving >98% O2 containing 250 ppm CO. Animals were monitored hourly and time of death was noted. Animals exposed to air or 250 ppm of CO in air had no untoward effects for up to 35 days, at which time the exposure was stopped. For p38 inhibition experiments, mice were treated with 20 mg/kg SB203580 (Calbiochem, San Diego, CA) i.p. 1 hour before exposure and daily thereafter. Animals were monitored for survival.
Carboxyhemoglobin Determination
To obtain COHb levels, mice were bled via the retroorbital sinus or via cardiac puncture at the time of sacrifice into a heparinized syringe and kept on ice until analysis. Blood samples were immediately analyzed by an OSM3 Hemoximeter (Radiometer Copenhagen, Copenhagen, Denmark) and levels were reported as %COHb.
Bronchoalveolar Lavage (BAL) Fluid Analysis
In a separate group of animals, the same exposure conditions were used as described above. All mice were removed from the corresponding exposure condition after 84 hours (when markers of injury are peaking) for bronchoalveolar lavage (BAL) fluid analysis. Briefly, the animals were removed individually from the chamber and anesthetized with an overdose of ketamine and sacrificed via transection of the heart. BAL (35 ml/kg) was performed four times with PBS (pH 7.4). Cell pellets were pooled from the lavages and resuspended in PBS. The supernatant from the first lavage was saved and frozen for protein determination. Cells counts were performed with a Neubauer hemocytometer (VWR, Boston, MA). For differential analysis, samples were cytocentrifuged and stained with Diff-Quik (Fisher Scientific, Pittsburgh, PA). Protein in each sample was determined by a standard Bradford assay (Bio-Rad, Hercules, CA).
Lung Tissue Injury Markers (Wet/Dry Ratio and Lipid Peroxidation)
Wet/dry ratios were determined in a separate group of unlavaged animals. Briefly, lungs from mice exposed for 84 hours in the same conditions as above were extracted, blotted gently, and immediately weighed. The tissue was then placed in a 50°C oven overnight and then weighed dry. The lipid peroxidation assay was performed as follows; lung samples were diluted fivefold (w/v) with 20 mmol/L Tris-HCl, pH 7.4, and then homogenized gently to prevent generation of foam and oxidation of the sample. The homogenate was then diluted another twofold (10-fold overall) and centrifuged 3,000 × g for 10 minutes at 4°C. End products of fatty acid peroxidation, malondialdehyde (MDA), and 4-hydroxyalkenals (4-HNE), were measured in a 200-μl aliquot of the supernatant using the Calbiochem Lipid Peroxidation assay kit (Calbiochem). The assay is based on extraction of fatty acids with acetonitrile and derivitization of MDA and 4-HNE with a chromagenic agent at 45°C The resulting chromophore was measured at 586 nm. Results are mean ± SE (n = 4) and are expressed as nanomoles of MDA plus 4-HNE (determined from standard curves) and standardized to milligrams of protein.
Cell Culture
A549 human alveolar epithelial cells were purchased from American Tissue Cell Culture (Rockville, MD). Cells were grown in Ham’s F-12 media containing 10% fetal bovine serum and gentamicin (100 μg/ml) in a humidified atmosphere of 5% CO2/balanced air or 250 ppm CO/5%CO2/balanced air. Cells were exposed for various time points and protein was extracted as described below. For survival experiments, cells at confluency were pretreated with SB203580 (10 μmol/L) 1 hour before exposure to a pretreatment with CO (250 ppm)/balanced air for 3 hours. Control cells were treated identically except they were exposed to 5%CO2/air. Following pretreatment, cells were exposed to approximately 95%O2/5%CO2 or 95%O2/5%CO2/250 ppm CO in airtight plexiglass chambers. Cells were kept under these conditions and monitored for cell death. Twice each day, the chamber was flushed with either mixture. On day 4 of exposure, the plates were removed, cells were counted and viability was assessed by trypan blue exclusion. For transient transfection experiments, cells were treated with the dominant negative mutants of p38β or MKK3 (kindly provided by Dr. J. Han). Briefly, cells were grown to approximately 50% to 75% confluency at which time each vector (1 μg) was transfected using FUGENE reagent as per the manufacturer’s directions (Roche, Indianapolis, IN). Following transfection, cells were allowed to proliferate 24 to 48 hours and then treated with either CO or CO/O2 as described above. Cells were exposed continuously for 4 days at which time viability was assessed.
