

Abstract
Fetal rat skin transitions from scarless fetal-type repair to adult-type repair with scar between day 16 (E16) and day 18 (E18) of gestation (term = 21.5 days). Deficient transforming growth factor (TGF)-β1 and -β2 injury response has been proposed as a mechanism for scarless fetal-type repair. However, previous fetal studies have inconsistently reported the degree of TGF-β induction after injury. To minimize developmental variables in fetal versus adult TGF-β regulation, we narrowed our study to wounded fetal animals. We hypothesize that TGF-β ligand and receptor expression will be differentially regulated during the transition from early gestation (E16) wounds manifesting scarless fetal-type repair to late gestation (E19) wounds manifesting adult-type repair with scar. In this study, decreased and rapidly cleared TGF-β1 and -β2 expression accompanied by increased and prolonged TGF-β3 levels in wounded E16 animals correlated with organized collagen deposition. In contrast, increased and prolonged TGF-β1 and -β2 expression accompanied by decreased and delayed TGF-β3 expression in wounded E19 animals correlated with disorganized collagen architecture. Similarly, expression of TGF-β receptors type I and II were also increased or prolonged in E19 animals. Our results implicate increased TGF-β1, -β2, and decreased TGF-β3 expression, as well as increased type I and II receptor expression in late gestation fetal scar formation.
Fetal rat skin transitions from scarless fetal-type repair to adult-type repair with scar between day 16 (E16) and day 18 (E18) of gestation (term = 21.5 days). 1 The transforming growth factor-β (TGF-β) family of molecules has been implicated in this ontogenetic transition. TGF-βs are multifunctional cytokines with widespread effects on cell growth and differentiation, embryogenesis, immune regulation, inflammation, and wound healing. 2 In terms of repair, TGF-β1 and TGF-β2 are known to promote scar, while TGF-β3 may reduce scar. 3,4 Exogenously added TGF-β1 is able to amplify its own expression and to induce scar in a normally scarless human model of fetal skin repair. 3 In addition, scar formation in adult wounds may be reduced by the early application of neutralizing antibodies specific for TGF-β1 and TGF-β2, or the exogenous addition of TGF-β3. 4,5
Deficient TGF-β1 and -β2 injury response has been proposed as a mechanism for scarless fetal-type repair. However, previous fetal studies have inconsistently reported the degree of TGF-β induction after injury. For instance, many studies found no TGF-β mRNA or protein induction in early gestation fetal mouse, 6 rabbit, 7 and human 3,8 wound models. In contrast, Martin et al 9 have demonstrated rapid TGF-β1 mRNA induction within 1 to 3 hours after wounding and rapid protein clearance to near background levels by 18 hours in a mouse model of embryonic limb repair. Moreover, research to date has primarily focused on differences between fetal and adult wounds and does not account for developmental changes in TGF-β expression. 10 To decrease the developmental variables in fetal versus adult TGF-β regulation, we narrowed our study to wounded fetal animals. We hypothesized that TGF-β ligand and receptor expression will be differentially regulated during the transition from scarless fetal-type repair to adult-type repair with scar in early and late gestation animals. In this study, we analyzed the expression of TGF-β1, -β2, and -β3 and type I, II, and III receptors (TGF-βRI, -βRII, and -βRIII, respectively) during cutaneous fetal repair by immunohistochemistry and reduced cycle-specific primer polymerase chain reaction (PCR). We showed that early (E16) and late (E19) gestation fetal rats exhibited markedly different patterns of TGF-β ligand and receptor expression. In addition, we demonstrated distinct differences between the collagen architecture of early and late gestation fetal wounds. Our result suggest that the ontogenetic transition from scarless fetal-type repair to adult-type repair with scar correlate with distinct patterns of TGF-β ligand and receptor expression and collagen deposition.
Materials and Methods
Animal Surgical Procedure
Female Sprague Dawley rats (∼300 grams) were mated with corresponding males. Detection of a vaginal plug as evidence of pregnancy was considered day 0.5 of gestation (term = 21.5 days). For creation of the fetal wounds, pregnant rats were anesthetized on days 16 and 19 of gestation. E19 fetal rats were chosen to avoid potential overlaps with the E16 to E18 transition period. Anesthesia consisted of 1% ketamine at a dose of 10 to 20 mg/kg and 0.1% xylazine at a dose of 0.3 mg/kg. The pregnant animals were shaved and a midline laparotomy performed. Each uterine segment was externalized and a 7–0 nylon purse-string suture was placed through all layers of the uterine wall on the non-placental surface. The myometrium and amniotic sac was then incised within the purse-string using microsurgical scissors. Subsequently, a 2-mm excisional wound was made on the dorsum of the fetuses by grasping the skin with microsurgical forceps and excising the skin with scissors. Blue or green vital stain was applied to the excisional sites for later wound identification. Warm sterile normal saline was then applied through the hysterotomy and the purse-string closed (Figure 1) ▶ . The maternal fascia and skin was then closed in two layers using 2–0 synthetic absorbable sutures. All animal protocols were performed under guidelines approved by the Animal Research Committee at UCLA (protocol number 99–055-02).
Figure 1.

Operative procedures. A: A small part of the anti-mesenteric surface of the uterus is incised and a purse-string suture is placed around the incision. B and C: A full-thickness wound is created on each embryo by excising a 2-mm disk of tissue. Blue or green vital stain is applied immediately after wounding for later wound identification.
Preparation of Fetal Tissue
For histology, E16 and E19 fetal wounds were harvested at 12, 24, 36, 48, and 72 hours postoperatively. Non-wounded skin from each of the wound harvest time points were used as controls (eg, E17 control skin for E16 + 24-hour wounds). A total of four animals from two separate pregnancies were used for each time point. All tissue specimens were fixed in 4% paraformaldehyde, dehydrated through graded ethanol, embedded in paraffin, and cut into 5-μm sections for hematoxylin and eosin (H&E) staining and immunohistochemistry, or into 7-μm sections for confocal microscopy.
For RNA analysis, E16 and E19 fetal wounds were harvested at 24 and 72 hours after injury. Non-wounded skin from each of the wound harvest time points were used as controls (eg, E19 control skin for E16 + 72-hour wounds). A total of 20 wounds were used for each time point. The isolated tissue was immediately frozen in liquid nitrogen and stored at −70°C until RNA extraction.
Confocal Laser Scanning Microscopy (CLSM)
For analysis of collagen architecture, sections from E16- and E19-wounded animals at 12, 36, 48, and 72 hours after injury were stained by the modified picrosirius red (PSR) techniques described by Dolber and Spach. 11 Briefly, sections of healing and completely healed wounds were deparaffinized in xylene and rehydrated with 100%, 95%, 70% ethanol, and distilled water. After a 1-minute treatment in 0.2% aqueous phosphomolybdic acid (PMA) to render the cytoplasm colorless, sections were stained in a 0.1% solution of sirius red F3BA (CT No. 35780; Pfaltz and Bauer, Stamford, CT) in saturated aqueous picric acid for 90 to 120 minutes. Sections were then washed in 0.01 N HCl for 2 minutes, dehydrated, cleaned, and mounted. All sections were examined on a Carl Zeiss LSM 310 Laser Scanning Confocal microscope which was equipped with two lasers, an external argon ion blue laser (488/514 nm) for excitation of green fluorophores and a helium-neon green laser (543 nm) for excitation of red fluorophores. The wound sites were first identified with a ×40 Plan-Neofluar multi-immersions objective (numerical aperture 0.75) by conventional light. The system was then switched to the frame mode and confocal pictures were generated with the 543-nm line of the helium-neon laser (attenuation = 1) with a Plan-Neofluar 63x/1.25 NA oil-immersion objective for the fluorescent imaging. The emission light beam was recorded by a photomultiplier after passing a pinhole aperture and emission filter. The depth of the optical section (confocal z resolution) was kept constant by holding the size of the aperture pinhole constant in the emission pathway. The scanning time setting for image collection was 8 seconds with average set to reduce photon noise. The bandwidth filter was set as 1 to clean up the images. Sections were scanned and collected with 0.5-μm increments to the entire section thickness of 7 μm. Data of frame-mode images were recorded and stored for 512 × 512 pixel images.
Total collagen density per healed wound site was calculated using Image Pro Plus by dividing total collagen surface area by total wound surface area (Media Cybernetics, Silver Spring, MD) for both E16 (n = 10) and E19 (n = 8) wounds at 72 hours after injury. For comparison, total collagen density in non-injured skin from age-mated controls [eg, E19 (E16 + 72 hours) and neonatal day 1 (E19 + 72 hours) animals] was also determined. Means and standard deviations (SD) were calculated and unpaired two-tailed Student’s t-test was performed to detect statistically significant differences in total collagen density. A P value of <0.05 was considered significant.
Immunohistochemistry
To determine TGF-β isoform expression, E16 and E19 animal wound sections were immunostained at 12, 24, 36, 48, and 72 hours after injury. After deparaffinization and rehydration of the tissue sections, endogenous peroxidase activity was quenched with 1% hydrogen peroxide (H2O2) for 30 minutes then 2% H2O2 for 15 minutes followed by rinses in phosphate-buffered saline (PBS), pH = 7.4. Sections were blocked with normal goat serum (initial concentration: 20 μg/μl; 1:50 dilution in PBS for final concentration: 0.4 μg/μl; Vector Laboratories, Burlingame, CA) overnight at 4°C in a humidified chamber to reduce background staining. The primary antibodies used for indirect immunostaining were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA) and are detailed in Table 1 ▶ . Initial concentration of all primary antibodies was 200 μg/ml and diluted 1:100 in PBS to a final concentration of 0.002 μg/μl. The only exception was TGF-β3 which was diluted 1:75 for a final concentration of 0.0027 μg/μl as no discernable staining was observed at 1:100 dilution. The tissue sections were incubated with primary antibodies for 1 hour at room temperature, followed by three rinses in PBS. Secondary antibodies consisting of biotinylated goat anti-rabbit IgG or biotinylated rabbit anti-goat IgG (for TGF-βRIII only) (1:50 dilution with PBS; Vector Laboratories, Burlingame, CA) were added for 1 hour at room temperature. After washing, the sections were further incubated with VECTASTAIN ABC reagent (1:50 dilution with PBS, avidin biotin immunoperoxidase labeling (ABC) kit, Vector Laboratories) for 30 minutes. Sections were developed with diaminobenzidine tetrahydrochloride (DAB) for 5 minutes, washed with distilled water, counter-stained with hematoxylin, and cover-slipped. Negative control sections were reacted with pre-immune rabbit serum or goat serum (for TGF-βRIII only) at 10-fold higher concentrations than the primary test antibodies (Vector Laboratories; initial concentration: 20 μg/μl; final concentration: 0.02 μg/μl after 1:1000 dilution). The intensity of immunostaining was semi-quantitatively analyzed by two blinded reviewers using the following criteria: <5% = no staining (“−”), 5 to <25% = minimal staining (“+”), 25 to 50% = moderate staining (“++”), >50% = strong staining (“+++”).
Table 1.
Description of Primary Antibodies Used for Indirect Immunostaining
| Antibody | Initial concentration | Origin | Epitope recognition | Final concentration (dilution in PBS) |
|---|---|---|---|---|
| TGF-β1 | 200 μg/ml | Rabbit polyclonal IgG | Extreme carboxy terminus of TGF-β1 (region of mature peptide) | 0.002 μg/μl (1:100) |
| TGF-β2 | 200 μg/ml | Rabbit polyclonal IgG | Extreme carboxy terminus of TGF-β2 (region of mature peptide) | 0.002 μg/μl (1:100) |
| TGF-β3 | 200 μg/ml | Rabbit polyclonal IgG | Extreme carboxy terminus of TGF-β3 (region of mature peptide) | 0.0027 μg/μl (1:75) |
| TGF-β RI (R-20) | 200 μg/ml | Rabbit polyclonal IgG | Carboxy terminus of TGF-β RI (region of mature peptide) | 0.002 μg/μl (1:100) |
| TGF-β RII (L-21) | 200 μg/ml | Rabbit polyclonal IgG | Amino terminus of TGF-β RII (region of signal and mature peptide) | 0.002 μg/μl (1:100) |
| TGF-β RII (H-567) | 200 μg/ml | Rabbit polyclonal IgG | Full length TGF-β RII (entire peptide) | 0.002 μg/μl (1:100) |
| TGF-β RIII (C-20) | 200 μg/ml | Goat polyclonal IgG | Carboxy terminus of TGF-β RIII (region of mature peptide) | 0.002 μg/μl (1:100) |
To further confirm the reliability of our observations, computerized immunolocalization intensity analyses was conducted on TGF-β1, -β2, and -β3-stained wound and control sections. Digital images (×200) of the dermal wound sites were taken under the same exposure conditions using a Zeiss Digital Microscope (Germany). The degree of positive immunostaining in dermal wound and control sections was calculated using an automatic image analyzer (KS300; Zeiss, Germany) as previously described. 12 Five sections were used for each experimental group. The results were graphically depicted as the mean ± the SD. Mean percent staining was compared using one way analysis of variance (ANOVA) methods on the wound minus control paired differences. Statistical significance was computed using the Tukey-Fisher LSD criterion based on the post-hoc t statistics. A P value of <0.05 was considered significant.
