

Abstract
To address the role of neutrophil elastase in pulmonary emphysema, neutrophil elastase-deficient mice and wild-type littermate controls were exposed to long-term cigarette smoke. Compared to wild-type littermates, mice that were deficient in neutrophil elastase were significantly protected (59%) from the development of emphysema. Previously, we demonstrated complete protection from emphysema in the absence of macrophage elastase. Further analysis revealed several interactions between these two elastases. Each elastase inactivated the endogenous inhibitor of the other, with neutrophil elastase degrading tissue inhibitor of metalloproteinase-1, and macrophage elastase degrading α-1-antitrypsin. Cigarette smoke-induced recruitment of both neutrophils and monocytes was impaired in the absence of neutrophil elastase. Moreover, there was less macrophage elastase activity secondary to decreased macrophage accumulation in neutrophil elastase-deficient mice. This study demonstrates a direct role for neutrophil elastase in emphysema and highlights the interdependence of the proteinases and inflammatory cells that mediate lung destruction in response to cigarette smoke.
Pulmonary emphysema is a main component of chronic obstructive pulmonary disease (COPD), currently the fourth leading cause of death in the United States and the only leading cause still increasing in prevalence. As cigarette smoking, the major risk factor for COPD, continues to be rampant worldwide, a major epidemic of COPD is looming. 1
According to the “elastase:antielastase” hypothesis, cigarette smoke causes inflammation and subsequent release of proteolytic enzymes into the lung in excess of their natural inhibitors. In the absence of normal repair, proteolysis leads to tissue destruction and airspace enlargement. This elastase:antielastase hypothesis has dominated COPD research for nearly four decades. In 1963, Laurell and Eriksson 2 reported their observation that patients lacking α-1-antitrypsin (α1 AT), the major inhibitor of neutrophil elastase (NE), developed early onset emphysema. Soon thereafter, Gross et al 3 instilled the elastase papain into rat lungs, resulting in emphysema. Subsequently, investigators were able to induce emphysema by instilling other elastolytic enzymes, including human neutrophil elastase, 4-6 into the airways of experimental animals. Together, these observations firmly established NE as the proteinase most likely responsible for tissue destruction in emphysema.
NE is a potent serine proteinase with a primary translational product of 267 amino acids and variable glycosylation, migrating with a molecular mass of 28 to 31 kd. 7 NE is synthesized in the promyelocytic stage of myeloid development and stored in large quantities in its active form in neutrophil azurophil granules. 8 Catalytic activity is conferred by the His-Asp-Ser triad which forms a charge relay system, resulting in a powerful nucleophile capable of attacking peptide bond substrates. NE has potent catalytic activity against a broad array of extracellular matrix substrates, including the highly resistant elastin that imparts structural stability to the lung.
However, macrophages are the primary inflammatory cell in the lower airspace under normal conditions, and particularly in response to long-term cigarette smoking. Macrophages also contain elastolytic enzymes, and we have previously demonstrated that gene-targeted mice deficient in macrophage elastase (MMP-12) are entirely protected from cigarette smoke-induced emphysema. 9 Taken together, the prominence of macrophage proteinases and the inconsistent linkage of NE to the “garden variety” emphysema 10 associated with cigarette smoking led some investigators to question the role of NE in this disease process.
To directly address the role of NE in emphysema, we generated mice deficient in NE by gene targeting, 11 and subjected them and their wild-type littermates to long-term cigarette smoke exposure. NE-deficient (NE−/−) mice were significantly protected from the development of emphysema. Further analysis revealed many interactions between NE and MMP-12, as well as between neutrophils and macrophages, which lead to enzymatic lung destruction.
Materials and Methods
NE and MMP-12 Gene-Targeted Mice
Both NE 11 and MMP-12 12 gene-targeted mice were previously generated in this laboratory, each by replacement of sequence from intron 1 and exon 2 with neomycin phosphotransferase cDNA driven by the phosphoglycerate kinase promoter (PGK-neo). These constructs underwent homologous recombination in RW4 ES cells, and correctly targeted clones were injected into C57BL/6J blastocysts and transmitted into the germlines of mice as previously described. 11,12 Mice homozygous for these mutations in a 129/Sv-C57BL/6J mixed background back-crossed at least eight generations into the C57BL/6J background were used for the current studies. The NE mutation did not alter expression of the closely linked gene encoding proteinase 3, nor did the MMP-12 mutation affect expression of neighboring MMP genes.