MAPK Western Blot
Assay kits were purchased from Cell Signaling (Beverly, MA) and used per manufacturer’s instructions. Briefly, at set time points, cells were removed from the exposure chamber, rinsed with cold PBS and then 200 μl of sodium dodecyl sulfate (SDS) sample buffer (62.5 mmol/L Tris-HCl (pH 6.8), 2% w/v SDS, 10% glycerol, 50 mmol/L dithiothreitol, and 0.1% w/v bromphenol blue) was added to each plate. For lung tissue, protein was extracted as described previously. 16 Briefly, 100 μg of protein in SDS sample buffer was electrophoresed, transferred and probed as described below. Cells were scraped and sonicated for 5 seconds. 20 μl of each sample was boiled for 5 minutes and then loaded into a 12% polyacrylamide gel and electrophoresed at 125 V for 90 minutes. The gel was transferred overnight at 20 V onto nitrocellulose membrane. Membranes were then incubated with blocking buffer [5% nonfat dry milk in TTBS (10% Tween in Tris-buffered saline) for 1 hour], washed with TTBS and then incubated overnight in the corresponding rabbit polyclonal primary antibody directed against either phosphorylated ERK, MKK3/6, p38, or JNK. The following day the membrane was washed in TTBS and proteins were visualized using horseradish peroxidase (HRP)-conjugated anti-rabbit IgG and the enhanced chemiluminescence assay (Amersham Life Sciences, Arlington Heights, IL) according to manufacturer’s instructions. All membranes were also stripped using a standard stripping solution (100 mmol/L β-mercaptoethanol, 2% SDS, and 62.5 MM Tris-HCl, pH 6.8) at 50°C. and then reprobed with rabbit polyclonal antibody targeting total nonphosphorylated ERK, p38, or JNK to assure equal loading of protein. For immunoprecipitation, monoclonal antibodies against p38α and p38β were used to precipitate each isoform as described and generated previously. 17 Samples were then electrophoresed, transferred, and probed per the MAPK Western blot protocol using a rabbit anti-p38α or anti-p38β polyclonal antibody. Rabbit polyclonal antibodies were detected using a commercial HRP-linked anti-rabbit secondary antibody (Cell Signaling, Beverly, MA) and visualized using the enhanced chemiluminescence assay as described above. To assure equal loading, the gel was stained with Coomassie blue.
RNase Protection Assay
Mice exposed to air, 250 ppm CO, O2, or O2 in the presence of 250 ppm for 84 hours were removed form the chamber individually, anesthetized, and the lungs were extracted and quick-frozen in liquid nitrogen. Total RNA was extracted using TRIZOL method (Invitrogen, Carlsbad, CA) and mRNA integrity verified by Northern blot analysis. RNase Protection Assay (RPA) was performed on samples using Pharmingen Kits and selected DNA templates. Briefly, 10 μg of each sample RNA was hybridized overnight to radiolabeled 32P-UTP DNA templates (BD Pharmingen, San Diego, CA) and then visualized on a 5% bis-acrylamide/urea gel run in 0.5X TBE. Gels were dried onto Whatman 3 mol/L filter paper and then exposed to autoradiograph film overnight. Each template set includes probes for L32 and GAPDH, which serve as internal standards to verify equal loading between sample lanes.
Statistical Analysis
Data are expressed as the mean ± SEM. Differences in measured variables between experimental and control group were assessed using Student’s t-test. Statistical calculations were performed on a Macintosh personal computer using the Statview II Statistical Package (Abacus Concepts, Berkeley, CA). Statistical difference was accepted at P < 0.05.
Results
CO Protects Against Lethal Hyperoxia in Mice
We used a clinically relevant in vivo model of oxidative stress to test the hypothesis that CO mediates cytoprotection against oxidant-induced lung injury. Animals exposed to >98% O2 undergo pathophysiological changes similar to those seen in acute lung injury or adult respiratory distress syndrome. 13,18 We and others have shown that mice exposed to hyperoxia develop measurable markers of lung injury by 64 to 72 hours, which increase in severity until death at 90 to 100 hours of hyperoxia exposure. 13 In this study, one group of mice was placed in hyperoxia and another was placed in hyperoxia in the presence of a low concentration of CO (250 ppm, 0.025%). The mice exposed to hyperoxia alone died between 90 and 100 hours, while 95% of mice exposed to hyperoxia in the presence of CO remained alive at 95 hours (Figure 1A) ▶ . Fifty percent of CO/O2 mice survived >128 hours of hyperoxia exposure (Figure 1A) ▶ .
Figure 1.
