

Abstract
Granulocyte macrophage-colony stimulating factor (GM-CSF) plays an important role in pulmonary homeostasis, with effects on both alveolar macrophages and alveolar epithelial cells. We hypothesized that overexpression of GM-CSF in the lung would protect mice from hyperoxic lung injury by limiting alveolar epithelial cell injury. Wild-type C57BL/6 mice and mutant mice in which GM-CSF was overexpressed in the lung under control of the SP-C promoter (SP-C-GM mice) were placed in >95% oxygen. Within 6 days, 100% of the wild-type mice had died, while 70% of the SP-C-GM mice remained alive after 10 days in hyperoxia. Histological assessment of the lungs at day 4 revealed less disruption of the alveolar wall in SP-C-GM mice compared to wild-type mice. The concentration of albumin in bronchoalveolar lavage fluid after 4 days in hyperoxia was significantly lower in SP-C-GM mice than in wild-type mice, indicating preservation of alveolar epithelial barrier properties in the SP-C-GM mice. Alveolar fluid clearance was preserved in SP-C-GM mice in hyperoxia, but decreased significantly in hyperoxia-exposed wild-type mice. Staining of lung tissue for caspase 3 demonstrated increased apoptosis in alveolar wall cells in wild-type mice in hyperoxia compared to mice in room air. In contrast, SP-C-GM mice exposed to hyperoxia demonstrated only modest increase in alveolar wall apoptosis compared to room air. Systemic treatment with GM-CSF (9 μg/kg/day) during 4 days of hyperoxic exposure resulted in decreased apoptosis in the lungs compared to placebo. In studies using isolated murine type II alveolar epithelial cells, treatment with GM-CSF greatly reduced apoptosis in response to suspension culture. In conclusion, overexpression of GM-CSF enhances survival of mice in hyperoxia; this effect may be explained by preservation of alveolar epithelial barrier function and fluid clearance, at least in part because of reduction in hyperoxia-induced apoptosis of cells in the alveolar wall.
Exposure of mammals to high concentrations of oxygen for prolonged periods results in noncardiogenic pulmonary edema, acute lung injury, and eventually, death. Hyperoxia-induced lung injury has become a well-recognized animal model for studies of the pathophysiology of acute lung injury. Studies of the mechanisms of hyperoxic lung injury have focused primarily on two cells. Early studies emphasized the role of pulmonary capillary endothelial cells. 1 After early endothelial cell injury, there is accumulation of inflammatory cells within the vascular space, and leak of protein-rich fluid into the interstitium. More recent studies have focused on the role of alveolar epithelial cells in these models. The alveolar epithelium provides a very tight barrier to fluid entry into the alveolar space. 2 Alveolar epithelial cells also actively remove salt and water from the distal airspaces to maintain gas exchange. 3 After prolonged hyperoxic stress alveolar epithelial cell function is severely impaired, with decreased barrier function, 4 diminished active fluid reabsorption capacity and Na/K ATPase activity, 5 and altered surfactant protein and lipid accumulation. 4,6,7 It is likely that both necrosis and apoptosis of alveolar wall cells contribute to hyperoxic lung injury. 8
The pathophysiological mechanisms involved in oxygen-induced lung injury are complex. 1 A variety of experimental manipulations can influence the outcome ofextended hyperoxia in animal models. Maneuvers that directly increase pulmonary antioxidant defenses 9-11 are protective; manipulations of pulmonary inflammation, including pretreatment with lipopolysaccharide, 12 tumor necrosis factor, 13 or interleukin (IL)-1, 14 or transgenic expression of IL-11 15 or IL-13 16 can protect rodents from death because of extended hyperoxia. Growth factors for alveolar epithelial cells 17 and pulmonary endothelial cells 16 also can render mice tolerant of levels of oxygen that are normally lethal.
Granulocyte macrophage-colony stimulating factor (GM-CSF) is a potent growth factor originally recognized for its effects on survival, proliferation, maturation, and differentiation of hematopoietic cells. It is now clear that pulmonary GM-CSF plays a central role in homeostasis in the normal lung. Gene-targeted mice deficient in GM-CSF (GM-CSF−/− mice) develop alveolar proteinosis 18 because of abnormal surfactant turnover. 19 These mice demonstrate increased susceptibility to pneumonia with bacteria 20 and fungi 21 because of impaired alveolar macrophage function. Expression of GM-CSF exclusively in the lungs of GM-CSF−/− mice is sufficient to restore normal surfactant metabolism and normal host defense function. 20-25
In addition to its predicted effects on alveolar macrophages, GM-CSF is a potent growth factor for alveolar epithelial cells. In vitro, GM-CSF is a mitogen for rat 26 or murine (unpublished observations) alveolar epithelial cells in primary culture. The present study was undertaken to determine whether overexpression of GM-CSF in the lung would impact the response of mice to hyperoxia. We now report that SP-C-GM mice are relatively tolerant of extended hyperoxia that is lethal to wild-type mice. This enhanced survival is a consequence of diminished apoptosis and preservation of alveolar epithelial cell function in the face of hyperoxic stress.
Materials and Methods
Animals
Wild-type C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). Bitransgenic mice in which GM-CSF is expressed exclusively in the lungs, were generated from GM-CSF-deficient mice by transgenic expression of a chimeric gene containing GM-CSF under the SP-C promoter (SP-C-GM mice). 22 The specificity of the SP-C promoter results in targeted expression of GM-CSF exclusively by type II alveolar epithelial cells of these SP-C-GM mice. Founder SP-C-GM mice were kindly provided by Dr. J Whitsett (Children’s Hospital, Cincinnati, OH). Although GM-CSF is not detectable by enzyme-linked immunosorbent assay (ELISA) in bronchoalveolar lavage (BAL) fluid of normal wild-type mice, BAL fluid GM-CSF concentration is >100 pg/ml in the SP-C-GM mice. 21 All mice were housed in microisolator cages under laminar flow hoods in an isolation room of the Animal Care Facilities at the University of Michigan and the Ann Arbor Veterans Affairs Medical Center. Mice were supplied with autoclaved bedding, food, and water. The Animal Care Committees at the University of Michigan and the Ann Arbor Veterans Affairs Medical Center approved all procedures.