Cigarette Smoke Exposure
Twenty-six NE−/− mice, 20 wild-type (NE+/+) littermates, and 12 MMP-12−/− mice, all 12 weeks of age, were subjected to two unfiltered cigarettes per day (University of Kentucky), 6 days per week for 6 months, with the use of a smoking apparatus as described. 9 Mice tolerated smoke exposure without toxicity; carboxyhemoglobin levels after two cigarettes were ∼10%. Twenty-four sham and non-smoke-exposed age-matched littermates were used as controls. Here and previously, sham chamber-exposed mice were equivalent to non-smoke-exposed mice.
Tissue Processing
Following smoke exposure, each mouse was killed and saline was perfused through the right ventricle to remove blood. The left lung was ligated and removed for protein and RNA analysis. The right lung was inflated by instilling 3% gluteraldehyde in 0.1 mol/L cacodylate buffer to 25 cm H20 (for 15 minutes), and then ligated and removed. Inflated lungs were fixed for 48 hours before embedding in paraffin. Serial sagittal sections were obtained for morphological studies and histological analysis.
Morphometry
Mid-sagittal sections stained with hematoxylin and eosin (H&E) were used to calculate the chord length as previously described. 9,13 All measurements were performed by a blinded investigator (S.D.S.), who calculated an average point count from 10 random fields at ×200 using NIH Image software. 13 Magnitude of changes were identical comparing manual intercepts [9] to NIH image results.
Immunohistochemistry
Alveolar and interstitial macrophages were quantified by counting macrophages identified by murine mac-3 (rat antibody to mouse at 1:500 dilution; Pharmigen, San Diego, CA) immunostaining, using the avidin-biotin horseradish peroxidase technique with 3,3′-diaminobenzadine as the chromogenic substance. 9,12 Neutrophils were identified by H&E stain. Cell counts were determined by averaging the counts of five high-powered fields of mid-sagittal slices.
Bronchoalveolar Lavage
Separate groups of smoke-exposed mice were used for these studies. Three separate 1-ml aliquots of saline were administered through an intravenous catheter placed in the trachea, and removed sequentially. Cell pellets were isolated and used to determine the total cell count, and aliquots were subjected to cytospin to determine differential cell counts. Bronchoalveolar lavage (BAL) is most reliable for quantifying neutrophil abundance, as neutrophils migrate directly from capillaries into the alveolar space, but less reliable for evaluating macrophage abundance, since many macrophages remain in the interstitial space.
Protein Analysis
Lungs were removed en bloc, perfused with saline until free of blood, then frozen and homogenized in 1 ml of 50 mmol/L Tris (pH 7.2), 5 mmol/L CaCl2, and 150 mmol/L NaCl. Samples were normalized for total lung protein content for further analysis. A monospecific polyclonal antibody directed against human α1AT (DiaSorin Ins., Stillwater, MN), but which also reacted against and was specific for mouse α1AT, was used at a 1:500 dilution for Western blot analysis with enhanced chemiluminescence. Findings among mice of similar genotypes and treatment showed similar results. Data from three pooled samples loaded with equal protein content are shown.
Zymography and Reverse Zymography
Total lung tissue extracts were prepared by perfusing (as above), removing the lungs en bloc, freezing them, and homogenizing them in 5 ml of 50 mmol/L Tris, 10 mmol/L CaCl2, and 150 mmol/L NaCl buffer. Following centrifugation, lyophilization, and equalization for total lung protein content, the samples were subjected to substrate 10% polyacrylamide gel electrophoresis using casein for zymography as described 12 and gelatin (1 mg/ml) for reverse zymography. After gel electrophoresis was performed at 4°C, gels were incubated in 2.5% (v/v) Triton for 30 minutes, then overnight in 50 mmol/L Tris (pH 8.0), 5 mmol/L CaCl, and 1 μmol/L ZnCl2 at 37°C. For reverse zymography, rat uterine explant conditioned medium was added to the Tris-Ca-Zn overnight buffer. As this medium has spontaneous gelatinase activity, proteinase inhibitors capable of blocking gelatin activation appear as dark blue bands following staining with 0.125% Coomassie blue. Again, results for mice of similar genotypes and treatment were equivalent. Data from pooled (three mice per lane) samples equally loaded by protein content are shown.