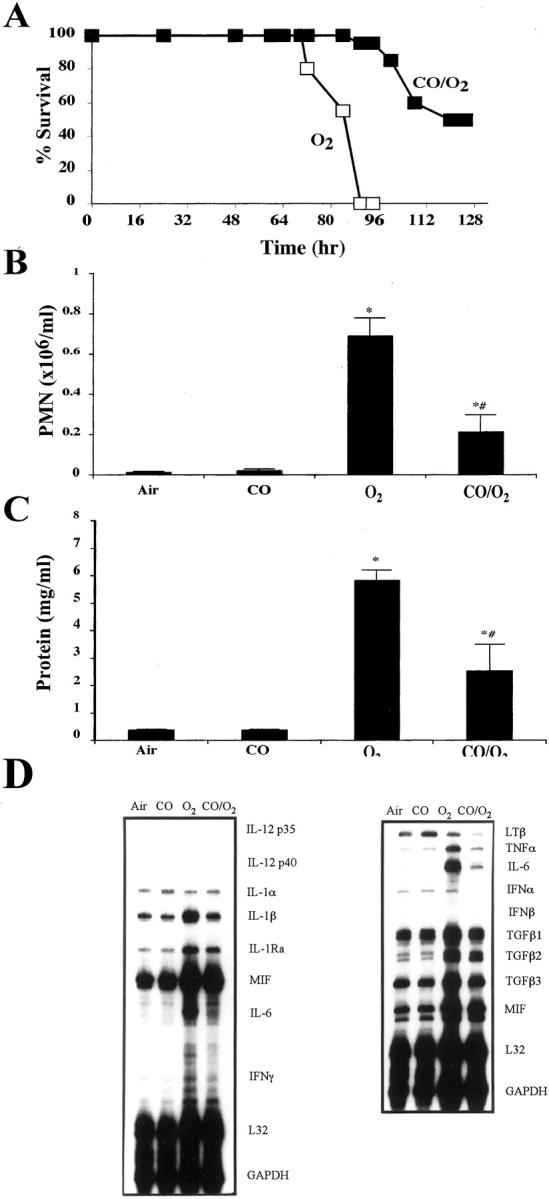
A: Effects of CO on survival following exposure to lethal hyperoxia. Mice in groups of 6 to 8 were exposed to >98% O2 or >98% O2 plus 250 ppm CO continuously and monitored for survival. Mice exposed to only O2 succumbed to O2 toxicity by 90 to 100 hours while >95% of those mice exposed in identical chambers to O2 in the presence of CO (250 ppm) were alive at 100 hours, with >50% alive at 128 hours of exposure. Data represent 20 to 25 animals per treatment group. B: Effects of CO on hyperoxia-induced PMN influx into the airways of mice. C: Effects of CO on hyperoxia-induced protein in the BAL. Mice were exposed continuously to either >98% O2 or >98% O2 plus 250 ppm CO. At 84 hours, a BAL was performed and PMN differential counts was determined. Total protein in the BAL was determined by Bradford assay. Data represent mean ± SEM of 8 to 10 mice per group. * P < 0.001 vs. air and CO. # P < 0.05 vs. CO/O2. D: Effects of CO (250 ppm) on hyperoxia-induced cytokine mRNA expression. Mice were exposed to >98% O2 in the presence or absence of CO (250 ppm). At 84 hours of exposure, the lungs were harvested and analyzed by RNase protection using the cytokine templates containing the above listed cytokines. L32 and GAPDH were used as loading controls. The blots are representative of three separate experiments in which the lungs from three mice were pooled for RNA extraction.
Carboxyhemoglobin Levels in Mice Exposed to Hyperoxia in the Presence and Absence of CO
Determination of COHb levels is tantamount to these studies as they are the accepted mode by which to assess CO exposure. The levels also allow us to ascertain the effects of high FiO2 on COHb levels in comparison to CO alone. Animals were exposed to either air, CO (250 ppm), O2 (>98%), or a mixture of CO (250 ppm) and O2 continuously for 70 hours. Those animals exposed to CO were pretreated with CO in air for 1 hour before being placed in the CO/O2 conditions. Blood samples were taken from the retroorbital sinus 24 hours into the exposure and again at 70 hours for COHb determination (Table 1) ▶ . The data clearly show the expected increase in COHb levels on exposure to CO which reaches a steady state by 24 hours and remains unchanged through the 70 hours exposure. The lower levels in the CO/O2 group (11% to 14%) versus the CO only group (32% to 25%) was predicted due to the presence of higher paO2 levels achieved with the higher FiO2 being delivered.
Table 1.
Carboxyhemoglobin (COHb) Levels in Mice Exposed to Hyperoxia in the Presence and Absence of CO
| Treatment group | COHb (24 hours) | COHb (72 hours) |
|---|---|---|
| Air | 4.1 ± 0.4 | 2.5 ± 0.5 |
| CO | 35 ± 2* | 31.6 ± 4* |
| O2 | 4.1 ± 0.5 | 2.2 ± 1 |
| CO/O2 | 14 ± 1† | 10.7 ± 0.8† |
*P < 0.001 vs. air and O2-exposed mice.
†P < 0.01 vs. CO alone treated mice.
Effects of CO (250 ppm) exposure on carboxyhemoglobin levels in mice in the presence and absence of 98% O2. A blood sample was taken at 24 hours and again at 70 hours of exposure and analyzed for COHb by a co-oximeter. Data represent mean ± SD of 4 animals/group.
CO Provides Cytoprotection against Hyperoxia-Induced Lung Injury
To further assess the beneficial effects of CO, we measured various indices of hyperoxia-induced lung injury. We evaluated hyperoxia-induced lung edema measured by wet/dry tissue ratio, protein accumulation in the airspace, and lung tissue lipid peroxidation, all of which are reliable markers of oxidant-induced lung injury. 13 Mice exposed to hyperoxia exhibited an increased wet/dry ratio and increased protein accumulation in the BAL, both of which were significantly reduced in mice exposed to hyperoxia in the presence of CO (Figure 1C ▶ ; Table 2 ▶ ). CO also prevented hyperoxia-induced increases in lipid peroxidation products, further providing evidence that CO provides cytoprotection against hyperoxia-induced lung tissue injury (Table 2) ▶ . Neither air nor CO alone had untoward effects on either of these markers.