In Vivo Exposure of Mice to Hyperoxia and Treatment with rmGM-CSF
For exposure to hyperoxia, mice in microisolator cages were placed in a Plexiglas chamber (Reming BioInstruments Inc., Redfield, NY). Mice continued to receive food and water ad libitum. Oxygen from an H cylinder was administered into the chamber under control of a Pro-ox device (Reming BioInstruments Inc.) that continuously measured oxygen tension and adjusted flow to maintain a steady-state fraction of oxygen of >95%. In selected experiments mice were treated with rmGM-CSF (9 μg/kg/day; R&D Systems, Minneapolis, MN) or an equal volume of mouse serum, given by subcutaneous injection. Mice were treated daily for 4 days during exposure to hyperoxia.
BAL Fluid Analysis
Mice were euthanized with pentobarbital and the lungs perfused via the right ventricle until the effluent was free of blood. The trachea was cannulated and the lungs were lavaged with a total of 3 ml of phosphate-buffered saline (PBS) in aliquots of 0.5 ml. The lavage aliquots for each mouse were pooled and the cell-free supernatant was collected after centrifugation (400 × g). The concentration of murine albumin in the BAL fluid was determined by ELISA (Bethyl Laboratories, Montgomery, TX).
Alveolar Fluid Resorption
The rate of alveolar fluid clearance was determined from the change throughout time in concentration of radiolabeled albumin that had been instilled into the distal airspaces via the trachea, using standard methods adapted for mice. 27,28 Baseline alveolar fluid clearance was determined in SP-C-GM and wild-type mice. Alveolar fluid clearance was also measured in both groups of mice after 80 hours of exposure to hyperoxia.
Preparation of Instillate
The instillate consisted of 5 g/100 ml bovine serum albumin (Sigma Chemical Co., St. Louis, MO) in Ringer’s lactate adjusted to 330 mOsm/kg H2O with NaCl to be isosmolar with mouse plasma, and 0.1 μCi of 131I-labeled albumin (Merck-Frost, Montreal, Canada) as the labeled alveolar protein tracer.
Surgical Preparation
Mice were euthanized by an overdose of pentobarbital sodium (200 mg/kg i.p.). The trachea was dissected and cannulated with a 20-gauge, trimmed Angiocath plastic needle (Becton Dickinson, Sandy, UT). The lungs were kept inflated with 5 cmH2O of continuous positive airway pressure and oxygenated with 100% oxygen throughout the experiment. The body temperature was maintained at 37 to 38°C with an external lamp, as in previous studies.
General Protocol
In all studies, 13 ml/kg of the instillate was delivered throughout 30 seconds into both lungs through the tracheal cannula. After 2 and 30 minutes, an alveolar fluid sample (0.05 to 0.10 ml) was aspirated with a 1-ml syringe directly connected to the 20-gauge angiocath. The aspirate was weighed and the radioactivity measured in a γ-counter. Alveolar fluid clearance (percent of instilled fluid volume) was calculated by measuring the increase in tracer-labeled albumin (131I-albumin) concentration in the instilled solution. Because the initial volume of the instilled solution and the initial and final radioactivity of the samples were known, alveolar fluid clearance could be determined by using the following mass-balance equation: alveolar fluid clearance = (1 − radioactivity in the instilled sample/radioactivity in the final sample) × 100, where alveolar fluid clearance is expressed as mean ± SD percentage of the initial volume of instillate that was cleared from the distal air spaces during the 30 minutes. Using this same protocol, pulmonary edema induced by hyperoxia-mediated acute lung injury was also quantified by calculating the decrease of the radioactivity (because of dilution) from the initial instillate in the sample collected 2 minutes after the instillation.
Lung Histology and Staining for Caspase 3
At appropriate time points, mice were euthanized with pentobarbital and the lungs perfused via the right ventricle until the effluent was free of blood. For selected photomicrographs, lungs were fixed in 2% glutaraldehyde, dehydrated in ethanol, and infiltrated and embedded in Polybed resin (Ernest F. Fullan, Inc., Schenectady, NY). Semithin (1 μm) plastic sections were prepared and stained with toluidine blue stain. For histological scoring of lung injury, the lungs were removed and inflated first with air, then with neutral buffered formalin. Paraffin-embedded tissue blocks were sectioned and stained with hematoxylin and eosin. The extent of histological injury was scored on a scale of 0, no injury; 1, minimal abnormality on searching; 2, mild alveolar wall edema, few red cells; 3, severe edema of the alveolar wall, mild alveolar exudates, red cells in the alveolar space; or 4, extensive alveolar exudates, obvious alveolar wall disruption. Sections were scored by an observer blinded to the identity of the sections. Injury scores are presented as median values. Staining for activated caspase 3 to detect apoptotic cells was performed according to the manufacturer’s protocol (R&D Systems) on deparaffinized lung sections that had been fixed in paraformaldehyde (4%).
Measurement of Relative Apoptosis and of Vascular Endothelial Growth Factor (VEGF) in Lung Homogenates
Mice were euthanized and the pulmonary circulation perfused with saline until the effluent was free of blood. The lungs were removed from the proximal airways with sharp scissors and homogenized using a Tissue Tearor (Biospec Products, Inc.). After removal of debris by low-speed centrifugation, histone-associated DNA was determined by ELISA (Cell Death ELISA; Roche, Indianapolis, IN) as a measure of apoptosis. VEGF protein in the lung homogenate was measured by ELISA (R&D Systems).