Reverse Transcriptase-Polymerase Chain Reaction
RNA isolation, reverse transcriptase-polymerase chain reaction (RT-PCR), and Southern blotting were performed as previously described. 13 At least two or more RT reactions from which at least four separate sets of cycle optimized PCR reactions were performed for each PCR primer set. Primer sequences, PCR reaction temperatures, and PCR cycle number were as previously described. 13
To compare general trends in mRNA expression for TGF-β ligands and receptors, membranes were analyzed by phosphorimagery. Densitometry values for each TGF-β ligand or receptor were corrected to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression at each time point and normalized by setting the highest value to one and graphically depicted as the mean ± the SD. Unpaired two-tailed Student’s t-test was performed to detect statistically significant differences in gene expression between wounded E16 and E19 animals and their non-wounded, age-matched controls. A P value of <0.05 was considered significant.
Results
Hematoxylin and Eosin Histology and Confocal Microscopy
E16 Fetal Wounds
H&E staining showed minimal inflammatory infiltrates in E16 fetuses 12 hours after wounding with some visible hemorrhage at the wound margin. Lymphocytes comprised the majority of the inflammatory cells present, followed by a few monocytes, neutrophils, and platelets (data not shown). At 24 hours after injury, there was minimally increased inflammation relative to the 12-hour wounds although increased neutrophil numbers were noted at the wound periphery (Figure 2, A and C) ▶ . Peripheral wound re-epithelialization was evident at 24 hours after injury with complete re-epithelialization by 72 hours (Figure 2, A, B, D, and E) ▶ . By 36 hours, neutrophil numbers declined dramatically while lymphocyte numbers decreased moderately (data not shown). Negligible inflammation was observed at 48 hours (data not shown) and none by 72 hours post-wounding (Figure 2, D and E) ▶ . Dermal cell numbers did not change significantly throughout. Hair follicle regeneration was apparent beginning at 48 hours. The wound was completely re-epithelialized by 72 hours and identified through the presence of blue vital stain (Figure 2, D and E) ▶ . Except for a minimal degree of epithelial hypertrophy, the healed wound was indistinguishable from non-wounded skin with complete restoration of normal skin architecture and hair follicles. Overall, E16 fetal wounds exhibited minimal inflammation and dermal cellularity at all time points assayed.
Figure 2.

H&E stain of wounded E16 rat skin. A: 24 hours post-injury; magnification, ×100. There is minimal inflammatory infiltrate. B: 24 hours post-injury; magnification, ×400. The presence of blue vital dye in hair follicles near the migrating epithelial edge suggests concurrent hair follicle regeneration with wound re-epithelialization (black open arrow). C: 24 hours post-injury; magnification, ×400. Neutrophils (black solid arrows) and lymphocytes (black open arrow) are the predominant cells of the wound periphery and the center of the wound, respectively. D: 72 hours post-injury; magnification, ×100. The wound is entirely healed with complete regeneration of the normal skin architecture. Normal distribution of hair follicles (black open arrows) are observed in the dermis. E: 72 hours post-injury; magnification, ×400. At higher magnification, the previous wound site, as indicated by the presence of blue vital stain in the dermis (black open arrows) is indistinguishable from the non-wounded skin. F: Confocal microscopic view of wounded E16 rat skin and control. (Fa through Fc). Fa: 48 hours post-injury; magnification, ×630. Note the organized appearance of the collagen fibers with a reticular lattice structure. Fb: 72 hours post-injury; magnification, ×630. The wound site is completely re-epithelialized with complete restoration of normal skin collagen architecture and hair follicle regeneration. Fc: Non-wounded E19 skin [ie, E16 + 72 hours]; magnification, ×630. No difference is observed between E16 skin, 72 hours post-wounding, and non-wounded E19 skin. e, epidermis; h, hair follicle; d, dermis. Bars: A and D, 200 μm; B, C, and E, 50 μm; Fa-Fc, 32 μm. G: There is no significant difference in total collagen density between E16 fetuses 72 hours post-injury and non-wounded E19 (E16 + 72 hours) fetuses (P >0.05).
Confocal microscopy revealed initial collagen fibril accumulation at the wound borders that reached the wound center by 48 hours after injury (Figure 2Fa) ▶ . After 72 hours, the collagen deposition pattern in completely repaired E16 wounds was comparable to normal, unwounded skin (Figure 2, Fb and Fc) ▶ . Both wounded and unwounded (control) dermis manifested similar-sized collagen bundles woven into a reticular, network-like pattern (Figure 2, Fb and Fc) ▶ . Fine, reticulating, and branching fibrils were found in the papillary dermis forming a subepidermal network (Figure 2Fb) ▶ . Digital imaging analysis verified that 72 hour post-injury E16 wounds and non-injured E19 (E16 + 72 hours) skin did not display significantly different collagen densities (P >0.05) (Figure 2G) ▶ .
E19 Fetal Wounds
In contrast to E16 wounds, H&E histology of E19 fetuses showed moderately increased (25 to 50%) inflammatory infiltrate at the wound edge and wound bed as early as 12 hours post-injury (data not shown). In addition, the preponderance of inflammatory cells were monocytes, with a few neutrophils, rather than lymphocytes (data not shown). Platelet numbers did not appear to be significantly increased relative to E16 wounds. By 24 hours post-injury, E19 wounds demonstrated strongly increased (>50%) inflammatory infiltrate that was primarily monocytic in origin as well as increased vascularity (Figure 3, A and C) ▶ . Similar to E16 wounds re-epithelialization was also noted beginning at 24 hours (Figure 3, A and B) ▶ . Increased dermal cells were observed initially at 24 hours post-injury and became more prominent at 36 hours (data not shown). Starting at 36 hours, inflammatory cell numbers decreased (data not shown) and had entirely disappeared by 72 hours (Figure 3, D and E) ▶ . E19 fetal wounds also re-epithelialized completely at 72 hours after injury. However, there was increased cellularity, neovascularity, and epithelial hypertrophy in the repaired wound along with disorganized collagen architecture and absent hair follicle regeneration (Figure 3, D and E) ▶ . Overall, E19 wounds exhibited increased and prolonged inflammation as well as increased dermal cellularity at all of the time points assayed.
Figure 3.
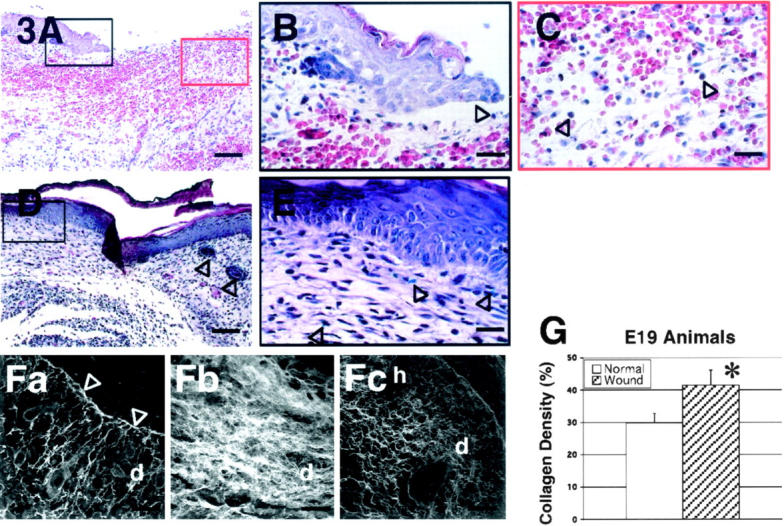
H&E stain of wounded E19 rat skin. A: 24 hours post-injury; magnification, ×100. There is moderate inflammatory infiltrate and increased red blood cells. B: 24 hours post-injury; magnification, ×400. Re-epithelialization is also noted at 24 hours after injury (black open arrow). C: 24 hours post-injury; magnification, ×400. Monocytes (black open arrows) comprises most of the inflammatory cells. D: 72 hours post-injury; magnification, ×100. The wound is completely re-epithelialized with increased cellularity and neovascularity. Hair follicles (black open arrows) are not observed in the repaired wound site (far left) compared with unwounded site (far right). E: 72 hours post-injury; magnification, ×400. At higher magnification, blue vital dye (black open arrows) within the repaired wound is visible. F: Confocal microscopic view of wounded E19 rat skin and control. (Fa through Fc). Fa: 48 hours post-injury; magnification, ×630. Large spaces among newly formed collagen fibers within the dermis are noticeable. A thin layer of dense collagen fibers is seen as basement membrane (white open arrows). Fb: 72 hours post-injury, magnification, ×630. Disorganized collagen deposition pattern with heterogeneously sized collagen fibers is apparent in the healed dermal scar tissue. Note the absence of hair follicle regeneration. Fc: Non-wounded neonatal day 1 (N1) skin [ie, E19 + 72 hours, E21 = term]; magnification, ×630. Non-wounded N1 skin exhibited an organized collagen deposition pattern that is significantly different from E19 skin, 72 hours post-wounding. e, epidermis; h, hair follicle; d, dermis. Bars: A and D, 200 μm; B, C, and E, 50 μm; Fa-Fc, 32 μm. G: There is significantly increased total collagen density in wounded E19 fetuses 72 hours post-injury relative to non-wounded N1 controls (P = 0.00043).
Confocal microscopy on 12-hour, E19 wounds also revealed initial peripheral wound collagen deposition (data not shown). By 48 hours, however, it was evident that the collagen architecture in E19 wounds exhibited larger interfibril distances than E16 wounds (Figure 3Fa) ▶ . In addition, a thin, dense aggregate of collagen fibrils was apparent under the newly forming epidermal layer (Figure 3Fa) ▶ . After 72 hours, the previous large, interfibril spaces were filled with a disorganized collection of dense, heterogeneous collagen fibrils in the completely healed wound (Figure 3Fb) ▶ . Digital imaging analysis verified that there was significantly increased collagen density in E19 wounds 72 hours after injury relative to non-wounded neonatal day 1 skin (E19 + 72 hours) (P = 0.00043) (Figure 3G) ▶ .
Immunohistochemistry and RT-PCR
TGF-β1
The specific intensities for TGF-β1 staining in the epidermis, dermis, and hair follicles are listed in Table 2 ▶ . Both E16 and E19-wounded fetuses exhibited increased epidermal TGF-β1 staining beginning 12 hours after injury (Table 2) ▶ . Epidermal TGF-β1 levels then peaked at 24 hours and returned to baseline levels by 36 hours in E16 animals (Figure 4, A to D ▶ ; Table 2 ▶ ), while TGF-β1 expression remained elevated to varying degrees in E19 animals until at least 72 hours (Figure 4, E to H ▶ ; Table 2 ▶ ). In E16-wounded fetuses TGF-β1 expression was up-regulated at 24 hours in the ECM and at 12 hours in dermal cells, but rapidly returned to minimal baseline levels by 36 hours post-injury (Figure 4, A to D ▶ ; Table 2 ▶ ). In contrast, E19-wounded fetuses showed elevated ECM and dermal TGF-β1 expression beginning 24 hours after injury that continued until 36 to 48 hours after injury, respectively (Figure 4, E to H ▶ ; Table 2 ▶ ). Interestingly, although E16-wounded fetuses contained far less inflammatory cells than E19-wounded fetuses, the intensity of TGF-β1 staining within inflammatory cells was greater at 24 hours in injured E16 animals than E19 animals (Figure 4C ▶ ; Table 2 ▶ ). Regenerated hair follicles in E16 animals did not demonstrate increased TGF-β1 expression (Table 2) ▶ . Computerized quantitation of dermal TGF-β1 immunostaining in E16- and E19-wounded fetuses generally correlated with visual quantitation (Figure 4I, a and b) ▶ . Dermal TGF-β1 was significantly elevated in both E16 and E19 animals at 24 hours after injury (P = 0.0000 and 0.0000, respectively). By 36 hours however, dermal TGF-β1 levels were indistinguishable from baseline in E16 wounds while E19 wounds had persistently elevated TGF-β1 (P = 0.0002). RT-PCR analysis revealed significantly increased steady-state TGF-β1 mRNA levels in wounded E19 relative to E16 animals at 24 hours (1.67-fold; P = 0.0353) and 72 hours (2.68-fold; P = 0.0006) after injury (Figure 4Ic) ▶ . When compared to non-wounded, gestation-matched controls, both E16 and E19 wounds demonstrated increased TGF-β1 expression at 24 hours after injury (P = 0.0346 and 0.0116, respectively) that returned to near baseline levels by 72 hours in E16 animals and slightly lower than baseline levels in E19 animals (P = 0.0338) (Figure 4Ic) ▶ .