Table 2.
Carbon Monoxide Reduces Markers of Inflammation in the Lung
| Treatment | Cells/ml × 106 | Wet/dry | MDA (nmol/mg) | |
|---|---|---|---|---|
| WBC | Mono | |||
| Air | 3.72 ± 0.60 | 3.71 ± 0.26 | 4.55 ± 0.01 | 0.45 ± 0.03 |
| CO | 3.55 ± 0.25 | 3.33 ± 0.24 | 4.54 ± 0.01 | 0.52 ± 0.01 |
| O2 | 8.89 ± 0.95* | 8.20 ± 1.06* | 7.14 ± 0.03* | 0.64 ± 0.05† |
| CO/O2 | 3.95 ± 0.56 | 3.74 ± 0.55 | 5.52 ± 0.04‡ | 0.47 ± 0.05 |
*P < 0.001 vs. air, CO, CO/O2.
†P < 0.03 vs. air, CO.
‡P < 0.02 vs. air, CO/O2.
Effects of CO (250 ppm) on hyperoxia-induced lung injury parameters. Mice were exposed continuously to >98% O2 in the presence or absence of CO. At 84 hours of exposure one group of animals was lavaged and cellular profiles and protein contents of the lavagates were determined. The lungs of a second group of mice were extracted and analyzed for wet/dry ratio and lipid peroxidation. Control animals were exposed either air or CO for 84 hours in identical exposure chambers. Values represent the mean ± SE of 4 to 12 animals per group.
Histological analyses showed marked differences in lung histology between the two treatment groups. Marked lung hemorrhage, edema, and fibrin deposition were observed in mice exposed to hyperoxia alone. In contrast, the lungs of mice exposed to hyperoxia in the presence of CO were normal macroscopically and microscopically (data not shown).
CO Inhibits Hyperoxia-Induced Neutrophil (PMN) Infiltration into the Airways
To further investigate the mechanism by which CO provides cytoprotection, we examined the inflammatory cell profile in the BAL fluid of animals exposed to hyperoxia in the presence of CO. It is well established that influx of inflammatory cells, particularly the neutrophil into the airways, plays an important role in mediating oxidant-induced lung injury in both animal models and in humans. 13,18 We hypothesized therefore that CO may attenuate hyperoxia-induced lung injury by inhibiting the neutrophil influx into the airspace. Animals exposed to hyperoxia had a marked increase in BAL neutrophils, while mice exposed to hyperoxia in the presence of CO exhibited significantly lower numbers of neutrophils recovered in the BAL (Figure 1B) ▶ .
CO Targets Inflammatory Cytokine Induction in Lung Tissue
Hyperoxia-induced expression of numerous pro-inflammatory cytokines including TNFα, IL-1β, and IL-6 were observed in lung tissues by 84 hours of hyperoxia exposure (Figure 1D) ▶ . Mice exposed to hyperoxia in the presence of CO exhibited marked attenuation of hyperoxia-induced TNFα, IL-1β, and IL-6 expression (Figure 1D) ▶ .
Hyperoxia Activates the MAP Kinase Pathway in Vivo
Lung tissues were obtained from mice after hyperoxia exposure, and analyzed for activation of the major mitogen-activated protein (MAP) kinases, including ERK1/ERK2, JNK, MKK3/6, and p38. Figure 2 ▶ demonstrates the activation of the ERK1/ERK2, JNK, MKK3/6, and p38 pathway by hyperoxia in a time-dependent manner (Figure 2A) ▶ .
Figure 2.

A: Hyperoxia activates the MAP kinase pathways in vivo in the lung. Mice were exposed continuously to hyperoxia and at 24, 48, and 72 hours, the lungs were removed and analyzed for the presence of phosphorylated ERK1/ERK2, JNK, p38, and MKK3/6 MAP kinases. Lanes 1 and 2, untreated air controls; 3 and 4, 24 hours hyperoxia; 5 and 6, 48 hyperoxia; 7 and 8, 72 hours hyperoxia. Total ERK1/ERK2, JNK, p38, and MKK3/6 immunoblotting were used as normalization controls. B: Effects of hyperoxia on p38 expression in vitro. A549 cells were exposed continuously to hyperoxia for 15 and 30 minutes to observe any effects on p38 phosphorylation versus untreated controls. Protein lysates were assayed by Western blot using an anti-phosphorylated p38 antibody. The membranes were then stripped and reprobed with an anti-p38 antibody to assess any differences in loading. C: Effects of CO on pan-p38 and MKK3 activation. A549 cells were exposed to CO (250 ppm) continuously for 0 and 30 minutes, and 1, 4, 8, 16, and 24 hours (lanes 1 to 7, respectively) and assayed for pan-p38 and MKK3 phosphorylation by Western blot versus untreated controls. D: A549 cells were exposed to either 5%CO2/air or 5%CO2/air/CO (250 ppm) for 24 hours and then assayed for p38α and p38β by Western blot following immunoprecipitation. Data are representative blots from three independent experiments.