Isolation and Culture of Murine Alveolar Epithelial Cells
Murine type II alveolar epithelial cells were isolated by the method of Corti and colleagues. 29 Mice were sedated with pentobarbital, secured to a dissecting board and exsanguinated by cutting the inferior vena cava. The anterior thoracic wall was removed and the left ventricle was cut with sharp scissors. The pulmonary vasculature was perfused with normal saline via a direct right ventricular puncture until the lungs were visually free of blood. The trachea was cannulated with a 20-gauge intravenous catheter secured with a suture. The lungs were filled with 1 to 2 ml of Dispase (Worthington Biochemical Corp., Lakewood, NJ) via a syringe connected to the tracheal catheter. Low-melt agarose (1%, 0.45 ml prewarmed to 45°C) was infused via the tracheal catheter and the lungs were suspended in ice-cold PBS for 2 minutes to allow the agarose to harden in the airways. The lungs then were immersed in Dispase (2 ml) at room temperature for 45 minutes. The lungs then were chilled again briefly in ice-cold PBS and transferred to a sterile Petri dish containing Dulbecco’s modified Eagle’s medium with 0.01% DNase. Using forceps, lung tissue was teased away from the airways, which were removed. The media and lung tissue were transferred to a trypsinizing flask and gently stirred with a magnetic stir bar for 10 minutes. The suspension was filtered successively through 100-, 40-, and 25-μm nylon mesh filters to create a single cell suspension. The cells were collected by centrifugation, counted, and resuspended in Dulbecco’s modified Eagle’s medium with biotinylated anti-CD-32 (FcγR) (0.65 μg/million cells) and anti-CD-45 (common leukocyte antigen) (1.5 μg/million cells). After incubation at 37°C for 30 minutes, the cells were pelleted, counted, resuspended in 7 ml of Dulbecco’s modified Eagle’s medium, and added to prewashed streptavidin-coated magnetic particles. The mixture was incubated for 30 minutes at room temperature with gentle rocking. The tube then was attached to a magnetic separator for 15 minutes to remove bone marrow-derived cells. Cells not bound with magnetic particles were recovered from the tube with a glass pipette, pelleted, and suspended in culture media. Viability was >97% by trypan blue exclusion. The cells were plated overnight in 60-mm culture plates. The nonadherent cells, including type II alveolar epithelial cells, were recovered and counted. Viability was >97% by trypan blue exclusion. These cells were >95% vimentin-negative. The percentage of vimentin-positive cells is similar to what we have achieved using a widely accepted isolation of rat type II cells described by Dobbs and colleagues. 30 For suspension culture, cells were suspended in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, penicillin, and streptomycin, and cultured in Teflon containers at 1 × 106 cells/ml at 37°C in an atmosphere of 7% CO2 in air.
Statistical Methods
Survival data are compared using chi-square analysis. In other experiments, data are expressed as mean ± SEM and compared by t-test (two groups), or by analysis of variance with the Neuman-Keuls multiple range test (more than two groups). Ordinal data are evaluated by the Kruskal-Wallis test. All tests are performed using the InStat software program (version 3.01; GraphPad Software, San Diego, CA). Data are considered statistically significant if P values are <0.05.
Results
Survival of Wild-Type and SP-C-GM Mice in Hyperoxia
When wild-type C57BL/6 mice were placed in an atmosphere of >95% oxygen, 100% of the mice had died by day 6 in hyperoxia (Figure 1) ▶ . Mice surviving beyond day 3 were clearly impaired, with limited movement, hunched backs, and burrowing behavior. In contrast, SP-C-GM mice demonstrated remarkable tolerance to hyperoxia. All of the SP-C-GM mice remained alive after 6 days in >95% oxygen, when control mice were all dead. After 10 days in hyperoxia, 70% of the SP-C-GM mice remained alive. The survivors were active, moving about the cages in no apparent distress, and continuing to take food and water. Thus, transgenic overexpression of GM-CSF in the alveolar space resulted in significantly enhanced tolerance of hyperoxic stress.
Figure 1.

Prolonged survival of SP-C-GM mice in hyperoxia. Wild-type (C57BL/6) mice and SP-C-GM mice overexpressing GM-CSF in the lungs were placed in an atmosphere of >95% oxygen. Survival was determined throughout 10 days. n = 15 mice in each group.
Pulmonary Histology of Wild-Type and SP-C-GM Mice Exposed to Hyperoxia
Photomicrographs of semithin sections of representative lungs from wild-type and SP-C-GM mice after 4 days in hyperoxia are shown in Figure 2 ▶ . As described previously by Huffman Reed and colleagues, 26 at baseline, the SP-C-GM mice had increased numbers of alveolar macrophages, and qualitatively increased numbers of type II alveolar epithelial cells compared to wild-type mice. After 4 days in hyperoxia wild-type mice developed modest alveolar exudate and some blebbing of cells along the alveolar wall, best seen under high magnification (Figure 2C) ▶ . These changes were less evident in SP-C-GM mice in hyperoxia (Figure 2D) ▶ . To provide a semiquantitative assessment of histological evidence of lung injury, paraffin sections were scored for parameters of lung injury. The median score for mice of both strains in normoxia was 0. Histological measures of injury after hyperoxia were relatively modest in wild-type mice, and were significantly reduced in SP-C-GM mice (Figure 2E) ▶ . Thus, overexpression of GM-CSF resulted in decreased histological evidence of lung injury after exposure to hyperoxia.
Figure 2.

Histological appearance of lungs after 4 days in hyperoxia. After 4 days in an atmosphere of >95% oxygen mice were euthanized and semithin sections of lung were prepared and stained with toluidine blue for light microscopy. A and C: Representative sections of lungs of wild-type mice. B and D: Representative sections from SP-C-GM mice. E: The extent of histological injury in the lungs of wild-type (cross-hatched bar) and SP-C-GM (solid bar) mice that had been in hyperoxia for 4 days was scored on paraffin-embedded sections, as described in Materials and Methods, on a scale of 0 to 4 by two observers blinded to the identity of the sections. The median score for mice of both strains in normoxia was 0. Ordinal data are presented as median values. *, P < 0.05 versus wild type by Student’s t-test. Original magnifications: ×40 (A and B); ×100 (C and D).
Leak of Albumin into the Alveolar Space after Exposure to Hyperoxia
Murine albumin was measured in BAL fluid from wild-type and SP-C-GM mice in normoxia and after exposure to hyperoxia (Figure 3) ▶ . In wild-type mice, albumin in the BAL fluid, as an indication of increased protein permeability across the alveolar epithelium, was significantly increased after 4 days in hyperoxia. In contrast, BAL fluid albumin remained near normoxic levels in SP-C-GM mice exposed to hyperoxia. Thus, hyperoxia resulted in noncardiogenic pulmonary edema in wild-type mice, while SP-C-GM mice were protected from protein leak across the alveolar wall in response to this stress.
