

Abstract
Previous co-immunoprecipitation studies have shown that endogenous PFK-M (phosphofructokinase, muscle-specific isoform) associates with caveolin (Cav)-3 under certain metabolic conditions. However, it remains unknown whether Cav-3 expression is required for the plasma membrane recruitment and caveolar targeting of PFK-M. Here, we demonstrate that recombinant expression of Cav-3 dramatically affects the subcellular localization of PFK-M, by targeting PFK-M to the plasma membrane, and by trans-locating PFK-M to caveolae-enriched membrane domains. In addition, we show that the membrane recruitment and caveolar targeting of PFK-M appears to be strictly dependent on the concentration of extracellular glucose. Interestingly, recombinant expression of PFK-M with three Cav-3 mutants [ΔTFT (63 to 65), P104L, and R26Q], which harbor the same mutations as seen in the human patients with Cav-3-related muscle diseases, causes a substantial reduction in PFK-M expression levels, and impedes the membrane recruitment of PFK-M. Analysis of skeletal muscle tissue samples from Cav-3(−/−) mice directly demonstrates that Cav-3 expression regulates the phenotypic behavior of PFK-M. More specifically, in Cav-3-null mice, PFK-M is no longer targeted to the plasma membrane, and is excluded from caveolar membrane domains. As such, our current results may be important in understanding the pathogenesis of Cav-3-related muscle diseases, such as limb-girdle muscular dystrophy-1C, distal myopathy, and rippling muscle disease, that are caused by mutations within the human Cav-3 gene.
Phosphofructokinase (PFK) is an enzyme of central importance in the regulation of carbohydrate metabolism. It catalyzes the rate-limiting and irreversible reaction in glycolysis, the conversion of fructose-6-phosphate to fructose-1,6-bisphosphate. Because PFK plays such a key role in the control of the glycolytic pathway, its activity appears to be tightly regulated and modulated by several allosteric effectors. 1 It is inhibited by high concentrations of ATP and citrate, and it is stimulated by AMP, Pi, fructose-2,6-bisphosphate, and one of the products of its own reaction, fructose-1,6-bisphosphate. Therefore, whenever the cell has high concentrations of ATP, or other sources of energy are available, such as citrate or long-chain fatty acids, PFK activity is inhibited and glycolysis is blocked. On the other hand, when the cell is in energy demand, PFK activity is stimulated. Thus, it is believed that, in resting cells, the enzyme is functionally inactive.
Three different PFK isoforms have been identified in mammals, termed 1) PFK-A or PFK-M (muscle-type), 2) PFK-B (PFK-L in human, liver-type), and 3) PFK-C or PFK-P (platelet-type)—each encoded by a different gene. 2,3 The protein products of these genes are differentially expressed during development and display distinct tissue specificity. These three isoforms can randomly associate to form homo- and hetero-oligomers, such that the mature PFK is a tetrameric enzyme complex of ∼340 kd. 4,5 Mature skeletal muscle expresses only the M subunit, and, therefore, contains exclusively the homotetramer M4. On the other hand, liver contains only the homotetramer L4. Erythrocytes express both the M and the L subunits, and random oligomerization gives rise to five isoenzymes, the homotetramers M4 and L4, and the three hybrid forms. 4,6
An inherited deficiency of the PFK-M subunit (glycogenosis type VII) 7 causes a syndrome, characterized by hemolysis, and by a significant myopathy with skeletal muscle weakness and cramps, exercise intolerance, and myoglobinuria. 8 The observed clinical symptoms in patients with PFK-M deficiency reflect the lack of PFK-M in muscle and the partial reduction of the enzyme in erythrocytes.
An increasing body of evidences pinpoints the association of PFK-M with cytoskeletal elements and with proteins involved in signal transduction processes, suggesting that PFK-M activity could be regulated and co-ordinated with other cellular processes in a very complex manner. In skeletal muscle, PFK-M binds to, and is inhibited by, tubulin and microtubules, indicating that the cytoskeleton may play a role in controlling the speed of glycolysis. 9 Moreover, PFK-M was shown to form a complex with creatine kinase at the sarcomeric I-band of skeletal muscle, and this coupling is thought to increase the efficiency of cellular metabolism. 10 In the myocardium, PFK-M interacts with phospholipase A2, suggesting a coordinated regulation of phospholipolysis and glycolysis. 11 PFK-M serves as a substrate for receptor tyrosine kinases, such as the insulin receptor (Ins-R) and epidermal growth factor-receptor (EGF-R) tyrosine kinases. 12,13 In addition, PFK-M was found to be associated with neuronal nitric oxide synthase in brain and skeletal muscle. 14 This interaction might be functionally relevant, as nitric oxide, the product of nitric oxide synthase activity, can regulate energy metabolism in normal muscle by stimulating exercise-induced glucose transport. 15
Throughout the last decade, an emerging role in orchestrating different signaling pathways has been attributed to 50- to 100-nm membrane invaginations of the plasma membrane, termed plasmalemmal caveolae. 16-18 Several proteins involved in signal transduction, including nitric oxide synthase isoforms, epidermal growth factor receptor, and insulin receptor, have been found to be concentrated in caveolar membrane domains. Compartmentalization within these cellular organelles appears to be essential in the regulation of the activation state of certain signaling molecules. The main structural elements of caveolae are a family of integral membrane proteins, termed caveolins. 19 In mammals, the caveolin gene family is composed of three members, termed caveolin (Cav)-1, Cav-2, and Cav-3. 19-22 Cav-1 and Cav-2 have similar tissue distributions, and are highly co-expressed in adipocytes, endothelial cells, pneumocytes, and fibroblasts, 23 whereas Cav-3 is muscle-specific, and is highly expressed in skeletal, cardiac, and smooth muscle cells. 21,24 Several studies have shown that Cav-1 and Cav-3, but not Cav-2, can induce the formation of the caveolae organelles, by a mechanism that involves their self-oligomerization properties. 25-29
Cav-3 and muscle caveolae appear to have specialized roles in skeletal muscle cells, other than cellular signaling compartmentalization. Immunohistochemical studies have shown that Cav-3 is localized at the plasma membrane of skeletal muscle fibers (sarcolemma). 24 Cav-3 expression is greatly induced during the differentiation of myoblasts to myotubes, such that fully differentiated skeletal muscle fibers show a high content of Cav-3 and Cav-3-generated caveolae. 22,24 At the level of the plasma membrane, Cav-3 associates with members of a protein complex that is thought to confer structural stability to the muscle cell membrane, the dystrophin-glycoprotein complex. 24,30 Cav-3 and dystrophin competitively bind to the same site on β-dystroglycan, suggesting that Cav-3 may have a role in regulating the membrane recruitment of dystrophin, and in the dynamic assembly of the dystrophin-glycoprotein complex. 30 Certain signaling molecules (such as Gi2α, Gβγ, c-Src, and other Src family kinases) are found to be enriched in muscle-derived caveolar membranes, corroborating the role of muscle cell caveolae in the compartmentalization and modulation of signal transduction processes. 24 In addition, Cav-3 interacts with nitric oxide synthase in skeletal muscle fibers, 31,32 a molecule that is important for the regulation of muscle contractility and exercise-induced glucose uptake.
An important role for Cav-3 in muscle physiology was clearly demonstrated by the finding that mutations in the Cav-3 gene are responsible for an autosomal dominant form of limb girdle muscular dystrophy-1C. 33 The main clinical features of these patients include calf hypertrophy and mild-to-moderate muscle weakness. After this initial report, other mutations in the Cav-3 gene have been associated with different clinical phenotypes, including idiopathic hyperCKemia, rippling muscle disease, and distal myopathy. 34-36 These phenotypes all share the down-regulation, to varying degrees (between 70 to 95%), of Cav-3 protein expression levels.
The role of Cav-3 in skeletal muscle functioning was further enlightened by the observation that PFK-M forms a tight complex with Cav-3 under certain metabolic conditions. 37 In C2C12 cells, as well as in skeletal muscle tissue lysates, the Cav-3/PFK-M interaction is favored by high concentrations of extracellular glucose, and is stimulated by known allosteric activators of PFK activity. 37 As Cav-3 associates with the enzymatically active form of PFK-M, these findings indicate that interaction of PFK-M with Cav-3 would be expected to recruit PFK-M to the muscle plasma membrane, and concentrate it within caveolar membrane domains. However, direct evidence for Cav-3-mediated recruitment of PFK-M to the plasma membrane is lacking. Thus, this hypothesis remains to be tested experimentally.
Here, we directly demonstrate that recombinant expression of Cav-3 regulates the subcellular distribution of PFK-M. We show that Cav-3 expression recruits PFK-M to the plasma membrane and caveolin-containing lipid raft microdomains. In contrast, expression of Cav-3 mutants that cause limb girdle muscular dystrophy and other muscle diseases prevents the plasma membrane targeting of PFK-M and induces the degradation of PFK-M by a proteasomal pathway. Finally, analysis of skeletal muscle tissue from Cav-3-deficient mice clearly demonstrates the importance of Cav-3 in the regulation of PFK-M behavior. Skeletal muscles from Cav-3-null mice show unperturbed expression levels of PFK-M, but significant changes in the subcellular localization of PFK-M. More specifically, PFK-M is no longer recruited to the plasma membrane, and is specifically excluded from lipid rafts/caveolar microdomains in Cav-3-null mice. Taken together, these results highlight the central role of muscle caveolae in the control of energy metabolism in skeletal muscle fibers and provide insight into the molecular mechanisms underlying muscle diseases in which Cav-3 expression is down-regulated.
Materials and Methods
Materials
Antibodies and their sources were as follows: anti-V5 monoclonal antibody (mAb) (Invitrogen, Carlsbad, CA); anti-Cav-3 mAb (clone 26) 24 (gift of Dr. Roberto Campos-Gonzalez; BD Transduction Laboratories, Inc.); rabbit anti-PFK-M polyclonal antibody (pAb) and guinea pig anti-PFK-M pAb (generated by Dr. Robert G. Kemp, University of Health Sciences, The Chicago Medical School, North Chicago, IL); anti-Cav-3 pAb (Affinity Bioreagents, Inc.); anti-Cav-1 pAb (Santa Cruz Biotechnology, Santa Cruz, CA); anti-actin mAb (clone AC-40) (Sigma Chemical Co., St. Louis, MO); anti-β-actin mAb (clone AC-15) (Sigma); proaerolysin (Protox Biotech, Inc., Victoria, Canada); anti-aerolysin polyclonal Ab (gift of Dr. J. Thomas Buckley, University of Victoria, Canada). A variety of other reagents were purchased commercially, as follows: cell-culture reagents were from Life Technologies, Inc. Grand Island, NY; the Effectene transfection reagent was from Qiagen, Valencia, CA; glutathione-agarose beads were from Amersham, Arlington Heights, IL; the proteasomal inhibitor, MG-132, was from Calbiochem, La Jolla, CA.
Expression Vectors
The cDNAs encoding Cav-3 and Cav-3 mutants (Cav-3 P104L, Cav-3 ΔTFT, and Cav-3 R26Q) were subcloned into pCAGGS, a mammalian expression vector driven by the cytomegalovirus promoter. 38,39 The cDNAs encoding human PFK-M, PFK-B, and PFK-P in the pEF6/V5 TOPO vector under the control of the human EF-1α promoter were purchased from Invitrogen (Genestorm Clones).
Cell Culture and Transfection
Cos-7 and 293T cells were grown in DME supplemented with 10% fetal bovine serum, 2 mmol/L glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies, Inc.). Cells (∼40 to 50% confluent) were transiently transfected using the Effectene transfection reagent, as per the manufacturer’s instructions, and analyzed 36 to 48 hours after transfection.