CO Extends Survival Specifically via the MKK3/p38 MAP Kinase Pathway
Since the major MAP kinases were all activated by hyperoxia, we used mice in which genes for the various MAP kinases were deleted to investigate whether CO was modulating the functional response through one of the MAP kinase pathways. We tested mice with a gene deletion for Mkk3. ERK1, ERK2, and p38 null mice are embryonic lethal. The mkk3(−/−) mice, a major upstream kinase regulating the p38 pathway, 19 were hypersusceptible to hyperoxic exposure, succumbing to lethal hyperoxia between 64 and 72 hours, whereas the control littermates died between 90 and 100 hours of exposure (Figure 3A) ▶ . These mice also had earlier signs of hyperoxic injury as determined by protein accumulation in the airspace. Mkk3(−/−) mice had a protein accumulation in the airspace of 2.96 ± 0.16 vs. hyperoxic-treated wild-type controls of 0.90 ± 0.11 mg/ml; P < 0.001 at the same time point. The presence of CO had no effect on hyperoxia-induced protein accumulation in the mkk3(−/−) mice (3.40 ± 0.23 mg/ml). The injury to the lungs of the mkk3(−/−) mice at 60 to 64 hours was similar by gross histological examination and BAL protein accumulation, as observed in control littermates at 85 to 90 hours of hyperoxia exposure. However, unlike control littermates, which exhibited increased numbers of leukocytes in the BAL, the mkk3(−/−) mice that had a similar injury at 64 hours exhibited no increase of leukocytes in the BAL (data not shown). Furthermore, while 250 ppm CO plus >98% O2 extended the survival of the control littermates, CO exerted no effect on extending the survival of the mkk3(−/−) mice (Figure 3A) ▶ . CO did protect and extend the survival of mice deficient in jnk2(−/−), in a similar fashion as that seen in control littermates (data not shown), demonstrating the specificity of the MKK3 signaling pathway in response to CO. To further confirm whether the p38 pathway downstream to MKK3 is involved, we used the selective p38 MAP kinase chemical inhibitor SB203580 (SB) that selectively inhibits the p38α and p38β isoforms, when administered at 20 mg/kg i.p. 20 Mice given SB and then exposed to hyperoxia in the presence and absence of CO (250 ppm) show similar effects as those observed with the mkk3(−/−) mice (data not shown). SB203580 administration prevented the protective effects otherwise observed in mice treated with CO. Mice treated with SB203580 alone, similarly to the mkk3(−/−) mice, succumbed to hyperoxic death earlier (72 to 80 hours) versus non-treated controls that died 95 to 100 hours after initiation of exposure (data not shown).
Figure 3.

A: Effects of CO on survival in mkk3(−/−) mice following exposure to lethal hyperoxia. Null mice and wild-type littermates were exposed to >98% O2 or >98% O2 plus 250 ppm CO continuously and monitored for survival. Mkk3(−/−) mice were hypersusceptible to O2, succumbing between 65 and 72 hours versus wild-type littermates that died between 90 and 100 hours. Furthermore, survival of the mkk3(−/−) mice could not be extended by CO (250 ppm) as observed in wild-type littermate controls. Data represent 20 to 25 animals per treatment group. B: Effects of CO (250 ppm) on hyperoxia-induced cytokine mRNA expression. Mkk3(−/−) or control littermates were exposed to >98% O2 in the presence or absence of CO (250 ppm). At 65 hours of exposure, the lungs from the mkk3(−/−) and control littermates were harvested and analyzed by RNase protection using the cytokine templates containing the cytokines listed above. L32 and GAPDH are included as loading controls. The blots are representative of three separate experiments in which the lungs from three mice were pooled for RNA extraction.
CO Mediates Anti-Inflammatory Effects via the MKK3/p38 MAP Kinase Pathway in Vivo
In control littermate mice, CO-induced protection correlated with inhibition of the pro-inflammatory cytokines TNFα, IL-1β, and IL-6 (Figure 3B) ▶ . These same cytokines were measured by RPA in the lungs of the mkk3(−/−) mice. Cytokine expression was detected earlier in the mkk3(−/−) mice when compared to the control littermate mice exposed for the same amount of time (64 hours; Figure 3B ▶ ). The wild-type littermate controls do not exhibit pro-inflammatory cytokines after 64 hours of hyperoxia (Figure 3B) ▶ because, as described in Figure 1 ▶ , it takes a longer exposure (84 hours of hyperoxia exposure) to elicit the lung injury and inflammation with increased pro-inflammatory cytokines. In contrast, the mkk3(−/−) mice exhibited increased pro-inflammatory cytokines at 64 hours of hyperoxia, which correlated with their increased susceptibility to hyperoxic lung injury. Furthermore, none of these pro-inflammatory cytokines were inhibited in the mkk3(−/−) mice by CO as was otherwise observed in control littermates.