Table 2.
Relative Immunostaining Intensity of TGF-β Ligands in Wounded and Non-Wounded Fetal Skin
| After injury | 12 hours | 24 hours | 36 hours | 48 hours | 72 hours | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TGF-β1 | W | C | W | C | W | C | W | C | W | C | ||||||||||
| E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | |
| Epidermis | ||||||||||||||||||||
| Migrating epi. | ++ | ++ | N/A | N/A | +++ | +++ | N/A | N/A | ++ | +++ | N/A | N/A | + | +++ | N/A | N/A | N/A | N/A | N/A | N/A |
| Outer layer | ++ | ++ | + | + | +++ | +++ | + | + | ++ | +++ | ++ | ++ | + | ++ | + | ++ | + | ++ | + | + |
| Basal layer | ++ | ++ | + | + | +++ | +++ | − | + | ++ | +++ | +++ | ++ | + | ++ | + | + | + | ++ | − | − |
| Dermis | ||||||||||||||||||||
| ECM | + | + | + | + | ++ | ++ | + | + | + | ++ | + | + | + | ++ | + | ++ | + | + | − | + |
| Fibroblasts | ++ | + | − | + | ++ | ++ | + | + | + | ++ | + | + | + | ++ | + | + | − | + | + | + |
| Inflammatory cells* | + | ++ | + | + | +++ | ++ | + | + | ++ | ++ | + | + | ++ | ++ | + | ++ | + | + | + | + |
| Hair follicles | N/A | N/A | + | − | N/A | N/A | + | + | N/A | N/A | + | + | N/A | N/A | − | + | + | N/A | + | + |
| After injury | 12 hours | 24 hours | 36 hours | 48 hours | 72 hours | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TGF-β2 | W | C | W | C | W | C | W | C | W | C | ||||||||||
| E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | |
| Epidermis | ||||||||||||||||||||
| Migrating epi. | ++ | + | N/A | N/A | +++ | ++ | N/A | N/A | ++ | +++ | N/A | N/A | + | ++ | N/A | N/A | N/A | N/A | N/A | N/A |
| Outer layer | ++ | + | ++ | + | +++ | ++ | ++ | + | ++ | +++ | + | ++ | + | ++ | + | ++ | ++ | ++ | + | ++ |
| Basal layer | ++ | + | ++ | + | +++ | ++ | + | + | ++ | +++ | ++ | ++ | + | ++ | − | ++ | ++ | ++ | ++ | ++ |
| Dermis | ||||||||||||||||||||
| ECM | − | + | − | + | − | ++ | − | + | + | + | + | + | + | ++ | + | ++ | − | + | − | + |
| Fibroblasts | − | + | − | + | + | ++ | − | − | + | + | + | + | + | + | + | + | − | + | − | − |
| Inflammatory cells* | + | + | + | + | ++ | ++ | + | + | ++ | ++ | + | + | ++ | ++ | + | + | + | ++ | + | + |
| Hair follicles | N/A | N/A | + | + | N/A | N/A | + | + | N/A | N/A | + | + | N/A | N/A | + | + | + | N/A | + | + |
| After injury | 12 hours | 24 hours | 36 hours | 48 hours | 72 hours | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TGF-β3 | W | C | W | C | W | C | W | C | W | C | ||||||||||
| E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | |
| Epidermis | ||||||||||||||||||||
| Migrating epi. | + | ++ | N/A | N/A | ++ | + | N/A | N/A | ++ | ++ | N/A | N/A | ++ | +++ | N/A | N/A | N/A | N/A | N/A | N/A |
| Outer layer | + | ++ | + | + | ++ | + | + | + | ++ | ++ | + | ++ | ++ | +++ | ++ | + | ++ | + | ++ | + |
| Basal layer | + | ++ | + | + | ++ | + | + | + | ++ | ++ | + | + | ++ | +++ | ++ | − | + | + | ++ | − |
| Dermis | ||||||||||||||||||||
| ECM | − | + | − | + | ++ | + | + | + | ++ | + | + | + | + | ++ | + | ++ | + | + | + | + |
| Fibroblasts | ++ | − | + | − | ++ | − | − | − | ++ | + | − | + | + | ++ | + | + | + | − | + | − |
| Inflammatory cells* | + | ++ | + | + | +++ | + | + | + | +++ | ++ | + | + | + | + | + | + | + | + | + | + |
| Hair follicles | N/A | N/A | − | − | N/A | N/A | + | − | N/A | N/A | + | − | N/A | N/A | + | + | + | N/A | + | + |
W, Wound; C, Control; N/A, not applicable; −, no staining (<5%); +, minimal staining (<25%); ++, moderate staining (25% to 50%); +++, strong staining (>50%). Peak expression time points for a given tissue component are highlighted for wounded E16 (bold) and E19 (underlined) animals.
*In general, non-wounded control skin and E16-wounded fetuses contained very few inflammatory cells. The chart reflects the intensity of intracellular TGF-β staining and not the actual number of inflammatory cells present.
Figure 4.
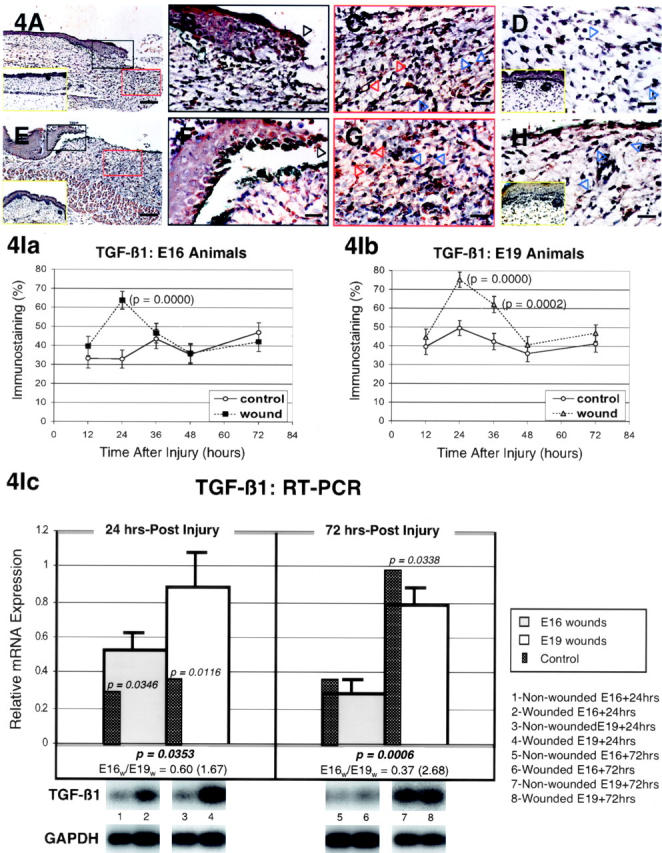
TGF-β1 expression in wounded E16 and E19 rat skin. A: Immunostained E16 skin, 24 hours post-injury; magnification, ×100. Increased TGF-β1 expression is evident in both wounded epidermal and dermal layers. Non-wounded E17 ]ie, E16 + 24 hours[ control skin is also shown (inset, A, yellow rectangle). B: Immunostained E16 skin, 24 hours post-injury; magnification, ×400. Higher magnification view of wounded epidermis demonstrating strong TGF-β1 staining in both outer and basal as well as migrating (black open arrow) epidermal layers after injury. C: Immunostained E16 skin, 24 hours post-injury; magnification, ×400. Moderately increased TGF-β1 staining is observed in dermal fibroblasts (blue open arrows) as well as the surrounding ECM after wounding. Strong TGF-β1 staining is present in inflammatory cells (red open arrows). D: Immunostained E16 skin, 36 hours post-injury; magnification, ×400. Minimal staining in dermal ECM and fibroblasts (blue open arrows), not significantly different from control, is observed 36 hours after wounding. Non-wounded E17.5 ]ie, E16 + 36 hours[ control skin is also shown (inset, D, yellow rectangle). E: Immunostained E19 skin, 24 hours post-injury; magnification, ×100. Similar to wounded E16 skin, wounded E19 skin also demonstrated increased epidermal and dermal TGF-β1 localization relative to non-wounded control skin. Non-wounded E20 ]ie, E19 + 24 hours[ control skin is also shown (inset, E, yellow rectangle). F: Immunostained E19 skin, 24 hours post-injury; magnification, ×400. Strong epidermal staining is also observed in the migrating (black open arrow) as well as the outer and basal layers after injury. G: Immunostained E19 skin, 24 hours post-injury; magnification, ×400. Inflammatory cells (red open arrows), fibroblasts (blue open arrows), and dermal ECM all demonstrate moderately increased TGF-β1 staining after injury. H: Immunostained E19 skin, 36 hours post-injury; magnification, ×400. In contrast to injured E16 skin, wounded E19 skin demonstrates persistent dermal fibroblast (blue open arrows) and ECM TGF-β1 staining 36 hours after injury. Non-wounded, E20.5 ]ie, E19 + 36 hours[ control skin (inset, H, yellow rectangle) exhibits minimal dermal TGF-β1 staining. Bars: A and E, 200 μm; B–D, F–H, 50 μm. I: Computerized quantitation of dermal TGF-β1 immunostaining in E16- and E19-wounded fetuses. Ia: Dermal TGF-β1 staining is significantly increased in E16 animals 24 hours after wounding relative to non-wounded controls (P = 0.0000). By 36 hours, however, no significant difference was noted between injured animals and controls (P >0.05). Ib: Dermal TGF-β1 staining is significantly increased in E19 animals at 24 hours (P = 0.0000) and at 36 hours (P = 0.0002) after wounding relative to non-wounded controls. Ic: RT-PCR of TGF-β1. The light gray bars represents E16 wounds while the white bars represents E19 wounds at 24 hours (left) and 72 hours (right) after injury. The narrow dark gray bars indicate relative mRNA levels in non-wounded, age-matched controls for each group of wounded animals. A P value, if present, next to the narrow dark gray bars, represents significant differences in relative mRNA expression between wounded E16 or E19 animals and their respective non-wounded controls at 24 and 72 hours post-injury. P values ≥0.05 are not shown. P values for significant differences between wounded E16 and E19 animal mRNA levels at 24 or 72 hours after injury, when present, are shown below the bar graphs as well as the corresponding ratio of E16 to E19 wound mRNA levels (inverse E19 to E16 ratio shown in parentheses). No ratios are shown for values ≥0.05. Also depicted below are representative Southern blots of with non-wounded, age-matched controls (left, odd numbers) and E16 wounds at 24 and 72 hours (2 and 6, respectively) and E19 wounds at 24 and 72 hours (4 and 8, respectively). The top series of blots represents TGF-β1; the bottom GAPDH.
TGF-β2
The specific intensities for TGF-β2 staining in the epidermis, dermis, and hair follicles are listed in Table 2 ▶ . Epidermal TGF-β2 expression increased in both E16 and E19 fetuses 24 hours after injury (Figure 5, A, B, E, and F ▶ ; Table 2 ▶ ). However, by 36 hours epidermal TGF-β2 levels declined markedly in E16-injured animals, while reaching maximal levels in E19-injured animals (Table 2) ▶ . For ECM TGF-β2 expression, up-regulation was present only at 24 hours in E19-wounded fetuses and not at all in E16-wounded fetuses (Figure 5, A, C, E, and G ▶ ; Table 2 ▶ ). Moreover, dermal cell TGF-β2 expression was more significantly increased in E19 relative to E16-wounded animals at 24 hours after injury (Figure 5, A, C, E, and G ▶ ; Table 2 ▶ ). The intensity of TGF-β2 staining within inflammatory cells was similar for both E16- and E19-wounded fetuses, although increased TGF-β2 expression persisted until at least 72 hours in E19-wounded animals (Table 2) ▶ . Like TGF-β1, TGF-β2 expression was not increased in regenerated hair follicles from E16-wounded fetuses (Table 2) ▶ . Computerized quantitation of dermal TGF-β2 immunostaining in E16 and E19-wounded fetuses generally correlated with visual quantitation (Figure 5I, a and b) ▶ . E16 animals did not demonstrate significant dermal TGF-β2 staining at any of the time points studied, while dermal TGF-β2 staining was significantly elevated in E19 animals at 24 hours after injury (P = 0.0000). Interestingly, RT-PCR analysis revealed significantly increased TGF-β2 mRNA levels in E16 relative to E19 wounds at 24 hours (2.11-fold; P = 0.0000) and 72 hours (2.52-fold; P = 0.0000) after injury (Figure 5Ic) ▶ . TGF-β2 expression was also increased 24 hours after injury in E16 animals relative to controls (P = 0.0002), while TGF-β2 expression did not change significantly in E19 animals at 24 hours post-wounding (Figure 5Ic) ▶ . By 72 hours, TGF-β2 levels in E16 wounds were not significantly different from controls, while E19 wounds actually demonstrated lower than control levels of TGF-β2 expression (P = 0.0346) (Figure 5Ic) ▶ .