Construction and Purification of GST Fusion Proteins
GST-Cav-3 and GST-Cav-1 expression constructs were as previously described. 30 Briefly, Cav-3 (residues 34 to 129) and Cav-1 (residues 1 to 178) were amplified and subcloned into the pGEX-4T vector. GST fusion protein constructs were transformed into Escherichia coli (BL21 strain; Novagen, Inc., Madison, WI). After induction of expression through addition of 0.5 mmol/L of isopropyl-β-d-thio-galactoside (Sigma), GST fusion proteins were affinity-purified on glutathione-agarose beads, using the detergent Sarcosyl for initial solubilization.
GST Pull-Down Assays
The pull-down assay using GST alone or GST-Cav-1/3 fusion proteins was essentially as previously described. 30 Briefly, 293T cells transiently overexpressing V5-tagged PFK-M were lysed in RIPA buffer [10 mmol/L Tris-HCl, pH 7.4, 300 mmol/L NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 1% sodium deoxycholate]. Precleared lysates were then diluted in Tween buffer (50 mmol/L Tris-HCl, pH 7.4, 1 mmol/L ethylenediaminetetraacetic acid, 100 mmol/L NaCl, 0.1% Tween-20, 1 mmol/L dithiothreitol, and protease inhibitors) and added to ∼100 μl of an equalized bead volume for overnight incubation at 4°C. After binding, the beads were extensively washed with phosphate-buffered saline (six times). Finally, the beads were resuspended in 3× sample buffer, boiled, and subjected to SDS-polyacrylamide gel electrophoresis (PAGE).
Immunoblot Analysis
Transfected cells were washed twice with phosphate-buffered saline (PBS), and lysed with hot sample buffer containing dithiothreitol. To prepare tissue lysates, mouse skeletal muscle tissue was harvested, minced with scissors, homogenized in a Polytron tissue grinder for 30 seconds at a medium range speed, using boiling lysis buffer (10 mmol/L Tris, pH 8; 1% SDS) containing protease inhibitors (Boehringer Mannheim, Indianapolis, IN). Protein concentrations were quantified using the BCA reagent (Pierce, Rockford, IL) and the volume required for 10 μg of protein was determined. Samples were separated by SDS-PAGE (12.5 or 10% acrylamide) and transferred to nitrocellulose. The nitrocellulose membranes were stained with Ponceau S (to visualize protein bands) followed by immunoblot analysis. All subsequent wash buffers contained 10 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 0.05% Tween-20, which was supplemented with 1% bovine serum albumin (BSA) and 4% nonfat dry milk (Carnation) for the blocking solution and 1% BSA for the antibody diluent. Horseradish peroxidase-conjugated secondary antibodies were used to visualize bound primary antibodies, with the Supersignal chemiluminescence substrate (Pierce).
Triton X-100 Insolubility Assay
Transfected Cos-7 cells were washed twice with ice cold PBS and a buffer containing ice-cold 25 mmol/L Mes, pH 6.5, 0.15 mol/L NaCl, 1% Triton X-100, and protease inhibitors was added to the cells on ice. 40 After a 30-minute incubation at 4°C without agitation, the soluble fraction was collected. The insoluble fraction was extracted using 1% SDS. Equal volumes of the soluble and insoluble fraction were resolved by SDS-PAGE (12.5% acrylamide) and analyzed by V5 or Cav-3 immunoblotting.
Purification of Caveolae-Enriched Membrane Fractions
Caveolae-enriched membrane fractions were purified essentially as we previously described. 41 Transfected Cos-7 cells were homogenized in MBS (25 mmol/L Mes, pH 6.5, 150 mmol/L NaCl) containing 1% Triton X-100 and solubilized by passage 10 times through a tight-fitting Dounce homogenizer. Cell lysates were mixed with an equal volume of 80% sucrose (prepared in MBS lacking Triton X-100), transferred to a ultracentrifuge tube, and overlaid with a discontinuous sucrose gradient (1.6 ml of 30% sucrose, 1.8 ml of 5% sucrose, both prepared in MBS, lacking detergent). The samples were then subjected to centrifugation at 200,000 × g (44,000 rpm in a Sorval rotor SW60) for 16 to 20 hours. A light-scattering band was observed at the 5/30% sucrose interface. Twelve 0.37-ml fractions were collected and 10 μg of each fraction were subjected to SDS-PAGE and subjected to immunoblotting with V5 or Cav-3 antibodies.
Immunofluorescence Analysis
This procedure was performed as we previously described. 38 Briefly, transfected Cos-7 cells were fixed for 30 minutes in PBS containing 2% paraformaldehyde and rinsed with PBS. The cells were then incubated in permeabilization buffer (PBS, 0.2% BSA, 0.1% Triton X-100) for 10 minutes, washed with PBS, and treated for 10 minutes with 25 mmol/L of NH4Cl in PBS to quench free aldehyde groups. Then, cells were incubated with the primary V5 monoclonal antibody. Bound primary antibody was visualized with a secondary antibody [fluorescein isothiocyanate-conjugated goat anti-mouse antibody (Jackson Immunochemicals, West Grove, PA)]. Cells were then washed with PBS (three times) and slides were mounted with Slow-Fade anti-fade reagent (Molecular Probes, Eugene, OR). A cooled charge-coupled device camera attached to an Olympus microscope was used for detection of bound secondary antibodies. For double-labeling experiments, the appropriate primary polyclonal antibody was incubated along with anti-V5 mAb and detected with a tetramethyl-rhodamine isothiocyanate-conjugated goat anti-rabbit (Jackson Immunochemicals). For double-labeling experiments with proaerolysin (to detect GPI-linked proteins), paraformaldehyde-fixed cells were incubated with 10−8 mol/L proaerolysin for 1 hour. After washing, cells were incubated with a monoclonal antibody directed against the V5-epitope tag and with a rabbit polyclonal antibody directed against proaerolysin, followed by incubation with the appropriate secondary antibodies.
Proteasomal Inhibitor Treatment
Twenty hours after transfection, Cos-7 cells were treated for 16 hours with normal growth media containing either vehicle alone [dimethyl sulfoxide ((DMSO)] or MG-132 (10 μmol/L). Proteasome inhibitors were dissolved in DMSO. 28
Immunostaining of Skeletal Muscle Sections
Gastrocnemius muscles were isolated from wild-type (WT) and Cav-3-null mice, rapidly frozen in liquid nitrogen-cooled isopentane and stored in liquid nitrogen. Unfixed frozen sections (6 μm thick) of skeletal muscle were blocked with 1% BSA, 10% horse serum, and 0.1% Triton X-100 for 1 hour at room temperature. Sections were then incubated with a given primary antibody diluted in PBS and 1% BSA. After three washes with PBS, sections were incubated with the appropriate secondary antibody (tetramethyl-rhodamine isothiocyanate-conjugated goat anti-guinea pig antibody or tetramethyl-rhodamine isothiocyanate-conjugated goat anti-rabbit antibody). Finally, the sections were washed three times with PBS and the slides were mounted with Slow-Fade anti-fade reagent. A cooled charge-coupled device camera attached to an Olympus microscope was used for detection of bound secondary antibodies.
Results
Recombinant Expression of Cav-3 Induces PFK-M Targeting to Caveolae-Enriched Microdomains and Recruitment to the Plasma Membrane
Recent evidence has demonstrated that under certain physiological conditions, endogenous PFK-M associates with Cav-3 as evidenced by co-immunoprecipitation studies in differentiated C2C12 cells (a well-studied skeletal myoblast cell line). 37 This interaction can be modulated by the presence of intracellular metabolites that are known allosteric activators or inhibitors of PFK activity. In addition, this complex formation can be driven by high concentrations of extracellular glucose, suggesting that it is the enzymatically active form of PFK that forms a complex with Cav-3. 37 These results suggest that, via the interaction with Cav-3, PFK-M would be recruited to the muscle cell plasma membrane, in proximity to which PFK-M would exert its activity. However, direct evidence supporting this hypothesis is lacking.
Here, we investigate the hypothesis that expression of Cav-3 is sufficient to recruit PFK-M to the plasma membrane and caveolar membrane microdomains. To this end, we used several complementary approaches, including a heterologous cellular expression system and a Cav-3-null mouse model. It should be noted that we used a cDNA that expresses PFK-M as a fusion protein carrying a V5 tag (a 14-amino acid epitope derived from P and V proteins of the paramyxovirus) at its C-terminus; as a consequence, we could easily detect PFK-M by using a V5 monoclonal probe.
First, to evaluate if we could detect complex formation between PFK-M and Cav-3 in a heterologous expression system, we attempted to reconstitute the interaction of PFK-M with Cav-3. We expressed and affinity-purified a GST fusion protein containing Cav-3 (termed GST-Cav-3). After immobilization on glutathione-agarose beads, GST-Cav-3 was incubated with lysates from 293T cells transiently overexpressing V5-tagged PFK-M. After extensive washing, the samples were resuspended in 3× sample buffer, boiled, and separated by SDS-PAGE. Blots were then immunostained with anti-V5 antibody. Figure 1 ▶ shows that Cav-3 binds specifically to PFK-M, as compared with GST alone or with beads alone. Importantly, the presence of the V5 epitope tag on PFK-M did not interfere with complex formation in this assay system.
Figure 1.

In vitro reconstitution of PFK-M binding to Cav-3. 293T cells were transiently transfected with the cDNA encoding PFK-M, containing the V5 epitope tag at its C-terminus. Cell lysates were incubated with affinity-purified GST alone or GST-Cav-3 (residues 34 to 129) immobilized on glutathione-agarose beads. After extensive washing, the beads were resuspended in 3× SDS-PAGE sample buffer, boiled, and separated by SDS-PAGE. The nitrocellulose membranes were subjected to Western blot analysis with the anti-V5 monoclonal antibody to detect PFK-M binding. Note that only GST-Cav-3 binds PFK-M; no binding was observed with GST alone or with the same incubation performed with beads alone.
We next evaluated whether the presence of Cav-3 might affect the biochemical properties of PFK-M. Previous studies have shown that the members of the caveolin family are normally targeted to liquid-ordered domains, which are membrane regions enriched in cholesterol and sphingolipids. 18,42 Such liquid-ordered membrane microdomains have been termed lipid rafts. 43 Their specific lipid composition renders these membrane microdomains more rigid than the surrounding phospholipid bilayer, and confers on them special biochemical properties. In particular, these membranes are resistant to solubilization by nonionic detergents at low temperatures, thereby facilitating their purification. 41,44-46
Because Cav-3 normally resides in such detergent-resistant membrane microdomains, it shares the property of being insoluble in nonionic detergents, such as Triton X-100. 21 Thus, we investigated whether the presence of Cav-3 would influence the ability of PFK-M to partition into a detergent-resistant membrane compartment. To test this hypothesis, we performed a well-established assay to determine Triton X-100 solubility, using Cos-7 cells transiently transfected with PFK-M alone or in combination with Cav-3.
Figure 2 ▶ shows that when expressed alone, PFK-M partitions equally between both the soluble and insoluble fractions. On the contrary, when PFK-M is co-expressed with Cav-3, PFK-M becomes exclusively Triton-insoluble. These results clearly indicate that in the presence of Cav-3, PFK-M changes its biochemical behavior and is shifted to the detergent-insoluble cellular fraction.
Figure 2.