CO Induces MKK3 and p38β while Decreasing the Expression of p38a
Lung epithelial cells (A549) exposed to CO (250 ppm) show a time-dependent increase in the activation of both p38 and MKK3 MAP kinases that peaks at 16 to 24 hours of exposure (Figure 2C) ▶ . To identify the specific p38 isoform involved, we used specific antibodies to immunoprecipitate each of the four p38 isoforms. There was specific activation of the p38β isoform, while there was a decrease in the activation of the p38α isoform (Figure 2D) ▶ . The gamma and delta isoforms were only minimally affected (data not shown).
CO Mediates Cytoprotective Effects via the MKK3/p38β MAP Kinase Pathway in Vitro
Epithelial cells (A549) when exposed to 95% O2 show a rapid increase in p38-phosphorylation (Figure 2B) ▶ that is maximal at 15 minutes. If the exposure is continued, the cells succumb to hyperoxia damage and necrosis after 4 days of exposure. Lee et al 21 demonstrated previously that hyperoxia-induced cell death can be inhibited by overexpression of HO-1. To test whether this protection was observed when CO supplanted HO-1, cells were exposed to 95% O2 in the presence of CO (250 ppm). We observed that after 4 days, the viability of O2-exposed cells had decreased to 20% while viability of those cells also treated with CO had decreased to 51% (Figure 4, A and B) ▶ . Furthermore, cells treated with CO in the presence of either SB203580 (10 μmol/L) or transiently transfected with dominant negative mutants of p38β or MKK3 showed that the cytoprotective effects of CO were ablated (Figure 4, A and B) ▶ . These findings support the paradigm that the MKK3/p38β pathway mediates CO-induced cytoprotection against hyperoxic stress.
Figure 4.

Effects of CO on survival of A549 cells exposed to O2. A549 cells were treated with the chemical inhibitor of p38β or SB203580 (10 μmol/L) (A) or transiently transfected with dominant negative mutants (DN) of p38β or MKK3 (1 μg/plate) (B) and then exposed to 95% O2 in the presence and absence of CO (250 ppm) for 4 days. pcDNA3 (1 μg/plate) was used as the vector control. After 4 days of exposure, cells were assessed for viability versus transfected and SB treated non-exposed cells. Results are expressed as a percentage of non-exposed cells from four replicate plates ± SD. There were no adverse effects of transfection or chemical treatment on viability in non-exposed cells. * P < 0.05 vs. O2-treated.
Discussion
There is strong evidence in the literature to support the emerging paradigm that HO-1 plays a vital role in maintaining cellular homeostasis in a variety of in vitro and in vivo models of oxidant cell and tissue injury. 7,21 Despite these compelling data, little is known regarding the mechanism(s) by which HO-1 mediates these cytoprotective effects. Many investigators have speculated that the byproducts released during heme catalysis by HO mediate these cytoprotective effects. Indeed, both ferritin and bilirubin have been shown to reduce the pro-oxidant state of the cell 22,23 and confer cytoprotection against oxidative stress. The gaseous molecule CO has been studied in both vascular and neuronal systems and has been shown to be an important signaling molecule. Until recently these were the extent of the functional studies of this gaseous molecule.
We have recently shown that CO also has the ability to provide cytoprotection against oxidative stress 10 ; administration of exogenous CO protected rats from hyperoxic lung injury and CO also had potent anti-inflammatory effects in LPS-treated macrophages and mice. 24 These data are supported by recent reports by Sato et al, 12 Fujita et al, 11 and Amersi et al, 25 demonstrating that CO at similar concentrations can impart potent anti-inflammatory effects. The mechanism by which CO mediates these cytoprotective effects is largely unknown. Thus, the major objective of this study was to delineate the mechanism(s) by which CO mediates the potent cytoprotective and anti-inflammatory effects against oxidative stress. We used a mouse model of oxidant-induced lung injury, and genetically altered mice to test the signaling pathways involved with the CO protective effects. Here in a clinically relevant model of acute lung injury in mice, we continue to provide such evidence and demonstrate, for the first time, potential molecular targets of CO. We show here that CO is cytoprotective as it attenuated hyperoxia-induced lung injury and increased survival of mice to lethal hyperoxia (Table 2 ▶ ; Figure 2, A and B ▶ ), but we also demonstrate that CO can confer potent anti-inflammatory effects in vivo, with inhibition of hyperoxia-induced TNFα, IL-1β, and IL-6. We have recently shown in vitro in cultured macrophages that CO at the same concentration used for this study also inhibited LPS-induced proinflammatory cytokines. 24 Moreover, using a well-described in vitro model of hyperoxic damage, we begin to demonstrate and unravel a potential cell signaling pathway affected by CO. These studies may lead to our understanding of how CO modulates downstream cytokine expression and subsequent tissue injury. The actions of CO clearly involve the MAP kinase signaling cascade, in particular the MKK3/p38β pathway. In support of this we describe that inhibition and/or deletion of the MKK3/p38β pathway not only modulated the expression of the inflammatory cytokines but also abrogated the CO-mediated cytoprotection against oxidant lung injury.