Figure 5.
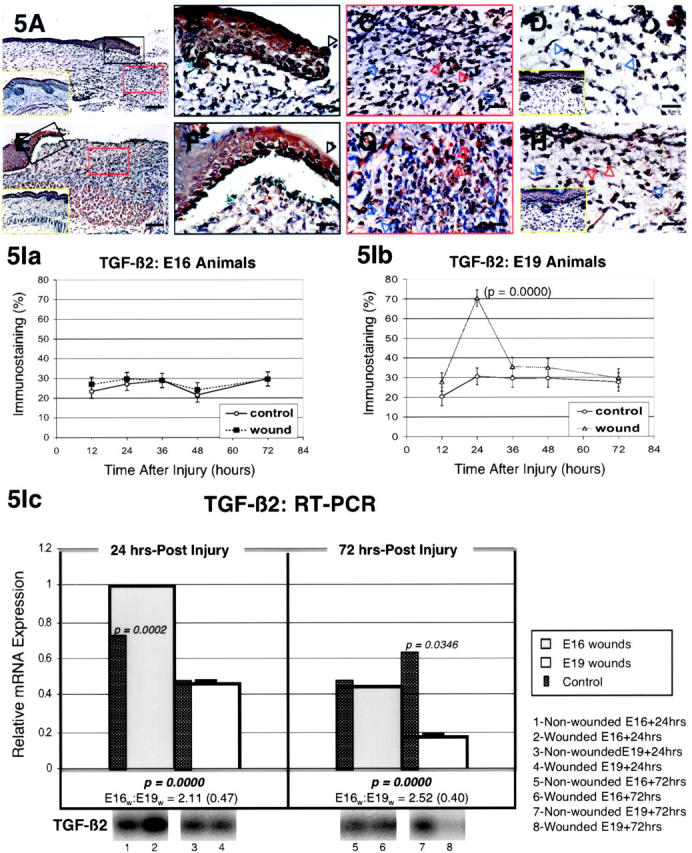
TGF-β2 immunostaining of wounded E16 and E19 rat skin. A: E16 skin, 24 hours post-injury; magnification, ×100. Up-regulated TGF-β2 expression is primarily present in epidermal cells and in dermal inflammatory cells. Non-wounded E17 [ie, E16 + 24 hours] control skin is also shown (inset, A, yellow rectangle). B: E16 skin, 24 hours post-injury; magnification, ×400. Strong TGF-β2 staining is present in outer and basal epidermal layers in addition to the migrating epidermis (black open arrow) after injury. C: E16 skin, 24 hours post-injury; magnification, ×400. Inflammatory cells (red open arrows) stain moderately for TGF-β2, while fibroblasts demonstrate minimal (blue open arrows) and ECM exhibit no TGF-β2 staining. D: E16 skin, 36 hours post-injury; magnification, ×400. Minimal TGF-β2 staining that is not significantly different from control is noted in fibroblasts (blue open arrows) and the ECM. Non-wounded E17.5 [ie, E16 + 36 hours] control skin is also shown (inset, D, yellow rectangle). E: E19 skin, 24 hours post-injury; magnification, ×100. Increased epidermal and dermal TGF-β2 localization relative to control skin are noted. Non-wounded E20 [ie, E19 + 24 hours] control skin is also shown (inset, E, yellow rectangle). F: E19 skin, 24 hours post-injury; magnification, ×400. Moderate TGF-β2 staining is present in outer and basal epidermal layers in addition to the migrating epidermis (black open arrow). G: E19 skin, 24 hours post-injury; magnification, ×400. Inflammatory cells (red open arrows), fibroblasts (blue open arrows), and ECM TGF-β2 expression are all increased to moderate levels after injury. H: E19 skin, 36 hours post-injury; magnification, ×400. Wounded E19 skin demonstrate persistent inflammatory cell TGF-β2 staining 36 hours after injury (red open arrows) whereas minimal TGF-β2 staining is observed in fibroblasts (blue open arrows) and the ECM. Non-wounded, E20.5 [ie, E19 + 36 hours] control skin (inset, H, yellow rectangle) exhibit minimal dermal TGF-β2 staining. Bars: A and E, 200 μm; B–D, F–H, 50 μm. I. Computerized quantitation of dermal TGF-β2 immunostaining in E16- and E19-wounded fetuses. Ia: Dermal TGF-β2 staining does not increase significantly in wounded E16 animals at any of the time points studied (P >0.05). Ib: In contrast, dermal TGF-β2 staining increases significantly in E19 animals 24 hours after wounding relative to non-wounded controls (P = 0.0000). Ic: RT-PCR of TGF-β2. Please refer to the Figure 4Ic ▶ legend for an explanation of the bar graphs and representative Southern blot data.
TGF-β3
The specific intensities for TGF-β3 staining in the epidermis, dermis, and hair follicles are listed in Table 2 ▶ . Epidermal TGF-β3 expression was continuously elevated in E16-wounded animals from 24 to 36 hours after injury, while E19-wounded animals demonstrated increased TGF-β3 expression at 12 hours, 36 hours (basal layer only), 48 hours, and 72 hours (basal layer only) (Figure 6, A, B, E, and F ▶ ; Table 2 ▶ ). Increased ECM TGF-β3 signals were observed in E16-wounded fetuses from 24 to 36 hours post-injury, while ECM TGF-β3 remained unchanged in E19-wounded fetuses (Figure 6, A, C, D, E, G, and H ▶ ; Table 2 ▶ ). Similarly, dermal cell TGF-β3 was up-regulated in E16-injured animals beginning 12 hours after injury and maximally up-regulated between 24 to 36 hours after injury (Figure 6, A, C, and D ▶ ; Table 2 ▶ ). In marked contrast, E19-injured animals exhibited only transiently increased dermal cell TGF-β3 expression at 48 hours after injury (Table 2) ▶ . Notably, TGF-β3 was more strongly expressed in inflammatory cells from E16 rather than E19 wounds (Figure 6, C and D ▶ ; Table 2 ▶ ). Regenerated hair follicles in E16 animals also did not exhibit increased TGF-β3 expression (Table 2) ▶ . Computerized quantitation of dermal TGF-β3 immunostaining in E16- and E19-wounded fetuses generally correlated with visual quantitation (Figure 6I, a and b) ▶ . TGF-β3 levels were significantly increased in E16 animals at 36 hours after injury (P = 0.0001) and not at all in E19 animals. RT-PCR analysis demonstrated significantly increased TGF-β3 transcripts for E16 wounds at 24 and 72 hours relative to E19 wounds (1.82-fold; P = 0.0481 and 2.45-fold; 0.0428, respectively) (Figure 6Ic) ▶ . In contrast, TGF-β3 expression did not vary significantly between age-matched controls and corresponding E16 and E19 wounds at 24 hours post-injury and in E16 wounds at 72 hours post-injury Figure 6Ic) ▶ . E19 wounds, however, demonstrated significant down-regulation of TGF-β3 transcripts at 72 hours after wounding (P = 0.0003) (Figure 6Ic) ▶ .
Figure 6.
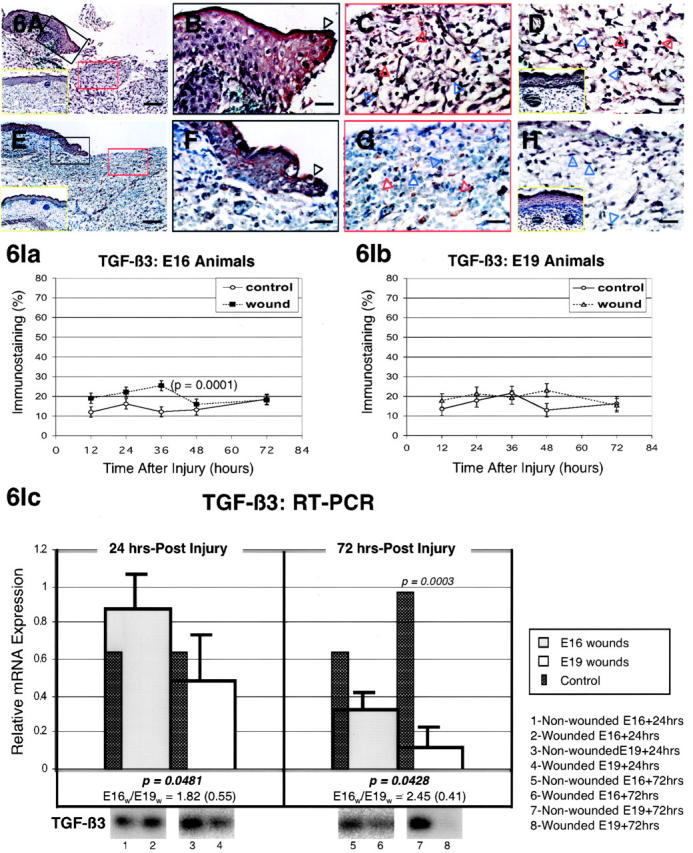
TGF-β3 immunostaining of wounded E16 and E19 rat skin. A: E16 skin, 24 hours post-injury; magnification, ×100. Up-regulated TGF-β3 expression is evident in both epidermal and dermal layers after wounding. Non-wounded E17 [ie, E16 + 24 hours] control skin is also shown (inset, A, yellow rectangle). B: E16 skin, 24 hours post-injury; magnification, ×400. Higher magnification view of wounded epidermis demonstrating strong TGF-β3 staining in both outer and basal as well as migrating (black open arrow) epidermal layers. C: E16 skin, 24 hours post-injury; magnification, ×400. Moderate TGF-β3 staining is observed in dermal fibroblasts (blue open arrows) as well as the surrounding ECM. Strong TGF-β3 staining is present in inflammatory cells (red open arrows). This is contrasted with the minimal degree of TGF-β3 staining in control skin (inset, A). D: E16 skin, 36 hours post-injury; magnification, ×400. Persistent TGF-β3 up-regulation is present in inflammatory cells (red open arrows), fibroblasts (blue open arrows), and the ECM 36 hours after wounding. Non-wounded E17.5 [ie, E16 + 36 hours] control skin is also shown (inset, D, yellow rectangle) demonstrating minimal to no TGF-β3 staining for the ECM and fibroblasts, respectively. E: E19 skin, 24 hours post-injury; magnification, ×100. In contrast, to wounded E16 skin, wounded E19 skin does not reveal increased epidermal or dermal TGF-β3 localization when compared with non-wounded control skin. Non-wounded E20 [ie, E19 + 24 hours] control skin is also shown (inset, E, yellow rectangle). F: E19 skin, 24 hours post-injury; magnification, ×400. Minimal intracellular epidermal staining is observed in the migrating (black open arrow) as well as the outer and basal layers. G: E19 skin, 24 hours post-injury; magnification, ×400. Inflammatory cells (red open arrows), fibroblasts (blue open arrows), and dermal ECM does not demonstrate any TGF-β3 up-regulation after injury. H: E19 skin, 36 hours post-injury; magnification, ×400. Wounded E19 skin demonstrate minimal dermal fibroblast (blue open arrows) and ECM TGF-β3 staining 36 hours after injury that is comparable to non-wounded control levels, while inflammatory cells (red open arrows) demonstrate moderately increased staining. Non-wounded, E20.5 [ie, E19 + 36 hours] control skin (inset, H, yellow rectangle) exhibit minimal dermal TGF-β3 staining. Bars: A and E, 200 μm; B–D, F–H, 50 μm. I: Computerized quantitation of dermal TGF-β3 immunostaining in E16- and E19-wounded fetuses. Ia: Dermal TGF-β3 staining is significantly increased in E16 animals 36 hours after wounding relative to non-wounded controls (P = 0.0001). Ib: Dermal TGF-β3 staining does not increase significantly in wounded E19 animals at any of the time points studied (P >0.05). Ic: RT-PCR of TGF-β3. Please refer to the Figure 4Ic ▶ legend for an explanation of the bar graphs and representative Southern blot data.