Results
NE-Deficient Mice Are Significantly Protected from Emphysema in Response to Long-Term Cigarette Smoke
To determine the contribution of NE to cigarette smoke-induced emphysema, we exposed groups of NE−/− and NE+/+ to cigarette smoke for a period of 6 months. (Note: 19 of 20 wild-type controls survived the cigarette smoke exposure protocol. There was no mortality in the other groups.) Age-matched sham and non-smoke-exposed littermates were used as controls. At the end of the 6 months, the mice were killed; their lungs were inflated to a fixed pressure, and H&E stained mid-sagittal sections were used to determine airspace enlargement. NE−/− smoke-exposed mice exhibited less airspace enlargement in response to cigarette smoke than NE+/+ mice (Figure 1) ▶ . Quantification of chord length, a measure of airspace size and hence emphysema, demonstrated 59% protection (P = 0.002) against emphysema in NE−/− mice compared to wild-type littermates (Table 1) ▶ . Similar to our previous findings, 9 a group of 12 MMP-12−/− mice (in the same genetic background as NE mice) failed to develop airspace enlargement in response to 6 months of cigarette smoke exposure (Table 1) ▶ .
Figure 1.
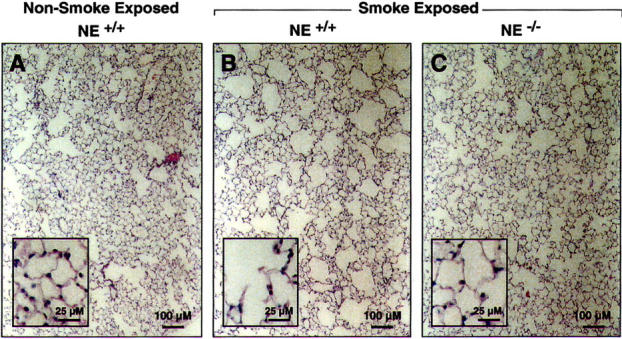
NE−/− mice have less cigarette smoke-induced emphysema than wild-type mice. Lungs from wild-type mice (NE+/+) and NE-deficient mice (NE−/−) were exposed to cigarette smoke for 6 months. Lungs from these mice and age-matched non-smoke-exposed control mice were inflated to 25 cm H2O and mid-sagittal slices stained with H&E as described in “Materials and Methods.” Note airspace enlargement in response to cigarette smoke exposure in NE+/+ mice and partial protection in NE−/− smoke-exposed mice.
Table 1.
Air-Space Enlargement in Response to Cigarette Smoke Exposure
| Group | n | CL* (S.E.) | % Increase with Sm | P value (vs NS) | P value (vs WT Sm) |
|---|---|---|---|---|---|
| WT (NS) | 24 | 30.2 (0.5) | — | — | <0.0001 |
| WT (Sm) | 19 | 36.8 (0.8) | 22 | <0.0001 | — |
| NE−/− (NS) | 14 | 30.4 (0.5) | — | — | |
| NE−/− (Sm) | 26 | 33.0 (0.6) | 9 | (0.1) | 0.002 |
| MMP-12−/− (NS) | 12 | 29.9 (0.4) | — | — | |
| MMP-12−/− (Sm) | 12 | 29.8 (0.5) | 0 | (0.5) | 0.001 |
*Chord Length (CL) measures alveolar diameter. Values shown are the mean and standard error (in parentheses). Groups of mice, with the number of mice in each group identified (n), were either non-smoke-exposed (NS) or exposed to cigarette smoke (Sm). Wild-type mice (WT) were 129/SvXC57BL/6, the background of both mutant genotypes. P values comparing different groups were derived from two-tailed Student’s t-test.