Furthermore, the studies by Sato et al 12 showing that CO can protect against cardiac xenograft rejection, and Brouard et al, 26 in a model of TNF-induced apoptosis in endothelial cells, reveal that CO can also prevent apoptosis through activation of the p38 pathway and give credence to the beneficial effects of CO exposure. In still other laboratories, Amersi et al 25 have shown that CO at 300 ppm can fully protect against cold liver ischemia/reperfusion transplant injury. It is becoming clear that CO is emerging as a functionally relevant cytoprotective, anti-inflammatory, and anti-apoptotic molecule.
CO can be highly toxic in vivo and has primarily been viewed as such. This toxicity is related directly to the formation of carboxyhemoglobin and the resulting anoxia given the higher binding-affinity of CO for heme compared with oxygen. The concentration of CO used in these experiments (<0.03%) is one-twentieth the lethal dose in rodents and humans, and when administered at this concentration had no untoward effects on the mice. This concentration is also 10-fold lower than that used in the measurement of lung diffusing capacity (DLCO) in humans, a standard pulmonary function test. Stupfel et al 27 previously demonstrated that the same concentration of CO used in our studies was well tolerated by rodents for greater than 6 months without obvious pathophysiological effects.
The precise molecular target(s) for CO is not clear at this time but our current knowledge that it preferentially binds heme moieties present in numerous proteins gives insight into potential target(s) of CO. For example, the activation of guanylyl cyclase (heme-containing molecule) by CO has been established as a key signaling pathway of CO in many systems, in particular the vascular and neuronal systems. In this model, CO is modulating pro-inflammatory cytokine production through the MAP kinases, so a plausible mechanism is an upstream intermediate involved in the modulation of these pathways. This speculation is supported by our observation that the protective effects of CO require new protein synthesis in a model of TNF-α-induced apoptosis (unpublished observations). Furthermore, the specificity of CO is exemplified here as observed in Figure 3 ▶ where it fails to modulate a number of other hyperoxia-induced cytokines such as the TGFβ family members. While some of these are also regulated by the MAP kinases they are also dependent on other transcriptional activators including the Stat and Smad family members. 28-30
Our study reveals a novel physiological function for the gaseous molecule CO in a model of acute lung injury. Despite the longstanding accepted paradigm that CO as present in our environment is toxic and even lethal, these data as well as more recent studies by other investigators suggest otherwise when administered at low concentrations. We demonstrate here in a model of acute lung injury, first that CO at low concentrations is anti-inflammatory and cytoprotective, second that it does so in part through inhibition of pro-inflammatory cytokines, and thirdly that it modulates these cytokines via the MKK3/p38β MAP kinase pathway. Lastly, we observed that the anti-inflammatory and cytoprotective effects of CO are mediated specifically by the MKK3/p38β pathway and do not involve the JNK pathway.
It is tempting to speculate that CO might be used therapeutically either by overexpressing HO-1, inducing it via genetic engineering, or by local administration of a low concentration of CO in areas of inflammation. CO might prove therapeutic not only in oxidant-induced lung injury, but also other inflammatory conditions.
Acknowledgments
We thank G. Aaron May for technical assistance with this work.
Footnotes
Address reprint requests to Augustine M. K. Choi, M.D., Section of Pulmonary and Critical Care Medicine, University of Pittsburgh School of Medicine, 3459 5th Avenue, Montifiore University Hospital 628NW, Pittsburgh, PA 15213. E-mail: choiam@msx.upmc.edu.
Supported by grants from the American Heart Association (0160332U); by an Atorvastatin Research Award (from the AHA) sponsored by Pfizer (awarded to L.E.O.); and by National Institutes of Health grant HL60234 (to A.M.K.C.).