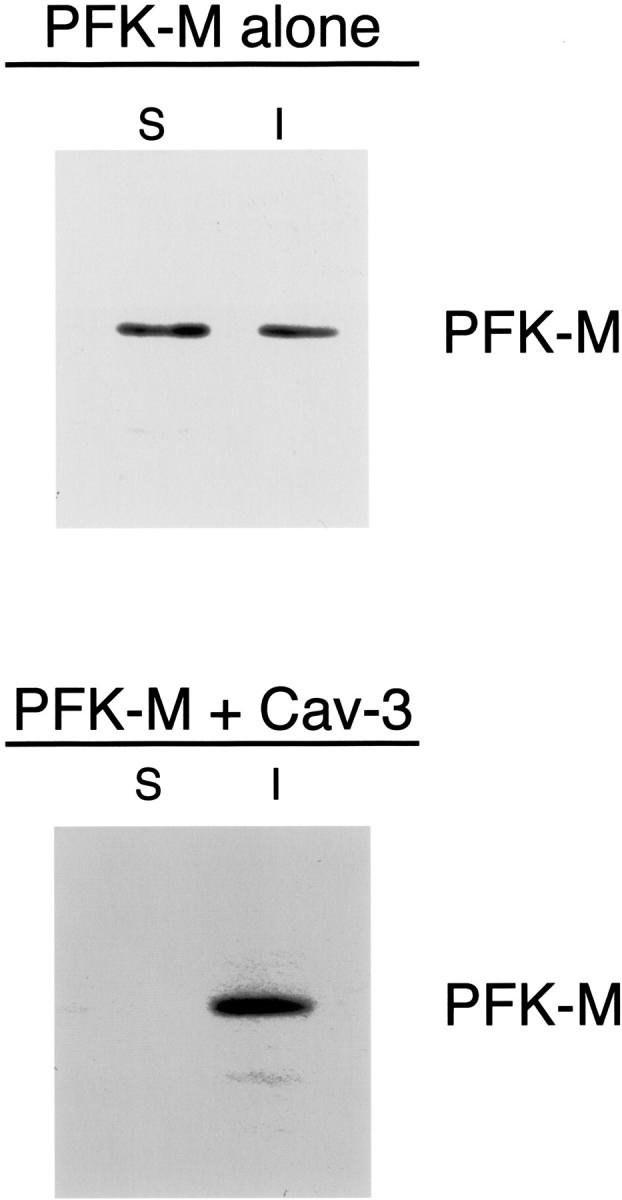
Recombinant expression of Cav-3 increases the detergent insolubility of PFK-M in cultured cells. Cos-7 cells were transiently transfected with the cDNA encoding V5-tagged PFK-M, alone or in combination with Cav-3. Thirty-six hours after transfection, the soluble fraction was extracted by incubating the cells with a buffer containing 1% Triton X-100. The insoluble fraction was collected using 1% SDS. Equal volumes of the soluble (S) and insoluble (I) fractions were resolved by SDS-PAGE and analyzed by V5 immunoblotting. Note that when expressed alone, PFK-M partitions both in the soluble and in the insoluble fractions. In contrast, when co-expressed with Cav-3, PFK-M is found only in the insoluble fraction.
We further investigated the influence of Cav-3 on the biochemical properties of PFK-M, by evaluating the subcellular fractionation of PFK-M. It is known that when Cav-3 is expressed, it is targeted to caveolae membranes that behave as detergent-resistant membrane microdomains. 24 Therefore, Cav-3 induces important changes in the morphology and function of this lipid microenvironment, and ultimately drives the formation of membrane invaginations, known as caveolae. To separate caveolae-enriched fractions from other membranous and cytosolic proteins, an established equilibrium sucrose density gradient system can be used. 41,44,47 Using this method, it is possible to distinguish the caveolae-enriched fractions (fractions 4 and 5) from other cellular components (fractions 8 to 12), by tracking the position of Cav-3. 24 To determine the subcellular localization of PFK-M, Cos-7 cells were transiently transfected with V5 tagged PFK-M, alone or in combination with Cav-3. Cells were then lysed at 4°C in the presence of Triton X-100, and were subsequently subjected to sucrose gradient ultracentrifugation.
Figure 3A ▶ shows that when Cos-7 cells are transiently transfected with PFK-M alone, PFK-M is excluded from these caveolae-enriched fractions. In striking contrast, when PFK-M is co-expressed with Cav-3, PFK-M co-fractionates with Cav-3 (fractions 4 and 5) and is exclusively incorporated into the caveolae-enriched membrane fractions (Figure 3B) ▶ . Taken together, these results indicate that expression of Cav-3 changes the biochemical behavior of PFK-M, and induces the targeting of PFK-M to detergent-resistant/caveolae-enriched membrane microdomains. In addition, these findings provide further support for the idea that PFK-M and Cav-3 form a tight complex in vivo.
Figure 3.

Recombinant expression of Cav-3 induces the targeting of PFK-M to caveolae-enriched membrane fractions. Caveolar microdomains were separated from the bulk of cellular membranes and cytosolic components using sucrose gradient fractionation. Cos-7 cells were transiently transfected with the cDNA encoding V5-tagged PFK-M, alone or in combination with Cav-3. Thirty-six hours after transfection, cells were lysed in a buffer containing 1% Triton X-100, and subjected to sucrose gradient ultracentrifugation. Twelve fractions were collected, and equal amounts of protein from each fraction (10 μg) were separated by SDS-PAGE and blotted onto nitrocellulose. The distribution of PFK-M and Cav-3 were analyzed by immunoblotting with V5 and Cav-3 antibodies, respectively. Note that when expressed alone, PFK-M is detected at the bottom of the gradient (A, fractions 8 and 12). In contrast, in the presence of Cav-3, PFK-M co-fractionates with Cav-3 (B, fractions 4 and 5), which marks the position of caveolar microdomains.
Given that, in presence of Cav-3, PFK-M changes its subcellular distribution as determined by biochemical assays, we next investigated whether recombinant expression of Cav-3 would modify the localization pattern of PFK-M. Cos-7 cells were transiently transfected with V5-tagged PFK-M, alone or in combination with Cav-3. Cells were then subjected to immunofluorescence analysis. The distribution of Cav-3 was visualized with a specific rabbit polyclonal antibody; the distribution of PFK-M was detected by using the V5 monoclonal antibody. Figure 4A ▶ shows that in cells expressing PFK-M alone, PFK-M is not targeted to the plasma membrane. Interestingly, recombinant expression of Cav-3 dramatically changes the distribution of PFK-M and induces the targeting of PFK-M to the plasma membrane (Figure 4B) ▶ .
Figure 4.
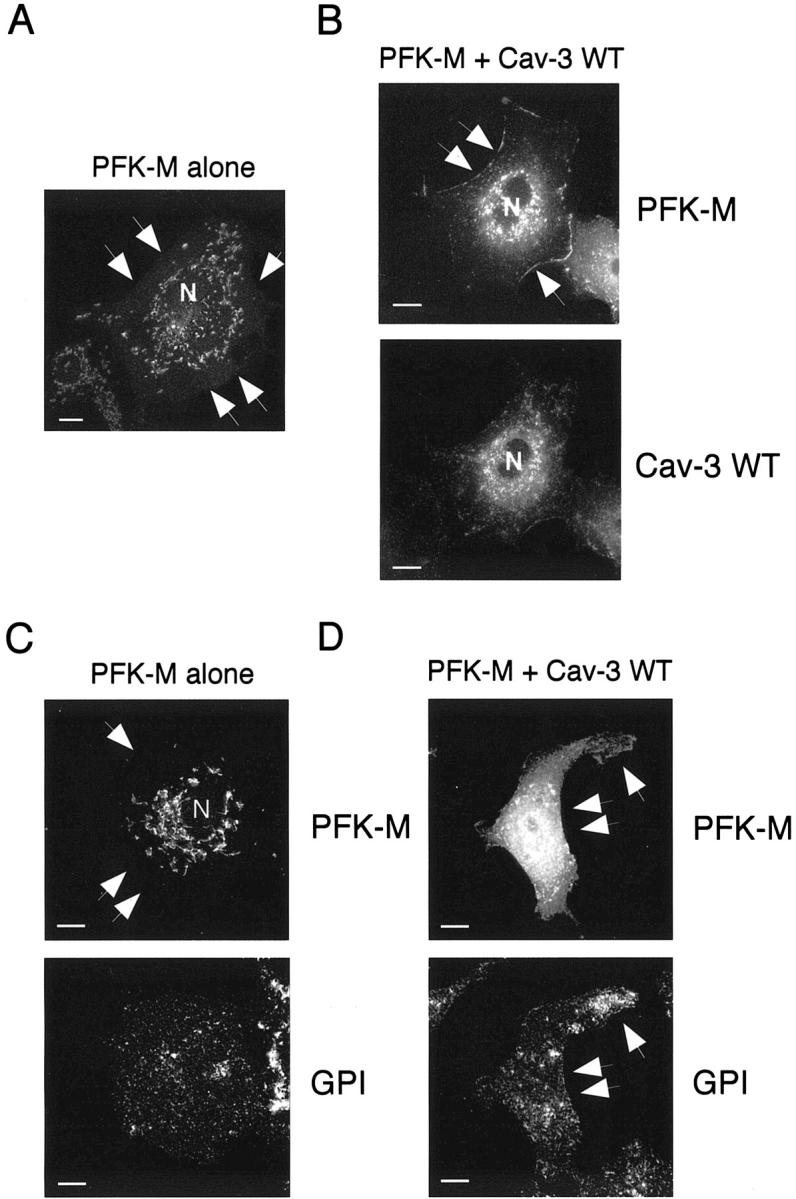
Caveolin-3 expression changes the subcellular distribution of PFK-M. Cos-7 cells were transfected with the cDNA encoding V5-tagged PFK-M, alone or in combination with Cav-3. Thirty-six hours after transfection, cells were formaldehyde-fixed and immunostained with a monoclonal antibody against the V5 epitope tag (A), or doubly immunostained with an anti-V5 mAb and an anti-Cav-3 pAb (B). Alternatively, cells transiently co-transfected with V5-tagged PFK-M and Cav-3 were doubly immunostained with an anti-V5 mAb and with proaerolysin for detecting GPI-anchored proteins (a marker of the plasma membrane). A: Cellular distribution of PFK-M alone. Note the intracellular perinuclear distribution of PFK-M, when it is expressed alone. N, Nucleus. B: Cellular distribution of PFK-M in the presence of Cav-3. Note that, when co-expressed with Cav-3, the localization of PFK-M shifts toward the plasma membrane (top). C: Intracellular distribution of PFK-M alone. Note that when expressed alone, the distribution of PFK-M overtly diverges from the distribution of a traditional plasma membrane marker, namely the distribution of GPI-anchored proteins. D: Membrane targeting of PFK-M in the presence of Cav-3 is illustrated by double labeling with a traditional marker of the plasma membrane, ie, GPI-anchored proteins. In A, B, C, and D arrows point at the plasma membrane. Scale bars, 10 μm.
To better visualize the targeting of PFK-M to the cell surface, we also performed double-labeling experiments with another well-established marker of the plasma membrane, namely GPI-anchored proteins. Cos-7 cells were transiently transfected with V5-tagged PFK-M, alone or in combination with Cav-3. Cells were then subjected to immunofluorescence analysis. The distribution of PFK-M was visualized using V5 monoclonal antibody. To detect the distribution of GPI-anchored proteins, we performed labeling using proaerolysin, a toxin that specifically recognizes the glycan core of the GPI anchor. 48
Figure 4C ▶ shows that in cells expressing PFK-M alone, PFK-M is localized intracellularly, whereas GPI-anchored proteins are localized to the plasma membrane. On the contrary, in cells co-expressing PFK-M and Cav-3, both PFK-M and GPI-anchored proteins are located at the plasma membrane (Figure 4D) ▶ .
PFK-M Membrane Recruitment and Caveolar Targeting Require Extracellular Glucose
Previous co-immunoprecipitation studies have shown that complex formation between endogenous PFK-M and Cav-3 in C2C12 cells is dependent on the presence of extracellular glucose. 37 In light of these findings, we next investigated whether PFK-M membrane recruitment was sensitive to the concentration of extracellular glucose.