NE Is Required for Neutrophil and Monocyte Recruitment in the Lungs in Response to Cigarette Smoke
While macrophages are far more prevalent than neutrophils both at baseline and in response to cigarette smoke, we did observe a fivefold increase in neutrophil counts in the BAL fluid of wild-type mice following acute (not shown) and long-term cigarette smoke exposure. In contrast, there was no excess accumulation of neutrophils in the BAL fluid of smoke-exposed NE−/− mice (Figure 2A) ▶ . Notably, total lung neutrophil abundance following smoke exposure was similar in NE−/− and NE+/+ mice. However, in mice lacking NE, most neutrophils remained in the blood vessels, unable to egress from the vasculature despite perfusing the right ventricle with saline before fixation (data not shown). This suggests a defect in neutrophil migration into the lung parenchyma in the absence of NE. With respect to macrophages, baseline numbers were unaffected by the genetic absence of NE. However, accumulation of macrophages in the lungs in response to long-term cigarette smoke was impaired ∼60% in NE−/− mice compared to wild-type NE+/+ mice (Figure 2B) ▶ . As previously shown, 9 macrophage accumulation was markedly decreased following cigarette smoke exposure in MMP-12−/− mice (Figure 2B) ▶ .
Figure 2.

Neutrophil elastase is involved in inflammatory cell recruitment in response cigarette smoke. A: Neutrophils that underwent transvascular migration to the alveolar space were quantified by bronchoalveolar lavage. Note, NE and MMP-12 did not affect recruitment in non-smoke-exposed mice (solid bars). However, NE but not MMP-12 is required for neutrophil migration in response to cigarette smoke (hatched bars). Statistically, there were more neutrophils in wild-type (WT) non-smoke-exposed (NS) versus smoke exposed (Sm)(P < 0.01), and in MMP-12−/− NS versus MMP-12−/− Sm (P < 0.01), but not NE−/− NS versus NE−/− Sm. However, NE Sm had significantly fewer neutrophils than WT Sm (P < 0.01). B: Tissue macrophage counts were assessed by immunhistochemical (mac-3) staining of mid-sagittal sections and equalized for alveolar tissue density. Note, cigarette smoke exposure increased macrophages in WT mice (WT NS versus WT Sm, P < 0.001) and NE−/− mice (NE−/− NS versus NE−/− Sm, P = 0.04), but not MMP-12−/− mice (MMP-12−/− NS versus MMP-12−/− Sm, P = 0.5). However, macrophage accumulation in response to cigarette smoke was inhibited 60% in NE−/− mice (WT Sm versus NE−/− Sm, P < 0.05), and 85% in MMP-12−/− mice (WT Sm versus MMP-12−/− Sm, P < 0.01). At least 10 mice were used for each group. Bars represent SE.
NE Is Required for the Presence and Activation of MMP-12
As shown by casein zymography (Figure 3 ▶ , lanes 1 and 2), and confirmed by Western blot (not shown), the fully processed, active 22-kd catalytic domain of MMP-12 was induced with smoking in the lungs of wild-type mice. In comparison, less active MMP-12 was present in lung homogenates of NE−/− mice (Figure 3 ▶ , lanes 3 and 4). This was due to decreased macrophage accumulation, as well as to less proteolytic activation of pro-MMP-12 (55 kd, Figure 3 ▶ , lane 4) in the absence of NE, and suggests that NE proteolytically activates MMP-12 in vivo.
Figure 3.

Proteinase activity in lungs of mice exposed to cigarette smoke. Perfused whole lung homogenates were subjected to casein zymography as described. Each lane represents equally pooled samples from three lungs of the given group. Note enhanced activity of fully processed MMP-12 (∼22 kd) in wild-type (WT) mice exposed to cigarette smoke (Sm) compared to non-smoke-exposed (NS). In the absence of NE (NE−/−), mice had markedly less active 22-kd MMP-12 and somewhat more pro-MMP-12 (migrating at ∼54 kd) in response to cigarette smoke than did wild-type mice (NE+/+). Also visible is a band migrating at ∼78 kd in all lanes that is likely an aspartyl proteinase based on inhibition pattern.
Neutrophil and Macrophage Elastase Enhance Each Other’s Potency by Degrading Each Other’s Inhibitor
We also observed interactions between the proteinases and their endogenous inhibitors. The tissue inhibitors of metalloproteinases (TIMPs), of which there are four, are the major endogenous inhibitors of MMP activity. Multiple cell types produce TIMPs locally within tissues, including the lung. Previously, it was demonstrated that NE degrades TIMP-1 in vitro. 14 To evaluate TIMP activity, we applied reverse zymography to lung tissue, and found enhanced TIMP activity only in smoke-exposed NE-deficient mice (Figure 4) ▶ . While the protected TIMP band at ∼28 kd could correspond to either TIMP-1 or glycosylated TIMP-3, RT-PCR demonstrated the production of only TIMP-1 in these samples. Of note, there are no mouse TIMP antibodies developed to distinguish the two. However, these findings are consistent with NE-mediated destruction of TIMP-1 in smoke-exposed lungs.