Figure 3.

Effect of overexpression of GM-CSF on BAL fluid albumin concentration in hyperoxia. Wild-type and SP-C-GM mice were placed in an atmosphere of >95% oxygen. After 2 or 4 days the concentration of murine albumin in BAL fluid was determined by ELISA. Cross-hatched bars, wild-type mice; solid bars, SP-C-GM mice. *, P < 0.05 versus wild type; n = 7 in each group.
Alveolar Fluid Clearance after Exposure to Hyperoxia
Having assessed the impact of overexpression of GM-CSF on protein permeability across the alveolar wall after exposure to hyperoxia, experiments were then performed to determine the effect of GM-CSF on the clearance of fluid from the distal airspaces of the lung. In normoxia, alveolar fluid clearance was not different in SP-C-GM mice compared to wild-type controls (Table 1) ▶ . After exposure to hyperoxia two differences were apparent. First, during the initial 2 minutes, when the tracer in the instillate was equilibrating with alveolar fluid, the tracer concentration decreased by 22 ± 13% in wild-type mice versus 1 ± 2% decrease in SP-C-GM mice (P < 0.002). The dilution of the tracer after instillation in the wild-type mice indicates that there was alveolar edema in the wild-type mice. In contrast, the lack of dilution in the SP-C-GM mice indicates that they were protected against the development of distal airspace edema. Second, in wild-type mice there was no net fluid clearance (zero of eight mice with positive clearance). In contrast, six of eight SP-C-GM mice demonstrated positive fluid clearance, providing evidence of submaximal, but net alveolar fluid clearance. Thus, overexpression of GM-CSF in the lung had no impact on baseline fluid resorption, but prevented alveolar edema and resulted in preservation of net alveolar fluid clearance in the face of the hyperoxic stress.
Table 1.
Net Alveolar Fluid Clearance in Wild-Type and SP-C-GM Mice
| Wild-type mice | SP-C-GM mice | |
|---|---|---|
| Normoxia | 14.6 ± 2.0 | 12.6 ± 3.0 |
| Hyperoxia | 0 | 5.2 ± 2.7* |
Alveolar fluid clearance was determined in mice in normoxia and after 80 hours of hyperoxia. Data are presented as mean percent fluid clearance per hour ± SD. There was no net clearance in wild-type mice in hyperoxia. Clearance in SP-C-GM mice in hyperoxia was calculated based on the mice that retailed positive fluid clearance (see text).
*P < 0.005.
Lung VEGF Expression in Wild-Type and SP-C-GM Mice
GM-CSF has been found to induce expression of VEGF in eosinophils in vitro. Furthermore, enhanced VEGF expression has been proposed as a mechanism of protection against hyperoxic injury in transgenic mice overexpressing IL-13. 16 Therefore, the expression of VEGF protein was measured in lung homogenates of wild-type and SP-C-GM mice in normoxia and after 4 days in hyperoxia. Overexpression of GM-CSF failed to augment VEGF expression in the lung in either atmosphere (data not shown), indicating that protection of SP-C-GM mice against hyperoxia is not mediated through effects on VEGF.
Apoptosis of Lung Cells after Exposure to Hyperoxia
Previous studies have suggested that apoptosis of cellular constituents of the alveolar wall may contribute to acute lung injury in response to hyperoxic exposure. 31 To provide a quantitative measure of relative lung apoptosis after hyperoxic stress, lungs from SP-C-GM mice and wild-type mice in normoxia or in hyperoxia for 4 days were harvested, and histone-associated DNA was measured on lung homogenates by ELISA. In wild-type mice, apoptosis in the lung increased significantly after hyperoxia (Figure 4) ▶ . In contrast, the relative quantity of histone-associated DNA did not increase significantly in SP-C-GM mice after exposure to hyperoxia. To determine whether overexpression of GM-CSF in the lung influenced hyperoxia-induced apoptosis of cells within the alveolar wall, lung sections were stained for caspase 3. Four days in hyperoxia resulted in increased numbers of caspase 3-positive cells in the alveolar wall of wild-type mice compared to mice in normoxia (Figure 5) ▶ . However, SP-C-GM mice exposed to hyperoxia demonstrated only modestly increased apoptosis in the alveolar wall compared to SP-C-GM mice in normoxia (Figure 5) ▶ . Together, these data indicate that hyperoxia-induced apoptosis was diminished significantly by overexpression of GM-CSF in the lung.
Figure 4.

Effect of overexpression of GM-CSF in the lung on lung apoptosis after hyperoxia. Lung homogenates were prepared from wild-type and SP-C-GM mice after 4 days in hyperoxia. The relative content of histone-associated DNA was determined using a Cell Death ELISA (Roche). Data are expressed as mean absorbance ± SEM. Cross-hatched bars, wild-type mice; solid bars, SP-C-GM mice. *, P < 0.05 versus wild type/hyperoxia; **, P < 0.05 versus wild type/normoxia, by analysis of variance. There is no difference between the wild-type/normoxia and SP-C-GM/hyperoxia groups (P > 0.1).
Figure 5.

Effect of overexpression of GM-CSF in the lung on apoptosis of cells in the alveolar wall after hyperoxia. Lung sections from wild-type and SP-C-GM mice in an atmosphere of >95% oxygen for 4 days were stained for activated caspase 3 as an indicator of apoptosis. Caspase 3+ cells appear brown in this procedure. A: Wild-type mice in normoxia; B: SP-C-GM mice in normoxia; C: wild-type mice in hyperoxia; D: SP-C-GM mice in hyperoxia. Original magnifications, ×100.
Effect of Acute Treatment with GM-CSF on Apoptosis in the Lung after Hyperoxia
Our studies in transgenic mice have demonstrated that prolonged exposure to supraphysiological concentrations of GM-CSF in the lung results in increased tolerance of hyperoxic stress compared to control mice. To determine whether acute treatment with GM-CSF would be sufficient to influence the response to hyperoxia, wild-type mice were inoculated subcutaneously with GM-CSF (9 μg/kg/day) daily, beginning with the day when the mice were placed in hyperoxia. Control mice received an identical volume of mouse serum. After 4 days, the mice were euthanized. Histone-associated DNA was measured in lung homogenates, while lung sections were stained for caspase 3. Treatment with GM-CSF significantly decreased relative apoptosis in lung homogenates (Figure 6) ▶ . Caspase 3 staining indicated that this change was a reflection of decreased numbers of apoptotic cells in the alveolar wall of hyperoxia-exposed mice treated with GM-CSF (Figure 7) ▶ . Thus, these results indicate that even short-term treatment with GM-CSF is sufficient to alter the response of the lung to hyperoxic stress.