TGF-β Type I Receptor
The specific intensities for TGF-β type I receptor (TGF-βRI) staining in the epidermis, dermis, and hair follicles are listed in Table 3 ▶ . In E16-wounded animals, increased TGF-βRI staining was evident 12 hours after injury in the epidermis and maximally expressed at 24 hours (Figure 7, A and B ▶ ; Table 3 ▶ ). E19-wounded animals on the other hand, exhibited increased epidermal TGF-βRI expression at 12 and 36 hours, but not at 24 hours after injury (Figure 7, E, F, and H ▶ ; Table 3 ▶ ). Neither E16- nor E19-injured animals exhibited any ECM staining, except for some minimal staining observed in E19 wounds at 36 hours post-injury (Figure 7, D and H ▶ ; Table 3 ▶ ). Dermal cell TGF-βRI expression increased at 24 hours only in E16 animals (Figure 7, A and C) ▶ and at 12 and 48 hours in E19 wounds (Table 3) ▶ . Elevated TGF-βRI expression was seen from 12 to 36 hours in both E16-and E19-injured fetal inflammatory cells (Figure 7, C and G ▶ ; Table 3 ▶ ). Regenerated hair follicles in E16 animals did not reveal increased type I receptor expression (Table 3) ▶ . RT-PCR analysis revealed a small, but statistically significant, increase in TGF-βRI mRNA at 24 hours after injury in E16 relative to E19 wounds (2.07-fold; P = 0.0272) (Figure 7I) ▶ . By 72 hours post-wounding, however, no significant difference was observed between E16 and E19 wounds for TGF-βRI expression. Neither E16 nor E19 wounds displayed any significant differences from age-matched controls at 24 hours after injury (Figure 7I) ▶ . At 72 hours after injury, E16 animals continued to show no difference between controls while E19 wounds demonstrated significantly decreased TGF-βRI transcripts (P = 0.0262) (Figure 7I) ▶ .
Table 3.
Relative Immunostaining Intensity of TGF-β Receptors in Wounded and Non-Wounded Fetal Skin
| After injury | 12 hours | 24 hours | 36 hours | 48 hours | 72 hours | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TGF-β RI | W | C | W | C | W | C | W | C | W | C | ||||||||||
| E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | |
| Epidermis | ||||||||||||||||||||
| Migrating epi. | ++ | ++ | N/A | N/A | +++ | +++ | N/A | N/A | + | ++ | N/A | N/A | + | + | N/A | N/A | N/A | N/A | N/A | N/A |
| Outer layer | ++ | ++ | + | + | +++ | ++ | ++ | ++ | + | ++ | + | + | + | + | + | + | + | + | + | + |
| Basal layer | ++ | ++ | + | + | +++ | ++ | ++ | +++ | + | ++ | + | + | + | + | ++ | ++ | + | ++ | + | ++ |
| Dermis | ||||||||||||||||||||
| ECM | − | − | − | − | − | − | − | − | − | + | − | − | − | − | − | − | − | − | − | |
| Fibroblasts | + | + | + | − | ++ | − | + | − | − | ++ | − | + | ++ | + | ++ | − | − | + | + | + |
| Inflammatory cells* | + | + | − | − | ++ | ++ | + | + | + | ++ | − | + | + | + | + | + | + | + | + | + |
| Hair follicles | N/A | N/A | − | − | N/A | N/A | + | + | N/A | N/A | − | + | N/A | N/A | − | − | + | N/A | + | + |
| After injury | 12 hours | 24 hours | 36 hours | 48 hours | 72 hours | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TGF-β RII | W | C | W | C | W | C | W | C | W | C | ||||||||||
| E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | |
| Epidermis | ||||||||||||||||||||
| Migrating epi. | + | + | N/A | N/A | + | ++ | N/A | N/A | + | ++ | N/A | N/A | + | + | N/A | N/A | N/A | N/A | N/A | N/A |
| Outer layer | + | + | + | + | + | ++ | + | + | + | ++ | + | + | + | + | + | + | + | + | + | + |
| Basal layer | + | + | + | + | + | ++ | + | + | + | ++ | + | + | + | + | + | − | + | + | + | + |
| Dermis | ||||||||||||||||||||
| ECM | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Fibroblasts | − | + | − | + | − | − | + | − | − | + | − | + | + | + | + | − | − | − | − | + |
| Inflammatory cells* | + | + | + | + | + | + | + | + | ++ | ++ | − | + | + | + | − | + | + | + | + | + |
| Hair follicles | N/A | N/A | + | − | N/A | N/A | + | − | N/A | N/A | − | + | N/A | N/A | + | + | − | N/A | − | + |
| After injury | 12 hours | 24 hours | 36 hours | 48 hours | 72 hours | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TGF-β RIII | W | C | W | C | W | C | W | C | W | C | ||||||||||
| E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | E16 | E19 | |
| Epidermis | ||||||||||||||||||||
| Migrating epi. | + | ++ | N/A | N/A | + | + | N/A | N/A | ++ | ++ | N/A | N/A | + | + | N/A | N/A | N/A | N/A | N/A | N/A |
| Outer layer | + | ++ | + | + | + | + | + | + | ++ | ++ | ++ | + | + | + | + | + | − | + | − | + |
| Basal layer | + | ++ | + | + | + | + | + | + | ++ | ++ | ++ | + | + | + | + | + | − | − | − | − |
| Dermis | ||||||||||||||||||||
| ECM | + | + | + | + | ++ | + | + | + | ++ | + | ++ | + | − | + | + | ++ | + | + | + | + |
| Fibroblasts | + | + | + | + | ++ | − | − | − | ++ | + | ++ | + | − | + | − | + | − | − | − | − |
| Inflammatory cells* | + | + | + | + | + | + | + | + | ++ | + | + | + | + | + | + | + | + | + | + | + |
| Hair follicles | N/A | N/A | − | − | N/A | N/A | − | − | N/A | N/A | − | − | N/A | N/A | − | − | − | N/A | − | − |
W, Wound; C, Control; N/A, not applicable; −, no staining (<5%); +, minimal staining (<25%); ++, moderate staining (25% to 50%); +++, strong staining (>50%). Peak expression time points for a given tissue component are highlighted for wounded E16 (bold) and E19 (underlined) animals.
*In general, non-wounded control skin and E16-wounded fetuses contained very few inflammatory cells. The chart reflects the intensity of intracellular TGF-β staining and not the actual number of inflammatory cells present.
Figure 7.
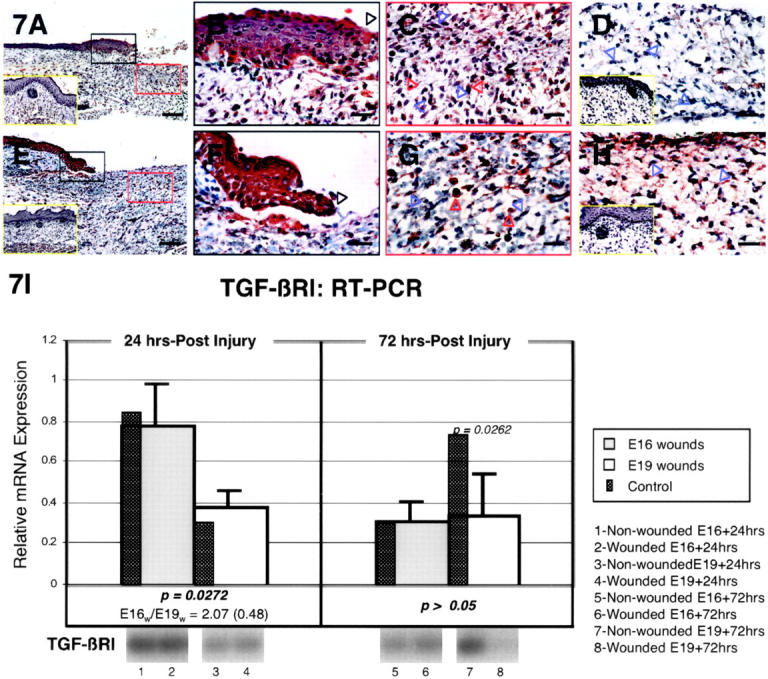
TGF-βRI immunostaining of wounded E16 and E19 rat skin. A: E16 skin, 24 hours post-injury; magnification, ×100. Up-regulated TGF-βRI expression is evident in both epidermal and dermal layers after injury. Non-wounded E17 [ie, E16 + 24 hours] control skin is also shown (inset, A, yellow rectangle). B: E16 skin, 24 hours post-injury; magnification, ×400. Higher magnification view of wounded epidermis demonstrating strong TGF-βRI staining in outer, basal and migrating (black open arrow) epidermal layers. C: E16 skin, 24 hours post-injury; magnification, ×400. Increased TGF-βRI staining from minimal to moderate levels is observed in dermal fibroblasts (blue open arrows) as well as in inflammatory cells (red open arrows) after injury. D: E16 skin, 36 hours post-injury; magnification, ×400. Except for some minimal TGF-βRI up-regulation in inflammatory cells, no epidermal or dermal components demonstrate any TGF-βRI expression above control non-wounded skin. Note the complete absence of TGF-βRI staining in fibroblasts (blue open arrows). Non-wounded E17.5 [ie, E16 + 36 hours] control skin is also shown (inset, D, yellow rectangle). E: E19 skin, 24 hours post-injury; magnification, ×100. Epidermal and dermal TGF-βRI localization is not increased relative to non-wounded control skin. Non-wounded E20 [ie, E19 + 24 hours] control skin is also shown (inset, E, yellow rectangle). F: E19 skin, 24 hours post-injury; magnification, ×400. Moderate epidermal staining is observed in the migrating (black open arrow) as well as the outer and basal layers. G: E19 skin, 24 hours post-injury; magnification, ×400. Inflammatory cells (red open arrows) show moderate TGF-βRI staining whereas fibroblasts (blue open arrows) and dermal ECM demonstrate no staining. H: E19 skin, 36 hours post-injury; magnification, ×400. Wounded E19 skin demonstrate up-regulated TGF-βRI expression in dermal fibroblasts (blue open arrows), inflammatory cells (red open arrows), and ECM. Non-wounded, E20.5 [ie, E19 + 36 hours] control skin (inset, H, yellow rectangle) exhibit minimal dermal TGF-βRI staining. Bars: A and E, 200 μm; B–D, F–H, 50 μm. I. RT-PCR of TGF-βRI. Please refer to the Figure 4Ic ▶ legend for an explanation of the bar graphs and representative Southern blot data.
TGF-β Type II Receptor
The specific intensities for TGF-β type II receptor (TGF-βRII) staining in the epidermis, dermis, and hair follicles using antibody (L-21) prepared from peptide human TGF-βRII sequences are listed in Table 3 ▶ . Antibody (H-567) prepared from full-length human TGF-βRII sequences demonstrated slightly increased staining intensity (less than 25% overall) on some sections relative to the L-21 antibody. However, staining intensities for both control and wound sections increased coordinately; and absolute differences between control and wound sections did not change significantly with either L-21 or H-567 antibody. Epidermal TGF-βRII levels remained unchanged in E16-wounded animals at all time points assayed (Figure 8, A and B ▶ ; Table 3 ▶ ). In contrast, epidermal TGF-βRII expression increased in E19-wounded fetuses from 24 to 48 hours after injury with peak expression between 24 to 36 hours (Figure 8, E and F ▶ ; Table 3 ▶ ). Elevated TGF-βRII expression was not present in the dermal cells of wounded E16 animals at any of the time points examined, although down-regulation was observed at 24 hours post-injury (Figure 8, C and D ▶ ; Table 3 ▶ ). E19-injured fetuses did not demonstrate any dermal TGF-βRII up-regulation until 48 hours after injury, but did exhibit higher basal levels of TGF-βRII than E16-injured animals at 12 and 36 hours after injury (Figure 8H ▶ ; Table 3 ▶ ). Inflammatory cell TGF-βRII expression was unchanged in either E16 or E19 animals in the first 24 hours after injury, but increased in E16-injured animals from 36 to 48 hours and in E19-injured animals at 36 hours only (Figure 8, C and G ▶ ; Table 3 ▶ ). TGF-βRII expression was not increased in the regenerated hair follicles of E16-injured animals (Table 3) ▶ . RT-PCR analysis did not reveal any significant changes in steady-state TGF-βRII mRNA levels between E16 and E19 wounds at either 24 or 72 hours after injury (Figure 8I) ▶ . In addition, TGF-βRII levels did not vary significantly from controls at 24 and 72 hours post-injury in E16 wounds, while E19 animals demonstrated significantly increased levels at 24 hours after injury (P = 0.0002), followed by significantly decreased levels at 72 hours (P = 0.0013) after wounding (Figure 8I) ▶ .
Figure 8.