Figure 4.

TIMP activity in response to cigarette smoke: evidence for NE-mediated TIMP degradation. Perfused whole lung extracts were subjected to reverse zymography. Protection from proteolytic activity is indicated by presence of gelatin (dark bands) at molecular migration of TIMPs on the blot. Note the presence of larger amounts of a 27.5-kd inhibitor, TIMP-1, in mice lacking NE. Again, each lane represents equally pooled samples from three lungs of the given genotype and exposure.
α1AT is produced predominantly in hepatocytes, released into the bloodstream, and deposited into extracellular spaces, including those in the lung, to protect against NE damage. In response to cigarette smoke, α1AT protein levels were at least as prominent in serum following cigarette smoke exposure in both wild-type and MMP-12-deficient mice (Figure 5 ▶ , bottom). In the lung, however, α1AT levels were elevated in MMP-12−/− mice, but reduced by 50% in wild-type mice, following exposure to cigarettes (Figure 5 ▶ , top).
Figure 5.

α1AT expression in response to cigarette smoke: evidence for MMP-12-mediated α1AT degradation. Serum and saline perfused (through right ventricle) lung tissue extracts were obtained following 6 months of cigarette smoke exposure (Sm). Age-matched sham or non-smoke-exposed (NS) mice were used as controls. Again, each lane represents equally pooled samples from three lungs. Equal amounts of protein were then subjected to Western analysis using an antibody specific for α1AT. Top: Blood serum α1AT levels were at least as prominent in cigarette smoke-exposed (Sm) lungs compared to non-smoke-exposed (NS) in both wild-type and MMP-12−/− mice. Bottom: Lung α1AT levels are decreased (by 50%) in response to cigarette smoke in wild-type as compared to MMP12−/− mice.
Discussion
The role of NE in emphysema has been a cornerstone of the elastase:antielastase hypothesis. However, studies in humans have inconsistently shown an association of neutrophils and NE with emphysema. While some groups have found NE localized in destructive areas, 15 others have demonstrated an inverse correlation between NE levels and airspace enlargement. 10 Indeed, it is difficult to determine the cause of a dynamic process when limited to static “snapshots” of the disease, often years after initiation.
One method to determine the relative contribution of an individual proteinase to the development of emphysema is to apply gene-targeted mice to a model that replicates the human disease. While humans and mice may differ considerably, exposure of mice to long-term cigarette smoke results in the inflammation and airspace enlargement characteristic of human emphysema. Using this model, we previously demonstrated that mice lacking the matrix metalloproteinase macrophage elastase (MMP-12) were entirely protected from airspace enlargement following cigarette smoke exposure. 9 These mice also had reduced macrophage accumulation in the lungs. Thus, in mice, macrophage accumulation and MMP-12 expression are sufficient to cause airspace enlargement.
Human COPD is more complicated, with recurrent airway infections and hence greater neutrophil involvement. However, we did not expect neutrophils and NE to play prominent roles in a model of emphysema characterized by cigarette smoke-induced chronic low-grade inflammation in mice that are housed in a sterile barrier facility. Yet, our findings demonstrate a significant role for NE in the development of emphysema in mice. This effect is multifactorial, involving NE in inflammatory cell recruitment, extracellular matrix proteolysis, inactivation of TIMP-1, and likely activation of MMP-12. In fact, we observed significant interactions between the NE and MMP-12 proteolytic systems, with each augmenting the other’s destructive capacity.
With respect to endogenous inhibitors, we observed disappearance of the serine proteinase inhibitor α1AT by an MMP, and disappearance of the MMP inhibitor TIMP-1 by a serine proteinase, in cigarette smoke-exposed lungs. On cleavage of a suicide bond by NE, α1AT changes from a stressed to a relaxed conformation, thereby trapping and inactivating NE. 16 This action accounts for >90% of the natural inhibition of NE, particularly in the lower airspace of the lung. MMPs also attack the reactive site of α1AT; however, in this case the molecule is inactivated without affecting MMP activity. 17,18 We have previously shown in vitro that MMP-12 is the most efficient MMP at degrading α1AT, being 10-fold more efficient than MMP-7 and over 100-fold more efficient than other MMPs. 19 Of note, as opposed to experiments in a test tube (with discrete, pure reagents and limited time of exposure) where discrete MMP-12-mediated α1AT breakdown products can be visualized, in vivo one can best appreciate disappearance of the intact α1AT (Figure 5) ▶ .