Cos-7 cells were transiently transfected with V5-tagged PFK-M, alone or in combination with Cav-3. After incubation for 1 hour either with high-glucose media (4.5 g of glucose per L), or with glucose-free media, cells were subjected to immunofluorescence analysis. The distribution of Cav-3 was visualized with a specific rabbit polyclonal antibody; the distribution of PFK-M was detected by using the V5 monoclonal antibody. Note that in cells expressing PFK-M alone, incubation with glucose-free media has no effect on the localization of PFK-M. Figure 5A ▶ shows that PFK-M displays an intracellular distribution, independently of extracellular glucose concentration.
Figure 5.

PFK-M membrane recruitment is dependent on the presence of extracellular glucose. Cos-7 cells were transfected with the cDNA encoding V5-tagged PFK-M, alone or in combination with Cav-3. Thirty-six hours after transfection, cells were incubated for 1 hour either with glucose-free media or with high-glucose media. Cells were then formaldehyde-fixed and immunostained with a monoclonal antibody directed against the V5 epitope tag (A) or doubly immunostained with anti-V5 mAb and anti-Cav-3 pAb (B and C). Note that glucose depletion of cells expressing PFK-M alone has no significant effect on the distribution of PFK-M (A, bottom), as compared to cells expressing PFK-M alone incubated in high-glucose media (A, top). However, incubation of cells expressing Cav-3 and PFK-M in glucose-free media for 1 hour clearly impedes the targeting of PFK-M to the plasma membrane (C). Under these conditions, the distribution of PFK-M remains perinuclear. In contrast, when cells expressing Cav-3 and PFK-M were incubated in high-glucose media, Cav-3 expression induces the membrane recruitment of PFK-M. (B). Arrows point at the plasma membrane. These results suggest that the membrane recruitment of PFK-M is dependent on the presence of extracellular glucose and on Cav-3 expression. N, Nucleus. Note that high-glucose media contains 4.5 g of glucose per L; this is the normal concentration of glucose that is routinely used in high-glucose DME for cell culture. Scale bars, 10 μm.
In contrast, depletion of extracellular glucose has a profound effect on the distribution of PFK-M in cells co-expressing PFK-M and Cav-3. Figure 5C ▶ shows that incubation of cells co-expressing PFK-M and Cav-3 in glucose-free media impedes the membrane targeting of PFK-M. However, no effect was observed on the distribution of Cav-3 under these experimental conditions (Figure 5C) ▶ . The membrane distribution of PFK-M in cells co-expressing PFK-M and Cav-3 incubated in high-glucose media is shown for comparison (Figure 5B) ▶ . These results suggest that the caveolin-mediated membrane recruitment of PFK-M requires high concentrations of extracellular glucose. Importantly, the effects of glucose-free media were completely reversible on readdition of high-glucose media (data not shown).
We next evaluated whether the concentration of extracellular glucose would affect the targeting of PFK-M to the caveolae membrane microdomains. To separate the caveolae-derived membranes from the other membranous and cytosolic cellular components, we performed sucrose gradient ultracentrifugation, as in Figure 3 ▶ above. Cos-7 cells expressing V5-tagged PFK-M and Cav-3 were incubated for 1 hour either with high-glucose media or with glucose-free media. Cells were then lysed with Triton X-100 at low temperatures and subjected to ultracentrifugation. Twelve fractions were collected and equal amounts of each fraction were separated by SDS-PAGE. The distribution of PFK-M and Cav-3 was determined by immunoblotting with V5 and Cav-3 antibodies, respectively.
Figure 6 ▶ , top, shows that when cells were incubated with high-glucose media, PFK-M co-fractionates with Cav-3 (fractions 4 and 5). In contrast, incubation of PFK-M- and Cav-3-expressing cells with glucose-free media impedes PFK-M targeting to the caveolar membrane microdomains (Figure 6 ▶ , bottom). Importantly, incubation of the cells expressing PFK-M alone with either high-glucose media or glucose-free media did not influence the caveolar targeting of PFK-M. Under these conditions, PFK-M is exclusively found in fractions 8 to 12, which are not considered to be caveolar fractions (data not shown). Taken together, these results suggest that the membrane recruitment and caveolar targeting of PFK-M are dependent on two concomitant factors, ie, Cav-3 expression and the presence of extracellular glucose.
Figure 6.

The caveolar targeting of PFK-M is dependent on the presence of extracellular glucose. Caveolar microdomains were separated from the bulk of cellular membranes and cytosolic components using sucrose gradient fractionations. Cos-7 cells were transfected with the cDNA encoding V5-tagged PFK-M in combination with Cav-3. Thirty-six hours after transfection cells were incubated for 1 hour either with glucose-free media or with high-glucose media. Then, cells were lysed in a buffer containing 1% Triton X-100 and subjected to sucrose gradient ultracentrifugation. Twelve fractions were collected and equal amounts of protein (10 μg) from each fraction were separated by SDS-PAGE and blotted onto nitrocellulose. The distribution of PFK-M and Cav-3 were analyzed by immunoblotting with V5 and Cav-3 antibodies, respectively. Note that when cells were incubated in high-glucose media, PFK-M is targeted to caveolar membrane domains (fractions 4 and 5; top). In striking contrast, incubation of cells with glucose-free media impedes the caveolar targeting of PFK-M, and under these conditions, PFK-M is excluded from the caveolar fractions (fractions 8 to 12; bottom). Importantly, the distribution of Cav-3 and its targeting to fractions 4 and 5 is not affected by varying the concentration of extracellular glucose. Similar experiments were performed on Cos-7 cells transfected with PFK-M alone. PFK-M was found to be excluded from the caveolar fractions when cells were incubated with high-glucose media or with glucose-free media (data not shown). These results indicate that the caveolar targeting of PFK-M requires both Cav-3 expression and extracellular glucose. Note that high-glucose media contains 4.5 g of glucose per L; this is the normal concentration of glucose that is routinely used in high-glucose DME for cell culture.
Cav-3 Mutants Cause the Degradation and Abnormal Localization of PFK-M
Mutations in the CAV-3 gene have been associated with a variety of muscle diseases, including autosomal dominant limb-girdle muscular dystrophy-1C, idiopathic hyperCKemia, rippling muscle disease, and distal myopathy. 33-36 Two heterozygous mutations were first identified as responsible for the limb-girdle muscular dystrophy-1C phenotype: a 9-bp microdeletion (ΔTFT) in the caveolin-scaffolding domain and a missense mutation (P104L) in the Cav-3 transmembrane domain. Both mutations lead to a ∼90 to 95% loss of Cav-3 protein expression. 33 The substitution of a glutamine for an arginine at residue 26 (R26Q) in the N-terminal domain accounts for the hyperCKemic phenotype, as well as for the rippling muscle disease and for the distal myopathy. 34-36 This mutation leads to a partial Cav-3 deficiency in muscle fibers. We previously generated the constructs harboring all three mutations, namely Cav-3 P104L, Cav-3 ΔTFT, and Cav-3 R26Q, and analyzed their phenotypic behavior using heterologous expression in NIH 3T3 cells. 38,39 The three mutants are expressed at much lower levels as compared to the WT, they are mostly retained at the level of a perinuclear compartment, and they are excluded from detergent-resistant/caveolae-enriched membrane fractions. 38,39
As the behavior of PFK-M is deeply influenced by the expression of WT Cav-3, we next investigated whether co-expression with Cav-3 mutants could exert any effects on the expression level and/or the localization pattern of PFK-M. Cos-7 cells were transiently transfected with V5-tagged PFK-M, alone or in combination with either Cav-3 WT, Cav-3 P104L, Cav-3 ΔTFT, or Cav-3 R26Q. We next assessed the expression level of PFK-M and Cav-3 by Western blot analysis with monoclonal antibodies against the V5-epitope and Cav-3, respectively.
Interestingly, recombinant expression of all three Cav-3 mutants causes a dramatic down-regulation of PFK-M protein levels (Figure 7 ▶ , top). However, no appreciable variations in PFK-M expression levels were observed when PFK-M was co-expressed with Cav-3 WT. As expected from previous results, 38,39 the middle panel of Figure 7 ▶ shows that the expression level of the Cav-3 mutants is greatly reduced as compared to Cav-3 WT. As a critical internal control, equal protein loading was assessed by Western blot analysis with anti-β-actin IgG (Figure 7 ▶ , bottom).
Figure 7.
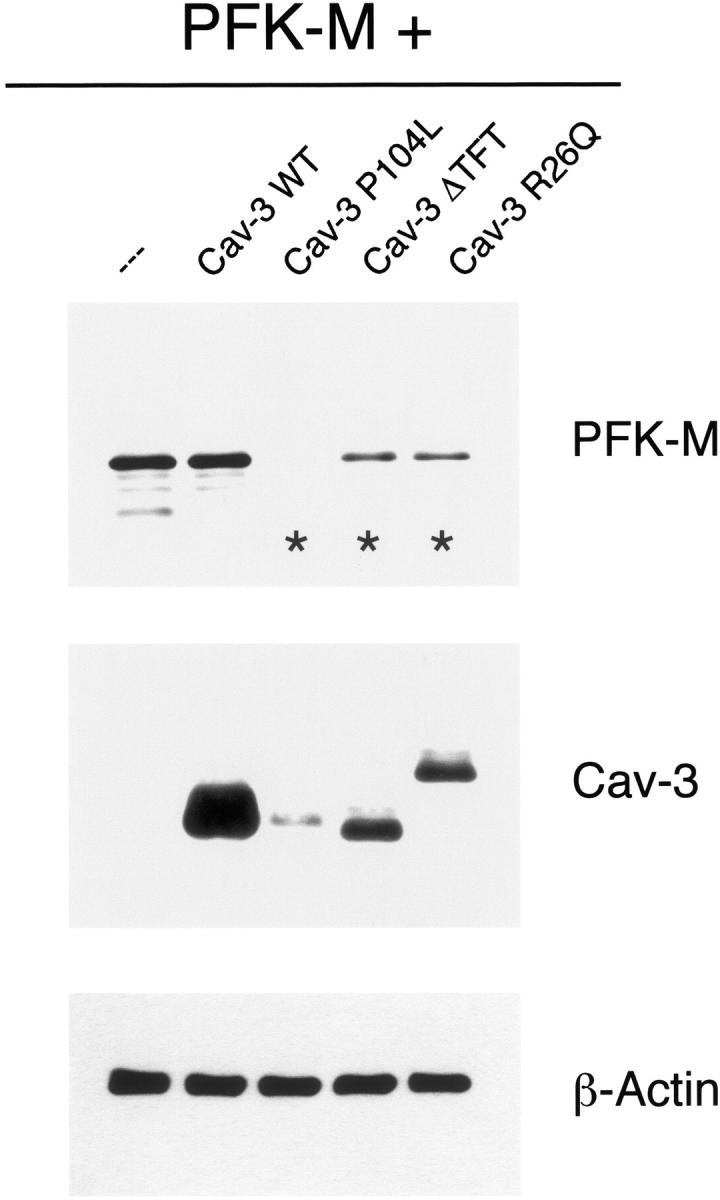
Pathogenic Cav-3 mutants down-regulate the expression of PFK-M. Cos-7 cells were transiently transfected with the cDNA encoding V5-tagged PFK-M, alone or in combination with Cav-3 WT or pathogenic Cav-3 mutants (Cav-3 P104L, Cav-3 ΔTFT, and Cav-3 R26Q). Thirty-six hours after transfection, cell lysates were prepared and resolved by SDS-PAGE. Transferred proteins were analyzed by immunoblotting, using either V5 or Cav-3 mAb probes. Note that PFK-M expression levels are greatly down-regulated when Cav-3 mutants are co-expressed (asterisks). In contrast, under the same experimental conditions, PFK-M expression levels remain unchanged in presence of Cav-3 WT. Western blot analysis with anti-β-actin IgG is shown as a control for equal loading.