Figure 6.

Effect of treatment with GM-CSF on lung apoptosis in response to hyperoxia. Wild-type (C57BL/6) mice were placed in 95% oxygen. One group of mice received recombinant murine GM-CSF (9 μg/kg/day) by subcutaneous injection daily, starting on the day of entry into hyperoxia (solid bar). The other group received placebo (cross-hatched bar). After 4 days, the relative content of histone-associated DNA in lung homogenates was determined by ELISA. Data are expressed as mean absorbance ± SEM. *, P < 0.05.
Figure 7.
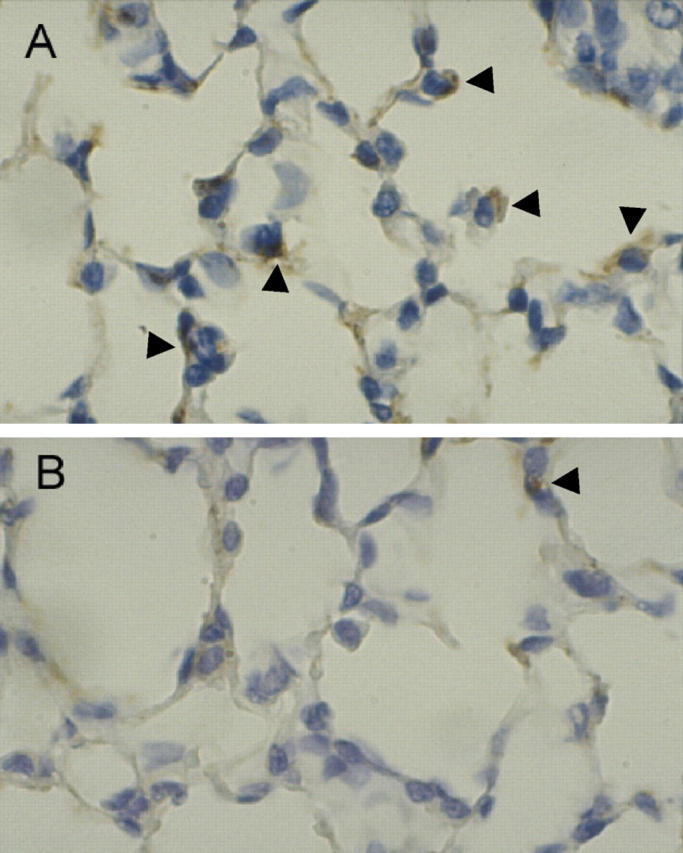
Effect of treatment with GM-CSF on apoptosis of cells in the alveolar wall after hyperoxia. C57BL/6 mice were placed in an atmosphere of >95% oxygen. Mice received either placebo (A) or rmGM-CSF (B; 9 μg/kg/day) daily by subcutaneous injection, beginning at the time of entry into hyperoxia. After 4 days, lung sections were stained for caspase 3 as an indicator of apoptosis. Caspase 3+ cells appear brown in this procedure (see arrowheads for examples). Original magnifications, ×100.
Effect of GM-CSF on Apoptosis of Murine Alveolar Epithelial Cells in Vitro
Having determined that treatment with GM-CSF resulted in reduced apoptosis of alveolar wall cells in response to hyperoxia, we determined that GM-CSF could directly protect murine alveolar epithelial cells from apoptosis. Suspension culture of cells that normally are adherent to a basement membrane is a well-recognized stress leading to apoptosis. 32 We confirmed that suspension culture induced apoptosis (anoikis) in highly purified cultures of murine type II cells. When GM-CSF (10 ng/ml) was added to the culture medium, apoptosis, as measured by ELISA for histone-associated DNA, was significantly reduced (Figure 8) ▶ . The proportion of cells staining positively for activated caspase 3 was similarly decreased (control, 61.7 ± 2.5%; GM-CSF treated, 33.2 ± 6.2%). This result suggests that the protection by GM-CSF against alveolar cell death in response to hyperoxia is at least in part a direct effect of GM-CSF on alveolar epithelial cells.
Figure 8.

Effect of in vitro treatment with GM-CSF on apoptosis of murine type II alveolar epithelial cells in suspension culture. Type II alveolar epithelial cells were isolated from wild-type mice and cultured in control medium or in medium supplemented with GM-CSF (10 ng/ml). After 48 hours, relative apoptosis was determined by measuring histone-associated DNA as described in Materials and Methods. Data are expressed as mean ± SEM from three independent experiments. *, P < 0.05.
Discussion
The major finding reported in this study is that overexpression of GM-CSF in the lungs of transgenic mice results in impressive resistance to acute lung injury caused by hyperoxia. This protective effect involves preservation of normal alveolar barrier function, ie, preservation of normal alveolar epithelial impermeability to protein, and preservation of submaximal alveolar fluid transport. Alveolar cell apoptosis induced by hyperoxia is significantly reduced in GM-CSF transgenic mice and by acute treatment with rmGM-CSF. Finally, in vitro treatment with GM-CSF resulted in decreased apoptosis of murine alveolar epithelial cells in suspension culture.
GM-CSF is a pleotropic cytokine that was originally identified from murine lung. It has been best known for its wide-ranging effects on bone marrow-derived cells. GM-CSF induces proliferation and maturation of mononuclear phagocytes and inhibits apoptosis of both mononuclear and polymorphonuclear leukocytes. More recently, it has been found that GM-CSF has significant effects on alveolar epithelial cells. GM-CSF induces proliferation in rat type II cells in vitro. 26 The effects of GM-CSF on alveolar epithelial cell differentiation and functional characteristics have not yet been determined.