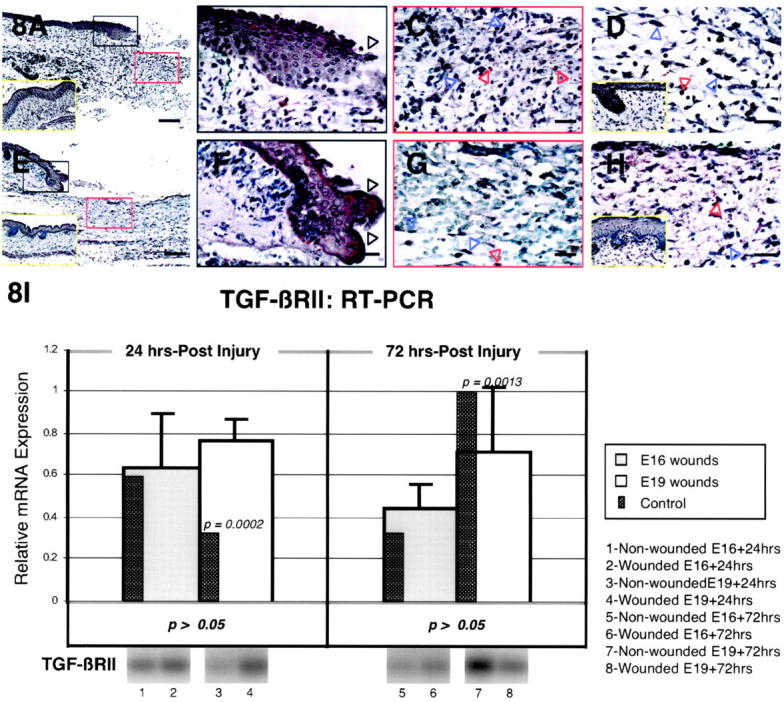
TGF-βRII immunostaining of wounded E16 and E19 rat skin. A: E16 skin, 24 hours post-injury; magnification, ×100. No TGF-βRII up-regulation is observed in comparison with non-wounded E17 [ie, E16 + 24 hours] control skin (inset, A, yellow rectangle). B: E16 skin, 24 hours post-injury; magnification, ×400. Higher magnification view of wounded epidermis demonstrating minimal TGF-βRII staining in outer, basal, and migrating (black open arrows) epidermal layers. C: E16 skin, 24 hours post-injury; magnification, ×400. Minimal TGF-βRII staining is observed in dermal fibroblasts (blue open arrows) as well as inflammatory cells (red open arrows). D: E16 skin, 36 hours post-injury; magnification, ×400. No TGF-βRII staining is observed in dermal ECM and fibroblasts (blue open arrows), whereas moderate staining is seen in inflammatory cells (red open arrow) 36 hours after wounding. Non-wounded E17.5 [ie, E16 + 36 hours] control skin is also shown (inset, D, yellow rectangle). E: E19 skin, 24 hours post-injury; magnification, ×100. Wounded E19 skin demonstrates epidermal TGF-βRII up-regulation in comparison with non-wounded control skin. Non-wounded E20 [ie, E19 + 24 hours] control skin is also shown (inset, E, yellow rectangle). F: E19 skin, 24 hours post-injury; magnification, ×400. Moderate epidermal staining is observed in the migrating (black open arrow) as well as in the outer and basal epidermal layers. G: E19 skin, 24 hours post-injury; magnification, ×400. Inflammatory cells (red open arrow) show minimal TGF-βRII staining whereas fibroblasts (blue open arrows) and dermal ECM demonstrate no staining. H: E19 skin, 36 hours post-injury; magnification, ×400. Dermal up-regulation of TGF-βRII receptor expression is observed solely in inflammatory cells (red open arrow). TGF-βRII receptor staining in fibroblasts (blue open arrow) and the ECM does not differ significantly from control skin. Non-wounded, E20.5 [ie, E19 + 36 hours] control skin (inset, H, yellow rectangle) exhibit minimal dermal TGF-βRII staining. Bars: A and E, 200 μm; B–D, F–H, 50 μm. I: RT-PCR of TGF-βRII. Please refer to the Figure 4Ic ▶ legend for an explanation of the bar graphs and representative Southern blot data.
TGF-β Type III Receptor
The specific intensities for TGF-β type III receptor (TGF-βRIII) staining in the epidermis, dermis, and hair follicles are listed in Table 3 ▶ . As for TGF-βRII, epidermal TGF-βRIII levels also remained unchanged in E16-wounded animals at all time points assayed (Figure 9, A and B ▶ ; Table 3 ▶ ). Meanwhile, E19-injured animals displayed TGF-βRIII up-regulation at 12 and 36 hours, but not at 24 hours after injury (Figure 9, E and F ▶ ; Table 3 ▶ ). ECM and dermal TGF-βRIII expression, on the other hand, increased to moderate levels in E16-wounded fetuses at 24 hours, but no up-regulation was observed in any of the E19-wounded fetuses (Figure 9, A, C, E, and G ▶ ; Table 3 ▶ ). At 48 hours post-injury, ECM TGF-βRIII was down-regulated in both E16 and E19-wounded animals (Table 3) ▶ . Inflammatory cell TGF-βRIII levels increased in E16-injured animals at 36 hours only (Figure 9D) ▶ , and not at all in E19-injured animals (Table 3) ▶ . TGF-βRIII expression remained unchanged in regenerated hair follicles (Table 3) ▶ . RT-PCR analysis revealed significantly increased steady-state TGF-βRIII mRNA levels for E19 wounds relative to E16 wounds at 24 hours after injury (2.63-fold; P = 0.0031) (Figure 9I) ▶ . When compared to age matched controls, however, neither E16 nor E19 animals demonstrated any difference at 24 hours after wounding, while both demonstrated significantly decreased TGF-βRIII expression at 72 hours after injury (P = 0.0100 and 0.0346), respectively) (Figure 9I) ▶ .
Figure 9.
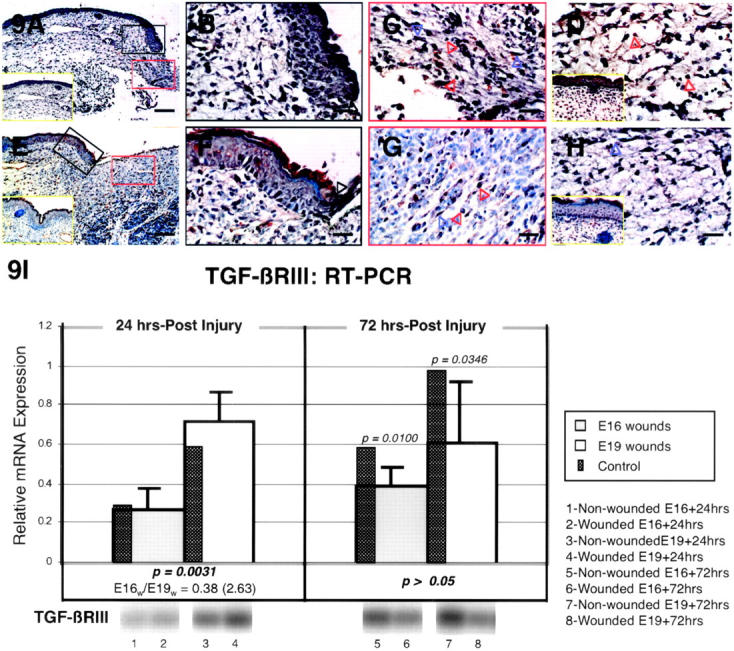
TGF-βRIII immunostaining of wounded E16 and E19 rat skin. A: E16 skin, 24 hours post-injury; magnification, ×100. Dermal TGF-βRIII expression is up-regulated while epidermal expression remain unchanged from non-wounded E17 skin levels [ie, E16 + 24 hours] (inset, A, yellow rectangle). B: E16 skin, 24 hours post-injury; magnification, ×400. Higher magnification view of wounded epidermis demonstrating minimal TGF-βRIII staining in outer, basal, and migrating (black open arrow) epidermal layers. C: E16 skin, 24 hours post-injury; magnification, ×400. Moderate TGF-βRIII staining is observed in dermal fibroblasts (blue open arrows) as well as in the ECM, while minimal staining is demonstrated in inflammatory cells (red open arrows). D: E16 skin, 36 hours post-injury; magnification, ×400. The moderate degree of TGF-βRIII up-regulation in dermal fibroblasts and ECM is not significantly different from non-wounded skin levels, although inflammatory cell (open red arrows) TGF-βRIII expression is increased relative to control E17.5 skin [ie, E16 + 36 hours] (inset, D, yellow rectangle). E: E19 skin, 24 hours post-injury; magnification, ×100. There is no significant difference in TGF-βRIII localization between injured and non-injured control skin. Non-wounded E20 [ie, E19 + 24 hours] control skin is also shown (inset, E, yellow rectangle). F: E19 skin, 24 hours post-injury; magnification, ×400. Similar to control skin, minimal epidermal staining is observed in the migrating (black open arrow) as well as in the outer and basal epidermal layers. G: E19 skin, 24 hours post-injury; magnification, ×400. Inflammatory cells (red open arrows) and dermal ECM for both wounded and non-wounded skin show minimal TGF-βRIII staining whereas fibroblasts (blue open arrow) demonstrate no staining. H: E19 skin, 36 hours post-injury; magnification, ×400. Dermal TGF-βRIII expression does not differ significantly from E20.5 [ie, E19 + 36 hours] control skin (inset, H, yellow rectangle). Bars: A and E, 200 μm; B–D, F–H, 50 μm. I: RT-PCR of TGF-βRIII. Please refer to the Figure 4Ic ▶ legend for an explanation of the bar graphs and representative Southern blot data.
Discussion
TGF-βs are a family of multifunctional cytokines with key roles in embryonic development and wound healing. While the biological functions of TGF-β1, -β2, and -β3 appear to be similar in most in vitro studies, murine knockouts of TGF-β1, -β2, or -β3 indicate isoform-specific functions for TGF-β. 14 In tissue repair, TGF-β1 and -β2 are known to promote scar, while TGF-β3 may reduce scar. 3,4
Although prior studies had reported a lack of TGF-β induction in fetal wounds, 3,6-8 our study showed an ontogenetic transition in the onset, degree, and duration of TGF-β induction that was accompanied by corresponding changes in collagen architecture. In addition, we observed a distinct difference in the inflammatory profile of E16- versus E19-injured animals. E16-injured animals exhibited decreased inflammatory cell numbers and a profile that was predominantly lymphocytes and neutrophils. E19-injured fetuses, on the other hand, exhibited increased inflammatory cell numbers and a profile that was predominantly monocytes with some neutrophils, in keeping with the post-injury profile observed by Hopkinson-Woolley et al in late gestation fetal mice 15 and by Levine et al 16 in adult animals.
Even though we did not detect major differences in TGF-β ligand or receptor expression between E16- and E19-wounded animals on an individual cellular basis, it is possible that the increased monocyte/macrophage population in E19 wounds, as well as in adult wounds, elaborate other factors involved in scar formation. 17 Given the central role of the macrophage in adult wound repair, the association between monocyte/macrophage appearance in E19 wounds and the transition to and adult-type repair phenotype may be more than coincidental. Interestingly, TGF-β1 has also been implicated in monocyte/macrophage 18,19 and lymphocyte 20,21 differentiation.
In terms of ligand expression, both E16- and E19-injured animals evidenced increased epidermal TGF-β1 immunostaining at 12 and 24 hours, which by 36 hours, had returned to control levels in E16 animals while E19 animals continued to exhibit increased TGF-β1 staining until 72 hours. Dermal TGF-β1 expression followed a similar pattern with E16-injured animals exhibiting increased ECM and fibroblast staining at 12 and 24 hours and a rapid return to baseline by 36 hours. E19-injured animals, on the other hand, demonstrated elevated fibroblast TGF-β1 staining until 48 hours. Rapid TGF-β1 induction and clearance by E16-injured animals concur with Martin et al’s study 9 showing rapid TGF-β1 induction and clearance in early gestation (E11.5) murine limb bud wounds. Meanwhile, RT-PCR analysis revealed increased TGF-β1 transcripts in both E16 and E19 animals at 24 hours after wounding with albeit lower expression (by 0.60-fold) in E16 animals. We hypothesize that while the initial inflammatory infiltrate may contain enough TGF-β1 to activate AP-1 mediated TGF-β1 autoinduction 22 in both E16 and E19 animals, the diminished inflammation characteristic of E16 wounds decreases the stimulus for further TGF-β1 autoinduction. In addition, continued TGF-β1 expression in E19 wounds may also reflect increased TGF-β1 gene transcription by more “adult-like” fibroblasts. 23 This suggests that persistent transcriptional up-regulation of TGF-β1 in E19 wounds is important for scar development.
Compared to TGF-β1, overall TGF-β2 staining was less, but still more predominant in E19 relative to E16 animals. Both wounded E16 and E19 animals exhibited increased epidermal TGF-β2 staining at 24 hours post-injury, but by 36 hours levels in E16-injured fetuses had significantly declined while TGF-β2 levels in E19-injured fetuses were maximal. In general, ECM and fibroblast TGF-β2 staining was more prominent in E19-wounded animals. Interestingly, RT-PCR demonstrated increased TGF-β2 transcripts in only E16 wounds at 24 hours post-injury. This suggests that the majority of TGF-β2 immunostaining in adult-type E19 wounds may originate from the inflammatory infiltrate rather than new transcription and that transcriptional up-regulation of TGF-β2 is not important for scar development. Comparison of fetal versus adult fibroblast TGF-β2 mRNA expression by Broker et al 23 also did not reveal increased TGF-β2 levels in adult fibroblasts which presumably would be more similar to the E19 fibroblasts.