We next explored the hypothesis that the lower protein expression of PFK-M was due to protein degradation, occurring through the proteasomal pathway. As a consequence, we analyzed the effect of proteasome inhibitor treatment on cells co-expressing PFK-M and Cav-3 mutants. Cos-7 cells were transiently co-transfected with V5-tagged PFK-M in combination with either Cav-3 P104L, Cav-3 ΔTFT, or Cav-3 R26Q. The expression levels of PFK-M were evaluated by Western blot analysis. Figure 8 ▶ shows that treatment for 16 hours with the proteasomal inhibitor MG-132 (10 μmol/L) is sufficient to rescue the expression level of PFK-M (Figure 8 ▶ , bottom). However, no effect was observed when cells were treated for the same time with the vehicle alone (DMSO) (Figure 8 ▶ , top). As a critical internal control, equal protein loading was also assessed by Western blot analysis with anti-β-actin IgG.
Figure 8.
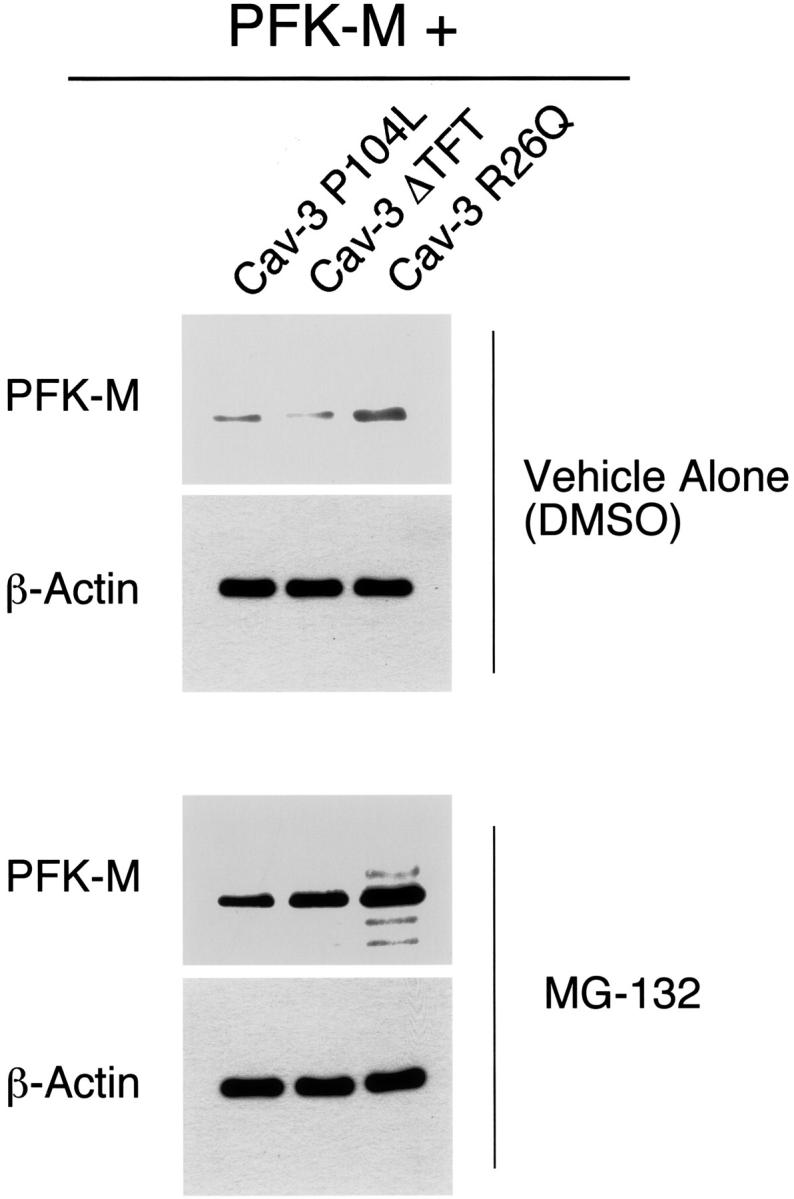
Treatment with the proteasome inhibitor MG-132 rescues the degradation of PFK-M induced by Cav-3 mutants. Cos-7 cells were transiently co-transfected with the cDNAs encoding V5-tagged PFK-M and pathogenic Cav-3 mutants (Cav-3 P104L, Cav-3 ΔTFT, and Cav-3 R26Q). Twenty hours after transfection, cells were incubated with the proteasomal inhibitor MG-132 (10 μmol/L) or with vehicle alone (DMSO). After 16 hours of treatment, the cells were lysed in hot sample buffer and proteins were resolved by SDS-PAGE. Transferred proteins were analyzed by V5 immunoblotting. Note that upon treatment with MG-132, PFK-M expression levels are greatly increased (bottom). In contrast, no effect was observed when cells were treated with vehicle alone (DMSO; top). Note that the V5 blots (top and bottom) were acquired with the same exposure time. Western blot analysis with anti-β-actin IgG is shown as a control for equal loading.
To evaluate the effect of Cav-3 mutant expression on the cellular distribution of PFK-M, we transiently co-transfected PFK-M with each one of the three mutants, and analyzed the cells by immunofluorescence microscopy. As shown in Figure 9 ▶ , recombinant expression of Cav-3 P104L (Figure 9A) ▶ , Cav-3 ΔTFT (Figure 9B) ▶ , and Cav-3 R26Q (Figure 9C) ▶ , impeded the targeting of PFK-M to the plasma membrane.
Figure 9.

Cav-3 mutants impede the recruitment of PFK-M to the plasma membrane. Cos-7 cells were co-transfected with the cDNA encoding V5-tagged PFK-M, in combination with each Cav-3 mutant. Thirty-six hours after transfection, the cells were formaldehyde-fixed and doubly immunostained with a monoclonal antibody directed against the V5 epitope and with a polyclonal antibody directed against Cav-3. Note that expression of Cav-3 mutants, namely Cav-3 P104L (A), Cav-3 ΔTFT (B), and Cav-3 R26Q (C) impedes the recruitment of PFK-M to the cell membrane. N, Nucleus. Scale bars, 10 μm.
In summary, we conclude that co-expression with Cav-3 mutants has a profound effect on the phenotypic behavior of PFK-M, as we observed that PFK-M is expressed at much lower levels, and is no longer targeted to the cell surface.
Caveolin-1 (Cav-1) Interacts with PFK-M and Induces the Membrane Recruitment of PFK-M
The caveolin gene family contains three members, ie, Cav-1, Cav-2, and Cav-3. Sequence alignment reveals that Cav-1 and Cav-3 are ∼65% identical and ∼85% similar. Given the high homology, we wondered whether Cav-1 could interact with PFK-M, and whether as a consequence of this binding PFK-M could be recruited to the plasma membrane. To test this hypothesis, we first performed a pull-down assay using an affinity-purified GST fusion protein containing the full-length sequence of Cav-1 (residues 1 to 178), GST-Cav-1. GST-Cav-1 was immobilized on glutathione-agarose beads, and incubated with lysates from 293T cells transiently overexpressing V5-tagged PFK-M. After extensive washing, the samples were resuspended in 3× sample buffer, boiled, and separated by SDS-PAGE. PFK-M binding was detected by immunoblotting with anti-V5 antibody. Figure 10A ▶ shows that Cav-1 interacts specifically PFK-M. No binding was detected in parallel experiments performed with GST alone or with beads alone.
Figure 10.

Cav-1 interacts with PFK-M and induces the membrane recruitment of PFK-M. A: GST pull-down assay. 293T cells were transiently transfected with the cDNA encoding PFK-M carrying the V5 epitope tag at its C-terminus. Cell lysates were then incubated with affinity-purified GST alone or GST-Cav-1 (residues 1 to 178) immobilized on glutathione-agarose beads. After extensive washing, the beads were resuspended in 3× SDS-PAGE sample buffer, boiled, and separated by SDS-PAGE. The nitrocellulose membranes were subjected to Western blot analysis with the anti-V5 monoclonal antibody. Note that only GST-Cav-1 binds PFK-M; no binding was observed with GST alone or with the same incubation performed with beads alone. B–C: Immunofluorescence. Cos-7 cells were transfected with the cDNA encoding V5-tagged PFK-M, alone or in combination with Cav-1. Thirty-six hours after transfection, cells were formaldehyde-fixed and immunostained with a monoclonal antibody directed against the V5 epitope (B), or doubly immunostained with the anti-V5 mAb and the anti-Cav-1 pAb (C). Note that when expressed alone, PFK-M is found mostly intracellularly in a perinuclear distribution. Interestingly, recombinant expression of Cav-1 induces the membrane recruitment of PFK-M. N, Nucleus. Scale bars, 10 μm.
We next investigated whether recombinant expression of Cav-1 could induce the membrane recruitment of PFK-M. Cos-7 cells were transiently transfected with V5-tagged PFK-M, alone or in combination with Cav-1. Cells were then subjected to immunofluorescence analysis. Figure 10B ▶ shows that when expressed alone, PFK-M mainly displays an intracellular pattern. Interestingly, recombinant expression of Cav-1 induces changes in the cellular distribution of PFK-M. Figure 10C ▶ shows that in cells co-transfected with PFK-M and Cav-1, PFK-M is recruited to the plasma membrane. Taken together, these results suggest that Cav-1 can interact with PFK-M, and can recruit PFK-M to the plasma membrane.
Recombinant Expression of Cav-3 Does Not Affect the Phenotypic Behavior of Other PFK Isoforms
Because there are three different isoforms of mammalian PFK, namely PFK-M, PFK-B, and PFK-P, 3 we wondered whether Cav-3 expression would also influence the phenotypic behavior of PFK-B and PFK-P. We first considered the detergent solubility properties of PFK-B and PFK-P. We performed an established assay to determine their Triton X-100 solubility using Cos-7 cells transiently transfected with PFK-B or PFK-P, each alone or in combination with Cav-3.
Figure 11A ▶ shows that when expressed alone, PFK-B partitions both in the soluble and insoluble fractions in an equivalent manner. Interestingly, Figure 11A ▶ shows that Cav-3 expression does not modify the biochemical behavior of PFK-B. Figure 11B ▶ shows that PFK-P is exclusively Triton-insoluble, and that its pattern is essentially identical when Cav-3 is expressed. These results clearly indicate that Cav-3 expression does not affect the detergent solubility of PFK-B and PFK-P.
Figure 11.