Studies using mutant mice deficient in GM-CSF or which express GM-CSF exclusively in the alveolar space have provided considerable insights into the role of GM-CSF in the lung. Mutant mice lacking GM-CSF develop a pathological pattern closely resembling human alveolar proteinosis. 18 Surfactant phospholipids and proteins accumulate in the alveolar space. Surfactant production is similar to that in wild-type mice, but turnover is greatly impaired because of decreased alveolar macrophage uptake and degradation of surfactant. 19 Further examination demonstrates that alveolar macrophage function is globally impaired, with diminished phagocytosis and killing of microbes, impaired tumor necrosis factor production, and altered integrin expression. 24,25 Not surprisingly, these defects in pulmonary innate immunity result in increased susceptibility to pneumonia because of a variety of pathogens, including group B streptococcus, 20 Pneumocystis carinii, 21 and Klebsiella pneumoniae (unpublished observations). Targeted expression of GM-CSF exclusively in the alveolar space restores normal surfactant turnover 23 and alveolar macrophage functional activities for host defense. 24,25 Together these studies indicate that GM-CSF plays a critical role in pulmonary homeostasis. Our study is the first to demonstrate that GM-CSF can also protect the lung from acute injury that leads to respiratory failure and death.
High levels of inspired oxygen are injurious to the mammalian lung. 1 Normobaric hyperoxia results in acute lung injury and, ultimately, death in rats and mice. It is likely that observations from these models can be extended to humans, although issues of dose and duration remain unclear. The pathophysiological mechanisms involved in oxygen-induced lung injury are complex. 1 Hyperoxic stress results in damage to both pulmonary capillary endothelial cells and type I alveolar epithelial cells. These cells eventually undergo injury that is likely to involve both necrosis and apoptosis. 31 Hyperoxia leads to impaired alveolar epithelial cell function, with decreased barrier function, 4 diminished fluid reabsorption and Na/K ATPase activity, 5 and altered surfactant protein and lipid accumulation. 4,6,7
A variety of manipulations can influence the outcome of extended hyperoxia in animal models. Maneuvers that directly increase pulmonary antioxidant defenses lead to increased tolerance of hyperoxia. 9-11 Specific growth factors that induce proliferation of either alveolar epithelial cells or pulmonary endothelial cells can induce tolerance of hyperoxic stress. Thus, the administration of the alveolar epithelial cell mitogen, keratinocyte growth factor to support epithelial cell proliferation 17 or transgenic expression of IL-13, leading to induction of the endothelial cell mitogen, VEGF 16 both provide protection against oxygen-induced lung injury. Augmentation of alveolar fluid clearance by adenovirally mediated gene transfer to the lung of the gene for the β1 subunit of Na+/K+-ATPase limits hyperoxia-induced pulmonary edema and also results in prolonged survival of rats in hyperoxia. 33 Finally, manipulations of pulmonary inflammation, including pretreatment with lipopolysaccharide, 12 tumor necrosis factor, 13 or IL-1, 14 or transgenic expression of IL-11 15 or IL-13 16 can protect rodents from death because of extended hyperoxia.
The results of these studies suggest several mechanisms by which mice overexpressing GM-CSF in the lung may be protected from hyperoxia-induced lung injury. The barrier function of the alveolar wall for protein flux is disrupted in wild-type mice exposed to hyperoxia, but not in SP-C-GM mice. Both endothelial cells and epithelial cells contribute to this barrier function, although in the normal lung, the alveolar epithelium provides the tighter barrier to the flux of proteins across the alveolar wall. 2 Maintenance of low BAL fluid albumin despite hyperoxia suggests that alveolar epithelial injury has been significantly limited by the overexpression of GM-CSF. Measurements of intact alveolar fluid clearance from lungs after hyperoxia provide additional confirmation that alveolar epithelial cell function is preserved in SP-C-GM mice, but significantly disrupted in wild-type mice. However, normoxic alveolar fluid clearance was the same in wild-type and SP-C-GM mice. Thus, these results indicate that GM-CSF-induced tolerance of hyperoxia is a result of epithelial cell resistance to hyperoxic injury rather than a consequence of alterations in the basic alveolar cell transport capacity.
Apoptosis in the lung was increased in wild-type mice after 4 days in hyperoxia, and this increase was inhibited in SP-C-GM mice. Caspase 3 staining indicates that apoptosis of alveolar wall cells was induced by hyperoxia and decreased in turn in SP-C-GM mice in hyperoxia compared to similarly stressed wild-type mice. Although it is possible that GM-CSF overexpression would lead to increased VEGF 34 and subsequent endothelial cell protection, VEGF protein in whole lung homogenates was not increased in SP-C-GM mice compared to wild-type mice. Based on in vitro findings, it is possible that the tolerance of hyperoxia is a consequence of direct effects of GM-CSF on alveolar epithelial cell survival.
Interestingly, acute treatment with GM-CSF decreased lung cell apoptosis in response to hyperoxia. In transgenic mice overexpressing GM-CSF in the lungs from birth, lung cells are chronically exposed to high concentrations of this growth factor. This chronic exposure to an epithelial cell mitogen can induce pulmonary hyperplasia, 26 which might contribute to the tolerance of hyperoxia. However, we have found that short-term treatment of wild-type mice with GM-CSF is sufficient to alter alveolar cell apoptosis after hyperoxia in vivo. Furthermore, treatment with GM-CSF protected alveolar epithelial cells from apoptosis induced during suspension culture in vitro. This result confirms that GM-CSF can influence the behavior of normal alveolar cells in response to stress, beyond any long-term effects of GM-CSF on lung growth. This observation supports the hypothesis that GM-CSF may have a potential role as a therapeutic agent in clinical lung injury, as suggested by a recent clinic trial in patients with sepsis. 35
The mechanisms by which GM-CSF limits alveolar epithelial cell apoptosis are not yet defined. Studies using a variety of cell lines have determined that hyperoxia-induced apoptosis is regulated by members of the Bcl-2 family, does not depend on mitochondrial reactive oxygen species, and can be inhibited by overexpression of the anti-apoptotic molecule Bcl-XL. 36 Studies in macrophages and neutrophils have shown that one of the mechanisms by which GM-CSF can protect against apoptosis involves activation of the transcription factor Akt. 37,38 Furthermore, adenoviral transfer of an active form of Akt to the lung is sufficient to induce relative tolerance of mice for hyperoxia. 39 However, we have not found that GM-CSF induces Akt activation in murine alveolar epithelial cells in vitro (data not shown). Thus, the mechanism by which GM-CSF influences alveolar epithelial cell apoptosis is the subject of ongoing investigation.