In general, increased and sustained TGF-β1 and -β2 expression in E19-injured animals correlated closely with the observed transition from fetal-type to adult-type wound repair. Mechanistically, relatively higher TGF-β1 and -β2 levels in E19 wounds can account for overall matrix accumulation and scar formation. This is supported by the observation that TGF-β1 alone was enough to induce scar when added to a normally scarless human fetal wound model, 3 and that exogenous addition of neutralizing antibody to TGF-β1 and -β2 decreased scar formation in an adult wound model. 4,5 The orientation, spacing, and size of collagen bundles in anti-TGF-β1 and -β2 antibody treated wounds more closely resembled that of non-wounded skin. 4 In a similar fashion, our confocal data demonstrated a clear distinction in collagen architecture between E16 (low TGF-β1, -β2 expression) and E19 (high TGF-β1, -β2 expression) wounds.
The lack of TGF-β1 and TGF-β2 induction seen in other fetal wound studies may reflect inherent differences in animal wounds systems, antibody recognition epitopes, and time points analyzed. For example, Whitby and Ferguson 6 used non-sutured, 2-mm lip incisions in E16 mice as their model. And though they analyzed multiple time points (eg, 1, 6, 12, 18, 36, 48, and 72 hours), they used a non-isoform-specific antibody that recognized both TGF-β1 and -β2, and possibly -β3. In contrast, Nath 7 used sutured, 1-cm full-thickness incisions in E23 rabbits (term = 31 days) and immunostained for TGF-β1 and -β2 at 3, 5, and 7 days. The absence of TGF-β1 and-β2 in their study may reflect xenogeneic differences (eg, lack of wound contraction in rabbit wounds) or the lateness of the time points assayed. In addition, while their TGF-β2 antibody recognized both intracellular and extracellular forms, their TGF-β1 antibody recognized only intracellular ligand. Thus, the lack of wound TGF-β1 in Nath’s study 7 may also reflect inherent limitations in their TGF-β1 antibody, since we found definite evidence of ECM TGF-β1 expression. The TGF-β antibodies used in our study were designed using peptides corresponding to the extreme carboxy terminus of human TGF-β1, -β2, and -β3 precursor protein and should recognize both intracellular and extracellular forms of TGF-β. Lastly, Sullivan’s model 8 used full-thickness human fetal and adult skin implanted into nude mice. After 7 days, the implanted skin graft was incised and assayed for TGF-β1 and -β2 expression from 1 hour to 28 days. In their study, they show convincingly that TGF-β1 and -β2 are induced in adult, but not fetal wounds, and that the amniotic environment is not critical for scarless repair. 8 However, isolated human skin grafted into immunocompromised mice may mask moderate levels of TGF-β1 or -β2 induction that could potentially occur after injury to intact human fetuses. Although our model did not show a markedly significant difference between TGF-β1 or -β2 staining per inflammatory cell, the total number of inflammatory cells was markedly greater in wounded E19 relative to E16 animals. Thus, a considerable amount of wound TGF-β1 and -β2 were derived from immune cells, and immunocompromised animals would presumably express less TGF-β overall.
Interestingly, previous work by Shah et al 4 had suggested an anti-fibrotic role for TGF-β3 when applied to incisional adult rat wounds. Our immunohistochemistry results also support a possible anti-fibrotic role for TGF-β3. Although both E16- and E19-injured fetuses exhibited increased epidermal TGF-β3 staining, only E16 animals demonstrated elevated ECM and fibroblast TGF-β3 staining in the first 36 hours post-injury. In contrast, E19 fetuses did not exhibit increased ECM TGF-β3 staining at all, while fibroblast TGF-β3 staining was only transiently increased at 48 hours after injury. RT-PCR analysis revealed increased TGF-β3 expression in E16 wounds (1.82-fold) at 24 hours relative to E19 wounds. The delay in TGF-β3 induction that occurred in E19-wounded animals manifesting adult-type repair with scar, also paralleled the delay in TGF-β3 up-regulation reported in other models of adult repair. For instance, Frank et al 24 had demonstrated rapid TGF-β1 mRNA induction (peaks at 1 to 3 days) and delayed TGF-β3 induction (peaks at 7 days) in a 6-mm, full-thickness, excisional wound model in mice. We had also observed a similar pattern of delayed TGF-β3 transcript induction in a full-thickness, 1-cm, adult rat excisional wound model. 13 Thus TGF-β3, a potential anti-fibrotic molecule, was expressed more rapidly and to a greater extent in the epidermis, ECM, dermal cells, and inflammatory cells of E16-injured animals exhibiting scarless fetal-type repair, while TGF-β3 expression was generally delayed or diminished in E19 fetuses showing adult-type repair with scar. Interestingly, fetal platelets have been described as a major source of TGF-β3 and may account for the relative abundance of TGF-β3 in E16 wounds. 25
In disagreement with Shah et al, 4 Wu 26 reported that TGF-β3 treatment of rabbit dermal ear ulcers at 0 and 3 days after injury did not decrease scar formation. However, the TGF-β3 treatment schedule used in Wu’s study 26 (days 0 and 3) may not be as effective as the regimen used in Shah et al’s study 4 (days 0, 1, and 2). The early, consecutive TGF-β3 application in Shah et al’s model 4 more closely mimicked the early, sustained TGF-β3 expression that we observed, especially in dermal cells, of scarless E16 wounds.
The exact mechanisms for how TGF-β3 may reduce scar are unknown, and indeed, the exact function of the different isoforms in wound repair and development are still unclear. TGF-β3 null mice exhibit deficient palate and lung morphogenesis, indicating the importance of TGF-β3 in epithelial-mesenchymal interactions. 27 We hypothesize that increased levels of TGF-β3 expressed early in fetal wounds, or applied early to adult wounds, may effectively compete with TGF-β1 and -β2 for binding to the type II receptor. And because TGF-β3 may have intrinsically less effects on matrix production than TGF-β1 or -β2, 26 and correspondingly more effects on epidermal development and regeneration, 28 an “anti-scar” effect is observed following early TGF-β3 induction in fetal wounds or after early TGF-β3 application to adult wounds. Alternatively, TGF-β3 regulation of mesenchymal cell proliferation and migration may also promote repair. 25
In terms of the TGF-β receptors, overall immunostaining intensity was less relative to the ligands. For the TGF-βRI, increased epidermal and fibroblast staining was present in E16-injured animals for only the first 24 hours. Wounded E19 fetuses, on the other hand, demonstrated bimodally increased epidermal staining at 12 and 36 hours after injury and bimodally increased fibroblast staining at 12 and 36 to 48 hours after injury. RT-PCR analysis, however, revealed unchanged TGF-βRI transcript levels in both E16 and E19 animals at 24 hours after injury, suggesting that differential transcriptional control of TGF-βRI is not critical for scarless repair.
For TGF-βRII, increased immunostaining was completely absent in injured E16 epidermal cells and fibroblasts at all of the time points studied. Conversely, injured E19 animals exhibited persistently increased epidermal TGF-βRII staining from 24 to 48 hours and transiently increased fibroblast staining at 48 hours. RT-PCR data showed increased steady-state mRNA levels at 24 hours after injury in E19 but not E16 wounds, indicating that like TGF-β1, increased TGF-βRII mRNA levels may be important for scar formation.
Cowin et al 10 recently compared fetal versus adult mice TGF-β ligand and receptor expression in an unsutured, flank incisional wound model [2-mm linear (E16; term = 19 to 20 days) or 1-cm linear (adult), full thickness]. Consistent with our results, they also reported no increased TGF-βRII immunostaining in wounded E16 animals. However, in contrast to our data on E16-injured animals, they observed neither increased type I immunostaining nor any detectable type I or II receptor mRNA expression by non-isotopic in situ hybridization after E16 fetal skin injury. Different methodologies may explain in part the discrepancy in our data. For instance, RT-PCR is much more sensitive than in situ hybridization in detection of low copy transcripts. In addition, other authors using 35S-labeled cRNA probes have reported definite TGF-βRII mRNA levels in developing mouse skin although long exposure times (5 weeks) were required. 29
Although no other fetal wound studies are available for comparison, a previous study by Gold et al 30 using an adult sheep excisional wound model (6-mm diameter, full thickness) have shown delayed up-regulation of epidermal type I and II receptor protein by 3 to 5 days relative to TGF-β1, -β2, and -β3 ligands. In the same study, Gold et al 30 also reported elevated TGF-β ligand and receptor immunostaining in the adjacent wound epithelium, but no TGF-β ligand or receptor staining in the migrating epithelium until complete wound re-epithelialization. We did not observe any delayed TGF-βRI up-regulation for wounded E16 animals, or delayed TGF-βRI or TGF-βRII up-regulation for wounded E19 animals. Moreover, in our study we observed that all six TGF-β ligands and receptors were expressed in the migrating epithelium of E16- and E19-injured fetuses at levels comparable to the outer and basal layers of wound edge epithelium.
The discrepancy between our data and theirs may simply reflect inherent differences between fetal and adult models of repair. As expected from the major roles of TGF-β in embryogenesis, we have found basal TGF-β ligand and receptor levels to be uniformly higher in non-wounded fetal relative to adult skin (data not shown). Alternatively, differences in antibody recognition epitopes may also account for the difference in epithelial staining. Only one of their antibodies for TGF-βRI (R-20) was also used in our study. However, using an adult pig split-thickness wound model, Levine et al 16 have also shown abundant staining for TGF-β2 and -β3, but not TGF-β1, in the migrating epithelium. Thus, ligand expression in the migrating epithelium (at least for TGF-β2 and-β3) does not appear to be an exclusively fetal phenomenon.
Of note, neither Gold et al nor Levine et al 16,30 documented significant staining in adult stratum basalis after injury. Gold et al 30 found stratum basalis staining for type I and II receptors to be “often negative”, while Levine et al 16 showed negative staining for TGF-β1, but not -β2 or -β3 in the stratum basalis. Taken in total, their results suggest an overall deficiency of TGF-β1 and type I and II receptor expression in wounded adult stratum basalis. TGF-β1 has demonstrated a concomitant ability to inhibit keratinocyte proliferation and promote keratinocyte migration. 31,32 Others have postulated that migrating epidermal TGF-β1 expression may promote keratinocyte motility, while the lack of basal epidermal TGF-β1 expression may promote epithelial proliferation. 32 In contrast, our data consistently show TGF-β1, as well as -β2 and -β3, expression in the basal layer of wound epidermis. This suggests that re-epithelialization in fetal wounds may be more dependent on epithelial cell migration than proliferation in accordance with previous work by Ihara and Motobayashi, 33 or that fetal cells are inherently less responsive to the anti-proliferative effects of TGF-β1. Additionally, we have observed more epithelial hypertrophy in E19- relative to E16-wounded fetuses, and significantly more epithelial hypertrophy in neonatal and adult wounds (data not shown). Although E16- and E19-injured animals did not exhibit marked differences in basal epithelial TGF-β1 expression, it would be interesting to determine whether basal epidermal TGF-β1 levels correlate inversely with the degree of epithelial hypertrophy.
We are not aware of any previous fetal or adult wound studies on TGF-βRIII (betaglycan) protein. Unlike type I and II receptors, the type III receptor is a membrane-bound proteoglycan with no intrinsic signaling function, but it may modulate TGF-β access to the signaling receptor. 34 For instance, both TGF-β1 and -β3 bind to the type II receptor with similar affinities, while TGF-β2 cannot bind the type II receptor in the absence of betaglycan. 35 Thus, while bound betaglycan may increase overall TGF-β2 bioactivity, release of soluble betaglycan (non-membrane bound) can bind and possibly inhibit generalized TGF-β activity. 36 Our immunostaining data showed a temporally and spatially distinct pattern of TGF-βRIII expression with increased staining only in the ECM and fibroblasts of E16 animals at 24 hours after injury. Meanwhile, transiently increased staining was observed only in the epidermis of E19 animals at 36 hours after injury. RT-PCR revealed no significant increase at 24 hours after injury in either E16 or E19 animals although higher basal levels were noted in E19 animals. The significance of these findings is unclear, but increased soluble betaglycan in the ECM of E16-injured animals may contribute to scarless fetal repair by decreasing overall TGF-β bioavailability.