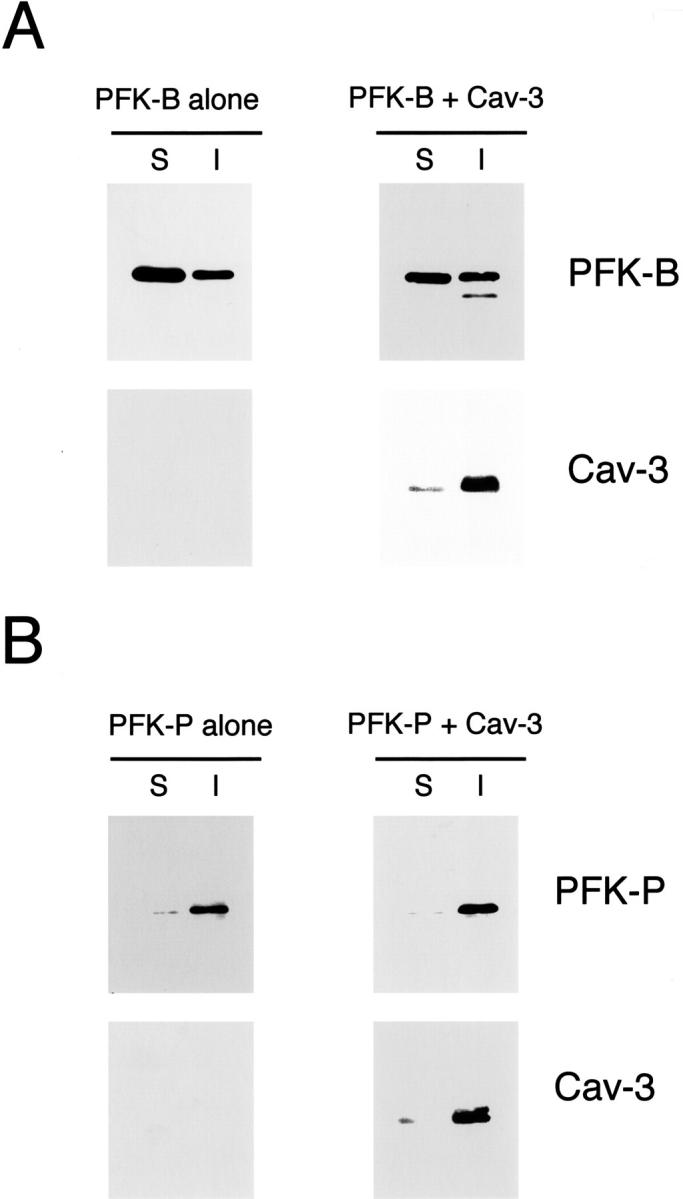
Cav-3 expression does not affect the detergent solubility of two other PFK isoforms, namely PFK-B and PFK-P. Cos-7 cells were transiently transfected either with the cDNA encoding V5-tagged PFK-B, alone or in combination with Cav-3 (A) or with the cDNA encoding V5-tagged PFK-P, alone or in combination with Cav-3 (B). Thirty-six hours after transfection, the soluble fraction was extracted by incubating cells with a buffer containing 1% Triton X-100. The insoluble fraction was collected using 1% SDS. Equal volumes of the soluble (S) and insoluble (I) fractions were resolved by SDS-PAGE and analyzed by immunoblotting with V5 or Cav-3 mAb probes. Note that Cav-3 expression does not affect the detergent solubility of the other two PFK isoforms, ie, PFK-B and PFK-P. PFK-B partitions both in the soluble and in the insoluble fractions, either when expressed alone or in combination with Cav-3 (A). B shows that PFK-P is mainly found in the insoluble fraction; the presence of Cav-3 does not affect this behavior.
We next investigated whether Cav-3 expression would induce PFK-B or PFK-P targeting to caveolae-enriched fractions. Cos-7 cells were transiently transfected with either PFK-B or PFK-P, alone or in combination with Cav-3. Cells were solubilized in the presence of Triton X-100, and subjected to sucrose gradient ultracentrifugation. Figure 12 ▶ shows that PFK-B is excluded from the caveolae-enriched fractions, and that expression of Cav-3 does not change this behavior. A similar outcome was observed when analyzing PFK-P. As shown in Figure 13 ▶ , Cav-3 expression does not affect the partial targeting of PFK-P to detergent-resistant membrane microdomains/caveolae-enriched fractions. Thus, we conclude that Cav-3 specifically affects the phenotypic behavior of PFK-M, but not PFK-B or PFK-P.
Figure 12.

PFK-B does not target to lipid rafts/caveolae-enriched fractions. To separate caveolar microdomains from the other membranous and cytosolic components, an established sucrose fractionation system was used. Cos-7 cells were transiently transfected with the cDNA encoding V5-tagged PFK-B, alone or in combination with Cav-3. Thirty-six hours after transfection, cells were lysed in a buffer containing 1% Triton X-100 and subjected to sucrose gradient centrifugation. Twelve fractions were collected and equal amounts of protein from each fraction were separated by SDS-PAGE and blotted onto nitrocellulose. The distribution of PFK-B and Cav-3 were analyzed by immunoblotting with V5 and Cav-3 antibodies, respectively. Note that when expressed alone, PFK-B is detected at the bottom of the gradient (fractions 9 to 12; top). Similarly, in the presence of Cav-3, PFK-B does not co-fractionate with Cav-3 (detected mainly in fractions 4 and 5), which marks the position of caveolar microdomains) (bottom).
Figure 13.

The expression of Cav-3 does not affect the targeting of PFK-P to lipid rafts/caveolae-enriched fractions. Lipid rafts/caveolae-enriched membranes were separated from other cellular components using an established sucrose density gradient system. In this fractionation scheme, immunoblotting with the anti-Cav-3 mAb probe can be used to track the position of lipid rafts/caveolae-originated membranes (fractions 4 and 5). Cos-7 cells were transiently transfected with the cDNA encoding V5-tagged PFK-P, alone or in combination with Cav-3. Thirty-six hours after transfection, cells were lysed in a buffer containing 1% Triton X-100 and subjected to sucrose gradient centrifugation. Twelve fractions were collected and equal amounts of protein from each fraction were separated by SDS-PAGE and blotted onto nitrocellulose. The distributions of PFK-P and Cav-3 were analyzed by immunoblotting with V5 and Cav-3 antibodies, respectively. Note that the targeting of PFK-P to lipid rafts/caveolae-enriched fractions is not affected by the presence of Cav-3. Interestingly, PFK-P is mostly excluded from lipid rafts/caveolae-enriched domains (fractions 4 and 5) and instead is concentrated at the bottom of the gradient (fractions 8 to 12).
In Skeletal Muscle from Cav-3-Deficient Mice, PFK-M Is Expressed at Normal Levels, but Is Mislocalized and Is Not Targeted to Caveolae-Enriched Microdomains
The above results suggest that recombinant expression of Cav-3 profoundly affects the phenotypic behavior of PFK-M, by inducing the targeting of PFK-M to caveolae-enriched fractions, and by recruiting PFK-M to the plasma membrane. In addition, recombinant expression of pathogenic Cav-3 mutants may annul these effects and may render PFK-M mostly cytoplasmic.
In light of these findings, we evaluated the fate of PFK-M in a mouse model in which Cav-3 has been genetically ablated. Using standard homologous recombination techniques, we and others have generated Cav-3-null mice. 27,49 Previous studies have shown that Cav-3-deficient mice reveal mild myopathic changes, with a complete loss of muscle caveolae, 49 and display alterations in targeting of the dystrophin-dystroglycan complex to lipid raft microdomains and abnormalities in the organization of the T-tubule system. 27 In addition, we have recently demonstrated that the GPI-linked proteins, a diversified group of molecules involved in signal transduction processes, manifest an abnormal localization pattern in Cav-3-null skeletal muscle fibers. 50
Lysates were prepared from skeletal muscle tissue biopsies derived from WT and Cav-3(−/−) mice and were subjected to SDS-PAGE. Immunoblot analysis using a rabbit PFK-M antibody that cross-reacts with all three PFK isoforms, reveals that PFK-M is expressed at normal levels in Cav-3-null mice (Figure 14A) ▶ . These results are consistent with our previous observations using a heterologous expression system demonstrating that Cav-3 WT expression does not have any effect on PFK-M protein levels (Figure 7) ▶ . Western blot analysis of the same samples was performed using a Cav-3-specific mAb probe and confirmed the absence of Cav-3 protein expression in the skeletal muscle samples from Cav-3-null mice (Figure 14A) ▶ . Immunoblotting with anti-actin IgG is shown as a control for equal protein loading (Figure 14A) ▶ .
Figure 14.

In skeletal muscle tissue from Cav-3(−/−) mice, PFK isoforms show normal expression levels but are mislocalized. A: Western blot analysis. Protein lysates were prepared from skeletal muscle biopsies of WT and Cav-3(−/−) mice [knockout (KO)] and separated by SDS-PAGE. The blots were subsequently subjected to immunoblotting using a rabbit polyclonal anti-PFK-M antibody that cross-reacts with all three PFK isoforms. Note that PFK-M is expressed at normal levels in the skeletal muscle tissue of Cav-3-deficient mice as compared with WT control littermate mice. Equal loading was assessed by immunoblotting with a monoclonal antibody directed against actin (clone AC-40). B: Immunohistochemistry. Skeletal muscle tissue frozen sections were prepared from muscle biopsies of WT and Cav-3 (−/−) mice (KO) and were immunostained with a rabbit polyclonal anti-PFK-M antibody that cross-reacts with all of the three PFK isoforms. PFK-M shows both an intracellular and membrane bound distribution in WT skeletal muscle fibers. In contrast, note that the absence of Cav-3 impedes the recruitment of PFK-M to the sarcolemma in Cav-3-deficient skeletal muscle. Also, in Cav-3(−/−) muscle, the overall intensity of immunostaining for PFK-M appears reduced; this is mostly likely because of that fact that PFK-M is soluble in the absence of Cav-3 (not membrane bound) and, as a consequence, is partially washed away during the immunostaining procedure (see Figure 15B ▶ ). In support of this assertion, total PFK-M levels are unaffected in Cav-3-null mice as determined by Western blot analysis (A) (see also Figure 15A ▶ ). Scale bars, 50 μm.
Next, frozen sections were prepared using skeletal muscle tissue derived from WT and Cav-3(−/−) mice and were probed with a PFK-M rabbit polyclonal antibody that cross-reacts with all of the three PFK isoforms. Figure 14B ▶ shows that in skeletal muscle fibers from WT mice, PFK-M localizes at the sarcolemma, as well as intracellularly. Interestingly, skeletal muscle fibers from Cav-3-null mice display a profound change in the localization of PFK-M. Figure 14B ▶ shows that PFK-M is no longer targeted to the sarcolemma of the skeletal muscle fibers of Cav-3-null mice, as compared to the WT littermates. To our knowledge, this is the first demonstration that PFK is localized to the plasma membrane (sarcolemma) in skeletal muscle cells or any cell type.
Also, in Cav-3(−/−) muscle, the overall intensity of immunostaining for PFK-M appears reduced; this is mostly likely because of that fact that PFK-M is soluble in the absence of Cav-3 (not membrane bound) and, as a consequence, is partially washed away during the immunostaining procedure (Figure 15B) ▶ . In support of this assertion, total PFK-M levels are unaffected in Cav-3-null mice as determined by Western blot analysis (Figures 14A and 15A) ▶ .
Figure 15.

PFK-M shows normal expression levels, but is mislocalized in skeletal muscle tissue from Cav-3(−/−) mice. A: Western blot analysis. Protein lysates were prepared from skeletal muscle biopsies of WT and Cav-3(−/−) mice (KO) and separated by SDS-PAGE. The blots were subsequently subjected to immunoblotting using a guinea pig polyclonal antibody specific for PFK-M. This antibody does not cross-react with other PFK isoforms. Note that the total content of PFK-M is not affected in skeletal muscle samples derived from of Cav-3 KO mice as compared to WT control mice. Equal loading was assessed by immunoblotting with a monoclonal antibody directed against actin (clone AC-40). B: Immunohistochemistry. Skeletal muscle tissue frozen sections were prepared from WT and Cav-3(−/−) mice (KO) and were immunostained with a guinea pig polyclonal antibody specific for PFK-M. Note that PFK-M is localized in a cytoplasmic compartment, as well as at the cell membrane of skeletal muscle fibers from WT mice, whereas it is mainly cytoplasmic in Cav-3-deficient skeletal muscle fibers. Scale bars, 50 μm.