In a mouse model of bullous pemphigoid characterized by neutrophil inflammation, neutrophil-derived MMP-9 inactivated α1AT, allowing NE to cause blister formation. 20 In the cigarette smoke-exposed mouse lung, where macrophage MMP-12 is the primary MMP and α1AT is abundant, MMP-12-mediated degradation of α1AT is dominant. It follows, therefore, that loss of α1AT would enhance NE-mediated lung destruction. Similarly, the serine proteinase NE is capable of cleaving and inactivating the MMP inhibitor TIMP-1 in vitro. 14 The current study demonstrates that TIMP-1 is the predominant TIMP expressed in the lung in response to cigarette smoke and disappearance of TIMP-1 in the presence of NE supports the concept that NE can degrade TIMP-1 in vivo, since enhanced TIMP-1 activity was only observed in smoke-exposed mice lacking NE.
NE also had dramatic effects on both neutrophil and macrophage accumulation in the lungs in response to cigarette smoke. In the absence of NE, neutrophils had difficulty escaping the capillary to the lung interstitium and alveolar space. This was not due to an inability of neutrophils to penetrate basement membranes in the absence of NE, for we have previously demonstrated normal chemotaxis and Matrigel invasion in NE−/− mice. 21 Importantly, others have failed to inhibit neutrophil migration using proteinase inhibitors of all classes. 22,23 Our results are consistent with a previously proposed concept that NE is required to dissociate the CD11/CD18-ICAM-1 complex tethering the neutrophil to the endothelium. 24
We also observed marked suppression of macrophage accumulation in the lungs in NE−/− mice exposed to cigarette smoke. In contrast to the neutrophil, the NE−/− peripheral blood monocyte does have impaired Matrigel penetration compared to wild-type controls (data not shown). This is consistent with previous findings that pro-inflammatory monocytes, those destined for transvascular migration into tissues, express significant amounts of NE. 25 It is plausible that surface-bound NE is required for the migration of these monocytes into the smoke-exposed lung. Here and previously, 9 we found that MMP-12-deficient mice also had reduced monocyte recruitment following smoke exposure. The attenuation of this response, however, was due not to the inability of monocytes to egress from the vasculature, but rather to the absence of chemotactic stimuli in the lung. This, we believe, is at least in part due to reduced MMP-12-mediated generation of elastin fragments, which are known monocyte chemokines. 4,26 A similar mechanism may be at work in NE−/− mice. That is, NE may generate or activate other, as yet undefined, chemokines responsible for monocyte migration in response to cigarette smoke.
In part due to the requirement of NE for macrophage accumulation in the lung, and in part to NE-mediated proteolytic activation of pro-MMP-12, there is less active MMP-12 in the lungs of smoke-exposed NE-deficient mice. Thus, protection from emphysema in NE−/− mice is likely secondary to NE’s elastolytic activity, and attributable to the requirement of NE for recruitment of monocytes and their proteinases. The combination of a model of cigarette smoke exposure with gene-targeted mice has allowed us to determine the contribution of both neutrophil elastase and macrophage elastase to emphysema, and identify interactions between these proteinases. While mouse models may not entirely predict disease pathways in humans, the data suggest that if one could safely inhibit either of these classes of proteinases, one may be able to prevent further lung destruction in many patients with emphysema.
Footnotes
Address reprint requests to Steven D. Shapiro, M.D., Parker B. Francis Professor of Medicine, Brigham and Women’s Hospital,Harvard Medical School, 15 Francis Street, Boston, MA 02115. E-mail: sshapiro@rics.bwh.harvard.edu.
Supported by the National Heart Lung and Blood Institute (S.D.S. and A.B.) and Glaxo Smith Kline Research Fellowship (to N.M.G.).