References
- 1.Otterbein LE, Soares MP, Yamashita K, Bach FH: Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol 2003, 24:449-455 [DOI] [PubMed] [Google Scholar]
- 2.McCoubrey WK, Huang TJ, Maines MD: Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem 1997, 247:725-732 [DOI] [PubMed] [Google Scholar]
- 3.Keyse SM, Tyrrell RM: Heme oxygenase, a 32 kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci USA 1989, 86:99-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soares MP, Lin Y, Anrather J, Csizmadia E, Takigami E, Sato K, Grey ST, Colvin RB, Choi AM, Poss KD, Bach FH: Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med 1998, 4:1073-1077 [DOI] [PubMed] [Google Scholar]
- 5.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME: Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest 1992, 90:267-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Otterbein L, Sylvester SL, Choi AM: Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am J Respir Cell Mol Biol 1995, 13:595-601 [DOI] [PubMed] [Google Scholar]
- 7.Poss KD, Tonegawa S: Reduced stress defense in heme oxygenase-deficient cells. Proc Natl Acad Sci USA 1997, 94:10925-10930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda HJ: Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. Clin Invest 1999, 103:129-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otterbein LE, Kolls JK, Mantell LL, Cook J, Alam J, Choi AM: Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest 1999, 103:1047-1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Otterbein LE, Mantell LL, Choi AMK: Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol 1999, 276:L688-L694 [DOI] [PubMed] [Google Scholar]
- 11.Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ: Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med 2001, 7:598-604 [DOI] [PubMed] [Google Scholar]
- 12.Sato K, Balla J, Otterbein LE, Smith RN, Brouard S, Lin Y, Csizmadia E, Sevigny J, Robson SC, Vercellotti G, Choi AM, Bach FH, Soares MPJ: Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse- to-rat cardiac transplants. Immunol 2001, 166:4185-4194 [DOI] [PubMed] [Google Scholar]
- 13.Clark JM, Lambertson CJ: Pulmonary oxygen toxicity : a review. Pharmacol Rev 1971, 23:37-133 [PubMed] [Google Scholar]
- 14.Lu H-T, Yang DD, Wysk M, Gatti E, Mellman I, Davis RJ, Flavell RA: Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J 1999, 18:1845-1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuan CY, Yang DD, Samanta DR, Davis RJ, Rakic P, Flavell RA: The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999, 22:667-676 [DOI] [PubMed] [Google Scholar]
- 16.Otterbein LE, Chin BY, Otterbein SL, Lowe VC, Fessler HE, Choi AMK: Mechanism of hemoglobin-induced protection against endotoxemia in rats: a ferritin-independent pathway. Am J Physiol 1997, 272:L268-L275 [DOI] [PubMed] [Google Scholar]
- 17.Fearns C, Kline L, Gram H, Di Padova F, Zurini M, Han J, Ulevitch RJJ: Coordinate activation of endogenous p38α, β, γ, and Δ by inflammatory stimuli. Leukoc Biol 2000, 67:705-711 [DOI] [PubMed] [Google Scholar]
- 18.Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD: Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Resp Crit Care Med 1994, 150:113-322 [DOI] [PubMed] [Google Scholar]
- 19.Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ: MKK3 and MKK6 regulation of gene expression is mediated by the p38 MAP kinase signal transduction pathway. Mol Cell Biol 1996, 16:1247-1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW: A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994, 372:739-746 [DOI] [PubMed] [Google Scholar]
- 21.Lee PJ, Alam J, Wiegand GW, Choi AMK: Overexpression of heme oxygenase-1 in human pulmonary epithelial cells results in cell growth arrest and increased resistance to hyperoxia. Proc Natl Acad Sci USA 1996, 93:10393-10398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stocker R, Glazer AN, Ames BN: Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci USA 1987, 84:5918-5922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vile GF, Tyrrell RM: Oxidative stress resulting from ultra violet A irradiation of human skin fibroblasts leads to a heme oxygenase-dependent increase in ferritin. J Biol Chem 1994, 268:14678-14681 [PubMed] [Google Scholar]
- 24.Otterbein LE, Bach FH, Alam J, Soares M, Lu HT, Wysk M, Davis RJ, Flavell RA, Choi AMK: Carbon monoxide mediates anti-inflammatory effects via the p38 mitogen-activated protein kinase pathway. Nat Med 2000, 6:422-428 [DOI] [PubMed] [Google Scholar]
- 25.Amersi F, Shen XD, Anselmo D, Melinek J, Iyer S, Southard DJ, Katori M, Volk HD, Busuttil RW, Buelow R, Kupiec-Weglinski JW: Ex vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology 2002, 35:815-823 [DOI] [PubMed] [Google Scholar]
- 26.Brouard S, Otterbein LE, Anrather J, Tobiasch E, Bach FH, Choi AMK, Soares MP: Carbon monoxide generated by heme oxygenase-1 suppresses endothelial cell apoptosis. J Exp Med 2000, 192:1015-1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stuepfel M, Bouley G: Physiological and biochemical effects on rats and mice exposed to small concentrations of carbon monoxide for long periods. Ann NY Acad Sci 1970, 174:342-368 [DOI] [PubMed] [Google Scholar]
- 28.Christian JL, Nakayama T: Can’t get no SMADisfaction: smad proteins as positive and negative regulators of TGF-β family signals. Bioessays 1999, 21:382-390 [DOI] [PubMed] [Google Scholar]
- 29.Visser JA, Themmen AP: Downstream factors in transforming growth factor-β family signaling. Mol Cell Endocrin 1998, 146:7-17 [DOI] [PubMed] [Google Scholar]
- 30.Hill CS: Signaling to the nucleus by members of the transforming growth factor-β (TGF-β) superfamily. Cell Signal 1996, 8:533-544 [DOI] [PubMed] [Google Scholar]