We next used a guinea pig polyclonal antibody that specifically recognizes only PFK-M, but not other PFK isoforms. Consistent with the above results, Western blot analysis on WT and Cav-3-null skeletal muscle tissue revealed that PFK-M expression levels are not altered in Cav-3-null mice, as compared to their WT counterparts (Figure 15A) ▶ . Immunoblot analysis of the same tissue lysates verified a complete absence of Cav-3 expression (Figure 15A) ▶ . However, immunostaining using the guinea pig anti-PFK-M probe revealed that the localization of PFK-M is severely affected in skeletal muscle fibers of Cav-3-deficient mice. As shown in Figure 15B ▶ , PFK-M is present in the cytosol, as well as at the sarcolemma, in the skeletal muscle fibers from WT mice. In striking contrast, PFK-M is primarily excluded from the sarcolemma and mainly found intracellularly in the skeletal muscle fibers from Cav-3-deficient mice.
To further explore the mislocalization of PFK-M observed in Cav-3-deficient skeletal muscle fibers, and to gain insight into the subcellular localization of PFK-M in Cav-3-deficient skeletal muscle fibers, we next subjected skeletal muscle tissue from WT and Cav-3-null mice to sucrose gradient ultracentrifugation. This procedure allows the separation of detergent-resistant membranes/caveolae-enriched fractions (fractions 4 to 5) from the bulk of cellular membranes and cytosolic proteins (fractions 8 to 12). The distribution of PFK-M was visualized using a specific guinea pig anti-PFK-M polyclonal antibody and the distribution of Cav-3 was detected using a specific anti-Cav-3 monoclonal antibody. Figure 16 ▶ shows that PFK-M is targeted to caveolar membrane microdomains in skeletal muscle tissue from WT mice. As predicted, PFK-M is selectively excluded from caveolae-enriched fractions in Cav-3-deficient mice. Taken together, these results indicate that, although the expression levels of PFK-M are not affected in Cav-3-null mice, the sarcolemmal distribution and caveolar targeting of PFK-M is severely compromised.
Figure 16.

PFK-M does not localize to lipid rafts/caveolae-enriched fractions in skeletal muscle tissue from Cav-3-deficient mice. Caveolar microdomains were separated from other membranous and cytosolic constituents using sucrose flotation gradients. Using this fractionation scheme, the caveolae-derived membranes (fractions 4 and 5) can be tracked by immunoblotting with a Cav-3 antibody. Skeletal muscle tissue samples from WT and Cav-3(−/−) mice were homogenized in lysis buffer containing 1% Triton X-100 and were subjected to sucrose gradient centrifugation. Twelve fractions were collected and 5 μg of each fraction was separated by SDS-PAGE and blotted onto nitrocellulose. The distribution of PFK-M and Cav-3 was analyzed by Western blotting using a polyclonal PFK-M antibody raised in guinea pig, and a monoclonal antibody probe specific for Cav-3, respectively. Note that in skeletal muscle from WT mice, a significant amount of PFK-M localizes to caveolae-enriched membrane fractions. In contrast, in Cav-3 KO mice, PFK-M is specifically excluded from the caveolae-enriched fractions (fractions 4 to 7) and is localized to the bottom of the gradient (fractions 9 to 12).
Discussion
Previous studies have demonstrated that, under certain metabolic conditions, endogenous PFK-M forms a complex with Cav-3 in differentiated C2C12 cells, as well as in mouse skeletal muscle tissue lysates. 37 This interaction appears to be sensitive to the concentration of extracellular glucose, such that it can occur when the extracellular glucose is elevated, whereas it is undetectable in the absence of glucose. Known activators of PFK activity stabilize endogenous Cav-3/PFK-M complex formation as measured by co-immunoprecipitation, suggesting that it is the active form of PFK that interacts with Cav-3. 37 Taken together, these findings suggest the hypothesis that extracellular glucose regulates the caveolar targeting of PFK-M to caveolae via interactions with Cav-3. However, direct experimental evidence supporting this hypothesis is lacking.
In this report, using heterologous expression systems, and by using a Cav-3-null mouse model, we show that expression of Cav-3 is necessary to target PFK-M to caveolar membrane microdomains. We show that recombinant expression of Cav-3 induces profound changes in the biochemical behavior of PFK-M, by targeting PFK-M to a detergent-resistant, caveolae-enriched membrane compartment, and the cellular distribution of PFK-M, by trans-locating PFK-M to the plasma membrane. Intriguingly, glucose depletion of cells co-expressing PFK-M and Cav-3 impedes PFK-M membrane recruitment and caveolar targeting. These findings suggest that Cav-3 expression and the presence of extracellular glucose are both necessary for delivering PFK-M to caveolar regions of the plasma membrane. These results are fully consistent with the idea that Cav-3 and PFK-M form a discrete complex in vivo. Additional insights were achieved by the recombinant co-expression of PFK-M with three pathogenic Cav-3 mutants, which harbor the same mutations seen in patients with muscle diseases. We used two constructs, Cav-3 P104L and Cav-3 ΔTFT, that carry the same mutations as in limb-girdle muscular dystrophy-1C patients, 33,38 whereas the third mutant, Cav-3 R26Q, is associated with idiopathic hyperCKemia, rippling muscle disease, and distal myopathy. 34-36,39 Surprisingly, when PFK-M is co-expressed with these Cav-3 mutants, PFK-M expression levels are greatly reduced. However, treatment with inhibitors of the proteasomal pathway, namely MG-132, restored PFK-M expression to normal levels. These results suggest that co-expression with Cav-3 mutants causes PFK-M protein degradation. In addition, recombinant co-expression with these pathogenic Cav-3 mutants impedes PFK-M targeting to the plasma membrane, and induces PFK-M retention in a perinuclear Golgi-like compartment. Interestingly, we show that Cav-1, a protein highly homologous to Cav-3, can also interact with PFK-M. In addition, recombinant expression of Cav-1 is sufficient to recruit PFK-M to the plasma membrane. We also evaluated the effects of Cav-3 expression on the behavior of the other two isoforms of PFK, namely PFK-B and PFK-P. However, PFK-B and PFK-P did not significantly change their detergent solubility properties or their subcellular distribution when co-expressed with Cav-3. As such, Cav-3 expression specifically affects PFK-M in an isoform-specific manner.
The evaluation of skeletal muscle fibers from Cav-3-deficient mice directly demonstrated the pivotal role of Cav-3 expression in regulating the subcellular distribution of PFK-M. Although the expression level of PFK-M remains unchanged in Cav-3-null skeletal muscle tissue as compared to WT littermate controls, the subcellular distribution was deeply affected by the absence of Cav-3. Immunofluorescence analysis and subcellular fractionation experiments revealed that, in Cav-3-deficient skeletal muscle fibers, PFK-M is no longer targeted to the muscle cell plasma membrane and is specifically excluded from lipid raft/caveolae-enriched fractions. One possible explanation for this shift in the distribution of PFK-M in Cav-3-null mice is low serum glucose levels. However, we have determined the serum glucose levels in Cav-3-null mice and they are normal, as compared with WT littermate controls (data not shown). In summary, we conclude that Cav-3 deficiency in mice causes a profound redistribution of PFK-M; these findings are fully complementary to what we have observed using a heterologous expression system.
The concept that Cav-3 coordinates the subcellular distribution of PFK-M, a key player of the glycolytic pathway, highlights the important role of muscle cell caveolae in the regulation of the energy metabolism. By interacting with Cav-3, PFK-M is recruited in a glucose-dependent manner to muscle caveolar membranes, in close proximity to the site of glucose uptake. In fact, previous studies have demonstrated that, in adipocytes, on insulin stimulation, a fraction of the insulin-responsive glucose transporter GLUT4 is rapidly trans-located to caveolar microdomains. 51-53 In addition, GLUT4 has been previously localized to caveolae domains in skeletal muscle fibers by immunoelectron microscopy. 54,55 Taken together, these findings strongly suggest that the glucose transporter and the key regulatory enzyme of glycolysis are simultaneously recruited to the plasma membrane, and locally concentrated in caveolar microdomains. This compartmentalization would render the usage of glucose far more efficient and rapid. As such, muscle caveolae would play a crucial role in the regulation of the energy metabolism of the cell.
The demonstration that PFK-M requires Cav-3 expression for cell membrane recruitment and caveolar targeting may be very important in elucidating the pathogenic mechanisms underlying muscle diseases that are caused by mutations within the Cav-3 gene. In these patients, almost complete loss of Cav-3 protein expression may cause an abnormal localization pattern of PFK-M, thereby impairing efficient glucose usage and muscle functioning. Studies on muscle biopsies from Cav-3-deficient patients will be extremely important to further elucidate this point.
Because we show that Cav-1 can bind PFK-M, and that recombinant expression of Cav-1 induces membrane targeting of PFK-M, it will be interesting to evaluate the distribution of PFK-M in smooth muscle cells from Cav-3-deficient mice, as well as from Cav-1-deficient mice. Smooth muscle cells are the only cell type that co-express both Cav-1 and Cav-3. As such, they may represent an ideal cell system to further investigate whether Cav-1 or Cav-3 can functionally complement each other in vivo in the membrane recruitment of PFK-M.
Acknowledgments
We thank Dr. Salvatore DiMauro (Columbia University, New York) for his helpful suggestions and for critical review of the manuscript and Dr. R. Campos-Gonzalez (BD-Transduction Laboratories/Pharmingen) for donating mAbs directed against Cav-3.
Footnotes
Address reprint requests to Michael P. Lisanti, Department of Molecular Pharmacology, Golding Bldg., Room 202, 1300 Morris Park Ave., Bronx, NY 10461. E-mail: lisanti@aecom.yu.edu.
Supported by grants from the National Institutes of Health, the Muscular Dystrophy Association, the American Heart Association, the Susan G. Komen Breast Cancer Foundation, and a Hirschl/Weil-Caulier career scientist award (all to M. P. L.). C. M. was supported by grants from Telethon-Italia and the Italian Ministry of Health (G. Gaslini Institute, Ricerca Finalizzata).