Observational data from patients with adult respiratory distress syndrome also support the hypothesis that GM-CSF provides protection for the alveolar space after acute lung injury. BAL fluid obtained during the first 3 days after patients had met criteria for adult respiratory distress syndrome contained significant amounts of GM-CSF. Most importantly, increased levels of GM-CSF were predictive of survival. 40 Whether increased GM-CSF is a marker of decreased alveolar epithelial cell injury or is a cause of decreased epithelial cell loss awaits further experiments. The data in these mouse lung injury studies indicate that GM-CSF may play a direct causal role.
In summary, overexpression of GM-CSF in the murine lung results in resistance to lethal acute lung injury because of hyperoxia. This protective effect is associated with preservation of alveolar barrier function and alveolar fluid transport capacity. The induction of pulmonary cell apoptosis by hyperoxia is greatly limited in SP-C-GM mice, suggesting that limitation of alveolar epithelial cell death induced by hyperoxia may be an important mechanism for contributing to maintenance of normal physiology and enhanced survival. These data suggest the potential for a novel therapy with GM-CSF in patients with acute lung injury.
Acknowledgments
We thank Jeffrey Whitsett and Jacqueline Reed (University of Cincinnati) for the SP-C-GM mice, Dr. Hedwig Murphy and the Pathology Department (Department of Veterans Affairs Medical Center, Ann Arbor) for technical assistance in processing lungs for semithin sections; and the other REAP investigators for useful discussions.
Footnotes
Address reprint requests to Robert Paine III, M.D., Pulmonary Section (111G), VAMC, 2215 Fuller Rd., Ann Arbor, MI 48105. E-mail: rpaine@umich.edu.
Supported by grants from the National Heart, Lung, and Blood Institute (HL64558 to R. P.; HL51856 and HL51854 to M. A. M.) and the Department of Veteran Affairs (VA Merit Review to R. P. and P. J. C.).
R. P. and P. J. C. are investigators in the Department of Veterans Affairs Research Enhancement Award Program, Pulmonary Section, Department of Veterans Affairs Medical Center, Ann Arbor.
References
- 1.Folz R, Piantadosi C, Crapo J: Oxygen toxicity. Crystal R West J Barnes P Weibel E eds. The Lung: Scientific Foundations. 1997:pp 2713-2722 Lippincott-Raven, Philadelphia
- 2.Gorin AB, Stewart PA: Differential permeability of endothelial and epithelial barriers to albumin flux. J Appl Physiol Respir Environ Exer Physiol 1979, 47:1315-1324 [DOI] [PubMed] [Google Scholar]
- 3.Matthay MA, Folkesson HG, Clerici C: Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev 2002, 82:569-600 [DOI] [PubMed] [Google Scholar]
- 4.Holm BA, Notter RH, Siegle J, Matalon S: Pulmonary physiological and surfactant changes during injury and recovery from hyperoxia. J Appl Physiol 1985, 59:1402-1409 [DOI] [PubMed] [Google Scholar]
- 5.Olivera WG, Ridge KM, Sznajder JI: Lung liquid clearance and Na,K-ATPase during acute hyperoxia and recovery in rats. Am J Respir Crit Care Med 1995, 152:1229-1234 [DOI] [PubMed] [Google Scholar]
- 6.Minoo P, King RJ, Coalson JJ: Surfactant proteins and lipids are regulated independently during hyperoxia. Am J Physiol 1992, 263:L291-L298 [DOI] [PubMed] [Google Scholar]
- 7.Nogee LM, Wispe JR, Clark JC, Weaver TE, Whitsett JA: Increased synthesis and mRNA of surfactant protein A in oxygen-exposed rats. Am J Respir Cell Mol Biol 1989, 1:119-125 [DOI] [PubMed] [Google Scholar]
- 8.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF: Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol 1998, 19:573-581 [DOI] [PubMed] [Google Scholar]
- 9.Ilizarov AM, Koo HC, Kazzaz JA, Mantell LL, Li Y, Bhapat R, Pollack S, Horowitz S, Davis JM: Overexpression of manganese superoxide dismutase protects lung epithelial cells against oxidant injury. Am J Respir Cell Mol Biol 2001, 24:436-441 [DOI] [PubMed] [Google Scholar]
- 10.Simonson SG, Welty-Wolf KE, Huang YC, Taylor DE, Kantrow SP, Carraway MS, Crapo JD, Piantadosi CA: Aerosolized manganese SOD decreases hyperoxic pulmonary injury in primates. I. Physiology and biochemistry. J Appl Physiol 1997, 83:550-558 [DOI] [PubMed] [Google Scholar]
- 11.Welty-Wolf KE SS, Huang YC, Kantrow SP, Carraway MS, Chang LY, Crapo JD, Piantadosi CA: Aerosolized manganese SOD decreases hyperoxic pulmonary injury in primates. II. Morphometric analysis. J Appl Physiol 1997, 83:559-568 [DOI] [PubMed] [Google Scholar]
- 12.Frank L, Summerville J, Massaro D: Protection from oxygen toxicity with endotoxin. Role of the endogenous antioxidant enzymes of the lung. J Clin Invest 1980, 65:1104-1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsan MF, White JE, Santana TA, Lee CY: Tracheal insufflation of tumor necrosis factor protects rats against oxygen toxicity. J Appl Physiol 1990, 68:1211-1219 [DOI] [PubMed] [Google Scholar]
- 14.Tsan MF, Lee CY, White JE: Interleukin 1 protects rats against oxygen toxicity. J Appl Physiol 1991, 71:688-697 [DOI] [PubMed] [Google Scholar]