Lastly, similar to previous immunohistochemistry studies on fetal mice by Pelton et al, 37 we also noted an overall pattern of more prominent TGF-β1, -β2, and -β3 staining in the epidermis rather than the dermis of non-wounded control animals. In addition, type I and type II receptor immunostaining were also more concentrated in the epidermis relative to the dermis, while type III receptor levels were more evenly distributed between the two tissue layers.
In summary, our results implicate increased TGF-β1, -β2, and decreased TGF-β3, as well as increased type I and II receptors in late gestation fetal scar formation. During scarless fetal repair, minimal induction and rapid clearance of pro-fibrotic molecules such as TGF-β1 and -β2, as well as type I, II receptors, in association with increased expression of anti-fibrotic molecules such as TGF-β3 and soluble betaglycan, may decrease overall TGF-β bioavailability. The diminished TGF-β bioactivity may in turn decrease ECM deposition and scar formation, and more importantly, prevent the AP-1 mediated autoinductive phenomenon that sustains TGF-β1 expression in adult wounds. 38 The ontogenetic transition in TGF-β regulation that we report here likely represents a key event in the phenotypic transition from a fetal-type to an adult-type wound repair response. In addition, we show that this phenotypic change occurs at defined time points in gestation and that it is intimately associated with collagen architecture. However, it is likely that other factors, besides TGF-β are involved. Recently we have determined that fibromodulin, a proteoglycan of the decorin family, is also up-regulated in scarless repair (manuscript in preparation). Fibromodulin binds both fibrillar collagens and TGF-β ligands and is important in regulating ECM assembly. 39-41 Thus, decreased TGF-β1 and -β2 coupled with increased fibromodulin in E16 fetal wounds may not only decrease the total amount of ECM synthesized, but it may also affect collagen fibrillogenesis, and more importantly, dictate the organized pattern of collagen deposition seen by confocal microscopy on repaired E16 wounds. Future strategies to minimize scar formation will likely use a multi-modality approach that combines anti-TGF-β1 and -β2 therapies with TGF-β3, soluble betaglycan, and fibromodulin
Footnotes
Address reprint requests to Kang Ting, D.M.D., D.M.Sc., UCLA School of Dentistry, Room 30–113 CHS, 10833 Le Conte Avenue, Los Angeles, CA 90095. E-mail: kting@ucla.edu.
Supported by the Wunderman Family Foundation, American Association of Orthodontists Foundation, Oral and Maxillofacial Surgery Foundation Grant, NIH/NIDCR DE10598, CRC/NIH RR00865, and NIDCR K23DE00422 and Smile Train Cleft Research Initiative and Plastic Surgery Education Foundation.
P. L. and K. T. are co-senior authors.
References
- 1.Ihara S, Motobayashi Y, Nagao E, Kistler A: Ontogenetic transition of wound healing pattern in rat skin occurring at the fetal stage. Development 1990, 110:671-680 [DOI] [PubMed] [Google Scholar]
- 2.Border WA, Noble NA, Ketteler M: TGF-β: a cytokine mediator of glomerulosclerosis and a target for therapeutic intervention. Kidney Int Suppl 1995, 49:S59-S61 [PubMed] [Google Scholar]
- 3.Lin RY, Sullivan KM, Argenta PA, Meuli M, Lorenz HP, Adzick NS: Exogenous transforming growth factor-β amplifies its own expression and induces scar formation in a model of human fetal skin repair. Ann Surg 1995, 222:146-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah M, Foreman DM, Ferguson MW: Neutralisation of TGF-β 1 and TGF-β 2 or exogenous addition of TGF-β 3 to cutaneous rat wounds reduces scarring. J Cell Sci 1995, 108:985-1002 [DOI] [PubMed] [Google Scholar]
- 5.Shah M, Foreman DM, Ferguson MW: Neutralising antibody to TGF-β 1, 2 reduces cutaneous scarring in adult rodents. J Cell Sci 1994, 107:1137-1157 [DOI] [PubMed] [Google Scholar]
- 6.Whitby DJ, Ferguson MW: Immunohistochemical localization of growth factors in fetal wound healing. Dev Biol 1991, 147:207-215 [DOI] [PubMed] [Google Scholar]
- 7.Nath RK, LaRegina M, Markham H, Ksander GA, Weeks PM: The expression of transforming growth factor type β in fetal and adult rabbit skin wounds. J Pediatr Surg 1994, 29:416-421 [DOI] [PubMed] [Google Scholar]
- 8.Sullivan KM, Lorenz HP, Meuli M, Lin RY, Adzick NS: A model of scarless human fetal wound repair is deficient in transforming growth factor β. J Pediatr Surg 1995, 30:198-202202–193 [DOI] [PubMed] [Google Scholar]
- 9.Martin P, Dickson MC, Millan FA, Akhurst RJ: Rapid induction and clearance of TGF β 1 is an early response to wounding in the mouse embryo. Dev Genet 1993, 14:225-238 [DOI] [PubMed] [Google Scholar]
- 10.Cowin AJ, Holmes TM, Brosnan P, Ferguson MW: Expression of TGF-β and its receptors in murine fetal and adult dermal wounds. Eur J Dermatol 2001, 11:424-431 [PubMed] [Google Scholar]
- 11.Dolber PC, Spach MS: Conventional and confocal fluorescence microscopy of collagen fibers in the heart. J Histochem Cytochem 1993, 41:465-469 [DOI] [PubMed] [Google Scholar]
- 12.Zoli M, Guidolin D, Agnati LF: Morphometric evaluation of populations of neuronal profiles (cell bodies, dendrites, and nerve terminals) in the central nervous system. Microsc Res Tech 1992, 21:315-337 [DOI] [PubMed] [Google Scholar]
- 13.Soo C, Hu FY, Zhang X, Wang Y, Beanes SR, Lorenz HP, Hedrick MH, Mackool RJ, Plaas A, Kim SJ, Longaker MT, Freymiller E, Ting K: Differential expression of fibromodulin, a transforming growth factor-β modulator, in fetal skin development and scarless repair. Am J Pathol 2000, 157:423-433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Letterio JJ, Bottinger EP: TGF-β knockout and dominant-negative receptor transgenic mice. Miner Electrolyte Metab 1998, 24:161-167 [DOI] [PubMed] [Google Scholar]
- 15.Hopkinson-Woolley J, Hughes D, Gordon S, Martin P: Macrophage recruitment during limb development and wound healing in the embryonic and foetal mouse. J Cell Sci 1994, 107:1159-1167 [DOI] [PubMed] [Google Scholar]
- 16.Levine JH, Moses HL, Gold LI, Nanney LB: Spatial and temporal patterns of immunoreactive transforming growth factor β 1, β 2, and β 3 during excisional wound repair. Am J Pathol 1993, 143:368-380 [PMC free article] [PubMed] [Google Scholar]
- 17.Cowin AJ, Brosnan MP, Holmes TM, Ferguson MW: Endogenous inflammatory response to dermal wound healing in the fetal and adult mouse. Dev Dyn 1998, 212:385-393 [DOI] [PubMed] [Google Scholar]
- 18.Turley JM, Falk LA, Ruscetti FW, Kasper JJ, Francomano T, Fu T, Bang OS, Birchenall-Roberts MC: Transforming growth factor β 1 functions in monocytic differentiation of hematopoietic cells through autocrine and paracrine mechanisms. Cell Growth Differ 1996, 7:1535-1544 [PubMed] [Google Scholar]
- 19.Pan Z, Hetherington CJ, Zhang DE: CCAAT/enhancer-binding protein activates the CD14 promoter and mediates transforming growth factor β signaling in monocyte development. J Biol Chem 1999, 274:23242-23248 [DOI] [PubMed] [Google Scholar]
- 20.Lebman DA, Edmiston JS: The role of TGF-β in growth, differentiation, and maturation of B lymphocytes. Microb Infect 1999, 1:1297-1304 [DOI] [PubMed] [Google Scholar]
- 21.Plum J, De Smedt M, Leclercq G, Vandekerckhove B: Influence of TGF-β on murine thymocyte development in fetal thymus organ culture. J Immunol 1995, 154:5789-5798 [PubMed] [Google Scholar]
- 22.Kim SJ, Angel P, Lafyatis R, Hattori K, Kim KY, Sporn MB, Karin M, Roberts AB: Autoinduction of transforming growth factor β 1 is mediated by the AP-1 complex. Mol Cell Biol 1990, 10:1492-1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broker BJ, Chakrabarti R, Blynman T, Roesler J, Wang MB, Srivatsan ES: Comparison of growth factor expression in fetal and adult fibroblasts: a preliminary report. Arch Otolaryngol Head Neck Surg 1999, 125:676-680 [DOI] [PubMed] [Google Scholar]
- 24.Frank S, Madlener M, Werner S: Transforming growth factors β1, β2, and β3 and their receptors are differentially regulated during normal and impaired wound healing. J Biol Chem 1996, 271:10188-10193 [DOI] [PubMed] [Google Scholar]
- 25.Kohama K, Nonaka K, Hosokawa R, Shum L, Ohishi M: TGF-β-3 promotes scarless repair of cleft lip in mouse fetuses. J Dent Res 2002, 81:688-694 [DOI] [PubMed] [Google Scholar]
- 26.Wu L, Siddiqui A, Morris DE, Cox DA, Roth SI, Mustoe TA: Transforming growth factor β 3 (TGF β 3) accelerates wound healing without alteration of scar prominence: histologic and competitive reverse-transcription-polymerase chain reaction studies. Arch Surg 1997, 132:753-760 [DOI] [PubMed] [Google Scholar]
- 27.Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J: Abnormal lung development and cleft palate in mice lacking TGF-β 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 1995, 11:415-421 [DOI] [PubMed] [Google Scholar]
- 28.Cox DA: Transforming growth factor-β 3. Cell Biol Int 1995, 19:357-371 [DOI] [PubMed] [Google Scholar]
- 29.Lawler S, Candia AF, Ebner R, Shum L, Lopez AR, Moses HL, Wright CV, Derynck R: The murine type II TGF-β receptor has a coincident embryonic expression and binding preference for TGF-β 1. Development 1994, 120:165-175 [DOI] [PubMed] [Google Scholar]
- 30.Gold LI, Sung JJ, Siebert JW, Longaker MT: Type I (RI) and type II (RII) receptors for transforming growth factor-β isoforms are expressed subsequent to transforming growth factor-β ligands during excisional wound repair. Am J Pathol 1997, 150:209-222 [PMC free article] [PubMed] [Google Scholar]
- 31.Moses HL: TGF-β regulation of epithelial cell proliferation. Mol Reprod Dev 1992, 32:179-184 [DOI] [PubMed] [Google Scholar]
- 32.Zambruno G, Marchisio PC, Marconi A, Vaschieri C, Melchiori A, Giannetti A, De Luca M: Transforming growth factor-β 1 modulates β 1 and β 5 integrin receptors and induces the de novo expression of the α v β 6 heterodimer in normal human keratinocytes: implications for wound healing. J Cell Biol 1995, 129:853-865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ihara S, Motobayashi Y: Wound closure in foetal rat skin. Development 1992, 114:573-582 [DOI] [PubMed] [Google Scholar]
- 34.Massague J: TGF-β signal transduction. Annu Rev Biochem 1998, 67:753-791 [DOI] [PubMed] [Google Scholar]
- 35.Lin HY, Moustakas A, Knaus P, Wells RG, Henis YI, Lodish HF: The soluble exoplasmic domain of the type II transforming growth factor (TGF)-β receptor: a heterogeneously glycosylated protein with high affinity and selectivity for TGF-β ligands. J Biol Chem 1995, 270:2747-2754 [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J: Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-β receptor system. Cell 1991, 67:785-795 [DOI] [PubMed] [Google Scholar]
- 37.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI: Immunohistochemical localization of TGF β 1, TGF β 2, and TGF β 3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol 1991, 115:1091-1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts AB: Molecular and cell biology of TGF-β. Miner Electrolyte Metab 1998, 24:111-119 [DOI] [PubMed] [Google Scholar]
- 39.Hedbom E, Heinegard D: Interaction of a 59-kDa connective tissue matrix protein with collagen I and collagen II. J Biol Chem 1989, 264:6898-6905 [PubMed] [Google Scholar]
- 40.Font B, Eichenberger D, Goldschmidt D, Boutillon MM, Hulmes DJ: Structural requirements for fibromodulin binding to collagen and the control of type I collagen fibrillogenesis: critical roles for disulphide bonding and the C-terminal region. Eur J Biochem 1998, 254:580-587 [DOI] [PubMed] [Google Scholar]
- 41.Hildebrand A, Romaris M, Rasmussen LM, Heinegard D, Twardzik DR, Border WA, Ruoslahti E: Interaction of the small interstitial proteoglycans biglycan, decorin, and fibromodulin with transforming growth factor β. Biochem J 1994, 302:527-534 [DOI] [PMC free article] [PubMed] [Google Scholar]