References
- 1.Kemp RG, Foe LG: Allosteric regulatory properties of muscle phosphofructokinase. Mol Cell Biochem 1983, 57:147-154 [DOI] [PubMed] [Google Scholar]
- 2.Tsai MY, Kemp RG: Hybridization of rabbit muscle and liver phosphofructokinases. Arch Biochem Biophys 1972, 150:407-411 [DOI] [PubMed] [Google Scholar]
- 3.Dunaway GA, Kasten TP, Sebo T, Trapp R: Analysis of the phosphofructokinase subunits and isoenzymes in human tissues. Biochem J 1988, 251:677-683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vora S, Seaman C, Durham S, Piomelli S: Isozymes of human phosphofructokinase: identification and subunit structural characterization of a new system. Proc Natl Acad Sci USA 1980, 77:62-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vora S: Isozymes of human phosphofructokinase in blood cells and cultured cell lines: molecular and genetic evidence for a trigenic system. Blood 1981, 57:724-732 [PubMed] [Google Scholar]
- 6.Vora S, Wims LA, Durham S, Morrison SL: Production and characterization of monoclonal antibodies to the subunits of human phosphofructokinase: new tools for the immunochemical and genetic analyses of isozymes. Blood 1981, 58:823-829 [PubMed] [Google Scholar]
- 7.Tarui S, Okuno G, Ikura Y, Tanaka T, Suda M: Phosphofructokinase deficiency in skeletal muscle, a new type of glycogenosis. Biochem Biophys Res Commun 1965, 19:517-523 [DOI] [PubMed] [Google Scholar]
- 8.Vora S, Davidson M, Seaman C, Miranda AF, Noble NA, Tanaka KR, Frenkel EP, DiMauro S: Heterogeneity of the molecular lesions in inherited phosphofructokinase deficiency. J Clin Invest 1983, 72:1995-2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehotzky A, Telegdi M, Liliom K, Ovadi J: Interaction of phosphofructokinase with tubulin and microtubules. Quantitative evaluation of the mutual effects. J Biol Chem 1993, 268:10888-10894 [PubMed] [Google Scholar]
- 10.Kraft T, Hornemann T, Stolz M, Nier V, Wallimann T: Coupling of creatine kinase to glycolytic enzymes at the sarcomeric I-band of skeletal muscle: a biochemical study in situ. J Muscle Res Cell Motil 2000, 21:691-703 [DOI] [PubMed] [Google Scholar]
- 11.Hazen SL, Gross RW: The specific association of a phosphofructokinase isoform with myocardial calcium-independent phospholipase A2. Implications for the coordinated regulation of phospholipolysis and glycolysis. J Biol Chem 1993, 268:9892-9900 [PubMed] [Google Scholar]
- 12.Reiss N, Kanety H, Schlessinger J: Five enzymes of the glycolytic pathway serve as substrates for purified epidermal-growth-factor-receptor kinase. Biochem J 1986, 239:691-697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sale EM, White MF, Kahn CR: Phosphorylation of glycolytic and gluconeogenic enzymes by the insulin receptor kinase. J Cell Biochem 1987, 33:15-26 [DOI] [PubMed] [Google Scholar]
- 14.Firestein BL, Bredt DS: Interaction of neuronal nitric-oxide synthase and phosphofructokinase-M. J Biol Chem 1999, 274:10545-10550 [DOI] [PubMed] [Google Scholar]
- 15.Roberts CK, Barnard RJ, Jasman A, Balon TW: Acute exercise increases nitric oxide synthase activity in skeletal muscle. Am J Physiol 1999, 277:E390-E394 [DOI] [PubMed] [Google Scholar]
- 16.Lisanti MP, Scherer P, Tang ZL, Sargiacomo M: Caveolae, caveolin and caveolin-rich membrane domains: a signalling hypothesis. Trends Cell Biol 1994, 4:231-235 [DOI] [PubMed] [Google Scholar]
- 17.Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE, Okamoto T, Lisanti MP: Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol 1999, 19:7289-7304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galbiati F, Razani B, Lisanti MP: Emerging themes in lipid rafts and caveolae. Cell 2001, 106:403-411 [DOI] [PubMed] [Google Scholar]
- 19.Glenney JR, Soppet D: Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in RSV-transformed fibroblasts. Proc Natl Acad Sci USA 1992, 89:10517-10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP: Identification, sequence and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci USA 1996, 93:131-135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang ZL, Scherer PE, Okamoto T, Song K, Chu C, Kohtz DS, Nishimoto I, Lodish HF, Lisanti MP: Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem 1996, 271:2255-2261 [DOI] [PubMed] [Google Scholar]
- 22.Way M, Parton R: M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett 1995, 376:108-112 [DOI] [PubMed] [Google Scholar]
- 23.Scherer PE, Lewis RY, Volonte D, Engelman JA, Galbiati F, Couet J, Kohtz DS, van Donselaar E, Peters P, Lisanti MP: Cell-type and tissue-specific expression of caveolin-2. Caveolins 1 and 2 co-localize and form a stable hetero-oligomeric complex in vivo. J Biol Chem 1997, 272:29337-29346 [DOI] [PubMed] [Google Scholar]
- 24.Song KS, Scherer PE, Tang Z-L, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS, Lisanti MP: Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem 1996, 271:15160-15165 [DOI] [PubMed] [Google Scholar]
- 25.Sargiacomo M, Scherer PE, Tang Z-L, Kubler E, Song KS, Sanders MC, Lisanti MP: Oligomeric structure of caveolin: implications for caveolae membrane organization. Proc Natl Acad Sci USA 1995, 92:9407-9411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monier S, Parton RG, Vogel F, Behlke J, Henske A, Kurzchalia T: VIP21-caveolin, a membrane protein constituent of the caveolar coat, oligomerizes in vivo and in vitro. Mol Biol Cell 1995, 6:911-927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, Hou HJ, Kneitz B, Edelmann W, Lisanti MP: Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and T-tubule abnormalities. J Biol Chem 2001, 276:21425-21433 [DOI] [PubMed] [Google Scholar]
- 28.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou HJ, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP: Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities J Biol Chem 2001, 276:38121-38138 [DOI] [PubMed] [Google Scholar]
- 29.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H, Jr, Christ GJ, Edelmann W, Lisanti MP: Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol 2002, 22:2329-2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sotgia F, Lee JK, Das K, Bedford M, Petrucci TC, Macioce P, Sargiacomo M, Bricarelli FD, Minetti C, Sudol M, Lisanti MP: Caveolin-3 directly interacts with the C-terminal tail of beta-dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem 2000, 275:38048-38058 [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Cardena G, Martasek P, Siler-Masters BS, Skidd PM, Couet JC, Li S, Lisanti MP, Sessa WC: Dissecting the interaction between nitric oxide synthase (NOS) and caveolin: functional significance of the NOS caveolin binding domain in vivo. J Biol Chem 1997, 272:25437-25440 [DOI] [PubMed] [Google Scholar]
- 32.Venema VJ, Ju H, Zou R, Venema RC: Interaction of neuronal nitric-oxide synthase with caveolin-3 in skeletal muscle. Identification of a novel caveolin scaffolding/inhibitory domain. J Biol Chem 1997, 272:28187-28190 [DOI] [PubMed] [Google Scholar]
- 33.Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M, Masetti E, Mazzocco M, Egeo A, Donati MA, Volonte D, Galbiati F, Cordone G, Bricarelli FD, Lisanti MP, Zara F: Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 1998, 18:365-368 [DOI] [PubMed] [Google Scholar]
- 34.Carbone I, Bruno C, Sotgia F, Bado M, Broda P, Masetti E, Panella A, Zara F, Bricarelli FD, Cordone G, Lisanti MP, Minetti C: Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 2000, 54:1373-1376 [DOI] [PubMed] [Google Scholar]
- 35.Betz RC, Schoser BG, Kasper D, Ricker K, Ramirez A, Stein V, Torbergsen T, Lee YA, Nothen MM, Wienker TF, Malin JP, Propping P, Reis A, Mortier W, Jentsch TJ, Vorgerd M, Kubisch C: Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet 2001, 28:218-219 [DOI] [PubMed] [Google Scholar]
- 36.Tateyama M, Aoki M, Nishino I, Hayashi YK, Sekiguchi S, Shiga Y, Takahashi T, Onodera Y, Haginoya K, Kobayashi K, Iinuma K, Nonaka I, Arahata K, Itoyama Y, Itoyoma Y: Mutation in the caveolin-3 gene causes a peculiar form of distal myopathy. Neurology 2002, 58:323-325 [DOI] [PubMed] [Google Scholar]
- 37.Scherer PE, Lisanti MP: Association of phosphofructokinase-M with caveolin-3 in differentiated skeletal myotubes: dynamic regulation by extracellular glucose and intracellular metabolites. J Biol Chem 1997, 272:20698-20705 [DOI] [PubMed] [Google Scholar]
- 38.Galbiati F, Volonte D, Minetti C, Chu JB, Lisanti MP: Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). J Biol Chem 1999, 274:25632-25641 [DOI] [PubMed] [Google Scholar]
- 39.Sotgia F, Woodman SE, Bonuccelli G, Capozza F, Minetti C, Scherer PE, Lisanti MP: Phenotypic behavior of caveolin-3 R26Q, a mutant associated with hyperCKemia, distal myopathy, and rippling muscle disease. Am J Physiol Cell Physiol 2003, 285:C1150-C1160 [DOI] [PubMed] [Google Scholar]
- 40.Song KS, Tang ZL, Li S, Lisanti MP: Mutational analysis of the properties of caveolin-1. A novel role for the C-terminal domain in mediating homotypic caveolin-caveolin interactions. J Biol Chem 1997, 272:4398-4403 [DOI] [PubMed] [Google Scholar]
- 41.Sargiacomo M, Sudol M, Tang Z-L, Lisanti MP: Signal transducing molecules and GPI-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol 1993, 122:789-807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li S, Galbiati F, Volonte D, Sargiacomo M, Engelman JA, Das K, Scherer PE, Lisanti MP: Mutational analysis of caveolin-induced vesicle formation. Expression of caveolin-1 recruits caveolin-2 to caveolae membranes. FEBS Lett 1998, 434:127-134 [DOI] [PubMed] [Google Scholar]
- 43.Simons K, Ikonen E: Functional rafts in cell membranes. Nature 1997, 387:569-572 [DOI] [PubMed] [Google Scholar]
- 44.Lisanti MP, Scherer PE, Vidugiriene J, Tang ZL, Hermanoski-Vosatka A, Tu Y-H, Cook RF, Sargiacomo M: Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol 1994, 126:111-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown DA, London E: Structure of detergent-resistant membrane domains: does phase separation occur in biological membranes? Biochem Biophys Res Commun 1997, 240:1-7 [DOI] [PubMed] [Google Scholar]
- 46.Brown DA, London E: Functions of lipid rafts in biological membranes. Ann Rev Cell Dev Biol 1998, 14:111-136 [DOI] [PubMed] [Google Scholar]
- 47.Li S, Okamoto T, Chun M, Sargiacomo M, Casanova JE, Hansen SH, Nishimoto I, Lisanti MP: Evidence for a regulated interaction between hetero-trimeric G proteins and caveolin. J Biol Chem 1995, 270:15693-15701 [DOI] [PubMed] [Google Scholar]
- 48.Diep DB, Nelson KL, Raja SM, Pleshak EN, Buckley JT: Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J Biol Chem 1998, 273:2355-2360 [DOI] [PubMed] [Google Scholar]
- 49.Hagiwara Y, Sasaoka T, Araishi K, Imamura M, Yorifuji H, Nonaka I, Ozawa E, Kikuchi T: Caveolin-3 deficiency causes muscle degeneration in mice. Hum Mol Genet 2000, 9:3047-3054 [DOI] [PubMed] [Google Scholar]
- 50.Sotgia F, Razani B, Bonuccelli G, Schubert W, Battista M, Lee H, Capozza F, Schubert AL, Minetti C, Buckley JT, Lisanti MP: Intracellular retention of glycosylphosphatidyl inositol-linked proteins in caveolin-deficient cells. Mol Cell Biol 2002, 22:3905-3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scherer PE, Lisanti MP, Baldini G, Sargiacomo M, Corley-Mastick C, Lodish HF: Induction of caveolin during adipogenesis and association of GLUT4 with caveolin-rich vesicles. J Cell Biol 1994, 127:1233-1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kandror KV, Stephens JM, Pilch PF: Expression and compartmentalization of caveolin in adipose cells: coordinate regulation with and structural segregation from GLUT4. J Cell Biol 1995, 129:999-1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karlsson M, Thorn H, Parpal S, Stralfors P, Gustavsson J: Insulin induces translocation of glucose transporter GLUT4 to plasma membrane caveolae in adipocytes. EMBO J 2002, 16:249-251 [DOI] [PubMed] [Google Scholar]
- 54.Friedman JE, Dudek RW, Whitehead DS, Downes DL, Frisell WR, Caro JF, Dohm GL: Immunolocalization of glucose transporter GLUT4 within human skeletal muscle. Diabetes 1991, 40:150-154 [DOI] [PubMed] [Google Scholar]
- 55.Dohm GL, Dolan PL, Frisell WR, Dudek RW: Role of transverse tubules in insulin stimulated muscle glucose transport. J Cell Biochem 1993, 52:1-7 [DOI] [PubMed] [Google Scholar]