- 15.Waxman AB, Einarsson O, Seres T, Knickelbein RG, Warshaw JB, Johnston R, Homer RJ, Elias JA: Targeted lung expression of interleukin-11 enhances murine tolerance of 100% oxygen and diminishes hyperoxia-induced DNA fragmentation. J Clin Invest 1998, 101:1970-1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corne J, Chupp G, Lee CG, Homer RJ, Zhu Z, Chen Q, Ma B, Du Y, Roux F, McArdle J, Waxman AB, Elias JA: IL-13 stimulates vascular endothelial cell growth factor and protects against hyperoxic acute lung injury. J Clin Invest 2000, 106:783-791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panos RJ BP, Simonet WS, Rubin JS, Smith LJ: Intratracheal instillation of keratinocyte growth factor decreases hyperoxia-induced mortality in rats. J Clin Invest 1995, 96:2026-2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, Mulligan RC: Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994, 264:713-716 [DOI] [PubMed] [Google Scholar]
- 19.Ikegami M, Ueda T, Hull W, Whitsett JA, Mulligan RC, Dranoff G, Jobe AH: Surfactant metabolism in transgenic mice after granulocyte macrophage-colony stimulating factor ablation. Am J Physiol 1996, 270:L650-L658 [DOI] [PubMed] [Google Scholar]
- 20.LeVine AM, Reed JA, Kurak KE, Cianciolo E, Whitsett JA: GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. J Clin Invest 1999, 103:563-569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paine R, III, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, Reed JA, Ross G, Whitsett JA, Beck JM: Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol 2000, 164:2602-2609 [DOI] [PubMed] [Google Scholar]
- 22.Huffman JA, Hull WM, Dranoff G, Mulligan RC, Whitsett JA: Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J Clin Invest 1996, 97:649-655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikegami M, Jobe AH, Huffman Reed JA, Whitsett JA: Surfactant metabolic consequences of overexpression of GM-CSF in the epithelium of GM-CSF-deficient mice. Am J Physiol 1997, 273:L709-L714 [DOI] [PubMed] [Google Scholar]
- 24.Paine R, III, Morris SB, Jin H, Wilcoxen SE, Phare SM, Moore BB, Coffey MJ, Toews GB: Impaired functional activity of alveolar macrophages from GM-CSF-deficient mice. Am J Physiol 2001, 281:L1210-L1218 [DOI] [PubMed] [Google Scholar]
- 25.Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC: GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU. 1. Immunity 2001, 15:557-567 [DOI] [PubMed] [Google Scholar]
- 26.Huffman Reed JA, Rice WR, Zsengeller ZK, Wert SE, Dranoff G, Whitsett JA: GM-CSF enhances lung growth and causes alveolar type II epithelial cell hyperplasia in transgenic mice. Am J Physiol 1997, 273:L715-L725 [DOI] [PubMed] [Google Scholar]
- 27.Fukuda N, Folkesson HG, Matthay MA: Relationship of interstitial fluid volume to alveolar fluid clearance in mice: ventilated vs. in situ studies. J Appl Physiol 2000, 89:672-679 [DOI] [PubMed] [Google Scholar]
- 28.Ma T, Fukuda N, Song Y, Matthay MA, Verkman AS: Lung fluid transport in aquaporin-5 knockout mice. J Clin Invest 2000, 105:93-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corti M, Brody AR, Harrison JH: Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol 1996, 14:309-315 [DOI] [PubMed] [Google Scholar]
- 30.Dobbs LG, Gonzalez R, Williams MC: An improved method for isolating type II cells in high yield and purity. Am Rev Respir Dis 1986, 134:141-145 [DOI] [PubMed] [Google Scholar]
- 31.Barazzone C, White CW: Mechanisms of cell injury and death in hyperoxia: role of cytokines and Bcl-2 family proteins. Am J Respir Cell Mol Biol 2000, 22:517-519 [DOI] [PubMed] [Google Scholar]
- 32.Frisch SM, Ruoslahti E: Integrins and anoikis. Curr Opin Cell Biol 1997, 9:701-706 [DOI] [PubMed] [Google Scholar]
- 33.Factor PDV, Saldias F, Brown LA, Sznajder JI.: Adenovirus-mediated transfer of an Na+/K+-ATPase beta1 subunit gene improves alveolar fluid clearance and survival in hyperoxic rats. Hum Gene Ther 2000, 11:2231-2242 [DOI] [PubMed] [Google Scholar]
- 34.Horiuchi T, Weller PF: Expression of vascular endothelial growth factor by human eosinophils: upregulation by granulocyte macrophage colony-stimulating factor and interleukin-5. Am J Respir Cell Mol Biol 1997, 17:70-77 [DOI] [PubMed] [Google Scholar]
- 35.Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson JW: A randomized phase II trial of granulocyte-macrophage colony-stimulating factor therapy in severe sepsis with respiratory dysfunction. Am J Respir Crit Care Med 2002, 166:138-143 [DOI] [PubMed] [Google Scholar]
- 36.Budinger GR, Tso M, McClintock DS, Dean DA, Sznajder JI, Chandel NS: Hyperoxia-induced apoptosis does not require mitochondrial reactive oxygen species and is regulated by Bcl-2 proteins. J Biol Chem 2002, 277:15654-15660 [DOI] [PubMed] [Google Scholar]
- 37.Goyal A, Wang Y, Graham MM, Doseff AI, Bhatt NY, Marsh CB: Monocyte survival factors induce Akt activation and suppress caspase-3. Am J Respir Cell Mol Biol 2002, 26:224-230 [DOI] [PubMed] [Google Scholar]
- 38.Klein JB, Rane MJ, Scherzer JA, Coxon PY, Kettritz R, Mathiesen JM, Buridi A, McLeish KR: Granulocyte-macrophage colony-stimulating factor delays neutrophil constitutive apoptosis through phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways. J Immunol 2000, 164:4286-4291 [DOI] [PubMed] [Google Scholar]
- 39.Lu YPL, Otterbein LE, Kureishi Y, Walsh K, Ray A, Ray P: Activated Akt protects the lung from oxidant-induced injury and delays death of mice. J Exp Med 2001, 193:545-549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matute-Bello G, Liles WC, Radella F, II, Steinberg KP, Ruzinski JT, Hudson LD, Martin TR: Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and granulocyte/macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med 2000, 28:1-7 [DOI] [PubMed] [Google Scholar]