References
- 1.MacKenzie TD, Bartecchi CE, Schrier RW: The human costs of tobacco abuse. N Engl J Med 1994, 330:975-980 [DOI] [PubMed] [Google Scholar]
- 2.Laurell CB, Eriksson S: The electrophoretic α-globulin pattern of serum in α-antitrypsin deficiency. Scand J Clin Invest 1963, 15:132-140 [Google Scholar]
- 3.Gross P, Pfitzer E, Tolker E, Babyak M, Kaschak M: Experimental emphysema: its production with papain in normal and silicotic rats. Arch Environ Health 1965, 11:50-58 [DOI] [PubMed] [Google Scholar]
- 4.Senior RM, Griffin GL, Mecham RP: Chemotactic activity of elastin-derived peptides. J Clin Invest 1980, 66:859-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janoff A, Sloan B, Weinbaum G, Damiano V, Sandhaus RA, Elias J, Kimbel P: Experimental emphysema induced with purified human neutrophil elastase: tissue localization of the instilled protease. Am Rev Respir Dis 1977, 115:461-478 [DOI] [PubMed] [Google Scholar]
- 6.Snider GL, Lucey EC, Christensen TG, Stone PJ, Calure JD, Cantanese A, Franzblaa C: Emphysema and bronchial secretory cell metaplasia induced in hamsters by human neutrophil products. Am Rev Respir Dis 1984, 129:155-160 [DOI] [PubMed] [Google Scholar]
- 7.Sinha S, Watorek W, Karr S, Giles J, Bode W, Travis J: Primary structure of human neutrophil elastase. Proc Natl Acad Sci USA 1987, 84:2228-2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi H, Nukiwa T, Yoshimura K, Quick CD, States DJ, Holmes MD, Ejsmh-Peng J, Knutsen T, Crystal RJ: Structure of the human neutrophil elastase gene. J Biol Chem 1988, 263:14739-14747 [PubMed] [Google Scholar]
- 9.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD: Macrophage elastase is required for cigarette smoke-induced emphysema in mice. Science 1997, 277:2002-2004 [DOI] [PubMed] [Google Scholar]
- 10.Eidelman D, Saetta MP, Ghezzo H, Wang N-S, Hoidal JR, King M, Cosio MG: Cellularity of the alveolar walls in smokers and its relation to lung destruction: functional implications. Am Rev Respir Dis 1990, 141:1547-1552 [DOI] [PubMed] [Google Scholar]
- 11.Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, Shapiro SD: Mice lacking neutrophil elastase reveal impaired host defense against gram-negative bacterial sepsis. Nat Med 1998, 4:615-618 [DOI] [PubMed] [Google Scholar]
- 12.Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD: Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci USA 1996, 93:3942-3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunnill MS: Quantitative methods in the study of pulmonary pathology. Thorax 1962, 17:320-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okada Y, Watanabe S, Nakanishi I, Kishi J, Hayakawa T, Watorek W, Travis J, Nagase H: Inactivation of tissue inhibitor of metalloproteinases by neutrophil elastase and other serine proteinases. FEBS Lett 1988, 229:157-160 [DOI] [PubMed] [Google Scholar]
- 15.Damiano VV, Tsang A, Kucich U, Abrams WR, Rosenbloom J, Kimbel P, Fallahnejed M, Weinbaum G: Immunolocalization of elastase in human emphysematous lungs. J Clin Invest 1986, 78:482-493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huntington J, Read R, Carrell R: Structure of a serpin-protease complex shows inhibition by deformation. Nature 2000, 407:923-926 [DOI] [PubMed] [Google Scholar]
- 17.Desrochers P, Jeffrey J, Weiss S: Interstitial collagenase (matrix metalloproteinase-1) expresses serpinase activity. J Clin Invest 1991, 87:2258-2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sires U, Murphy G, Baragi V, Fliszar C, Welgus H, Senior R: Matrilysin is much more efficient than other matrix metalloproteinases in the proteolytic inactivation of α 1-antitrypsin. Biochem Biophys Res Commun 1994, 204:613-620 [DOI] [PubMed] [Google Scholar]
- 19.Gronski TJ, Martin R, Kobayashi DK, Walsh BC, Holman MC, Van Wart HE, Shapiro SD: Hydrolysis of a broad spectrum of extracellular matrix proteins by human macrophage elastase. J Biol Chem 1997, 272:12189-12194 [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Zhou X, Shapiro S, Shipley J, Twinning S, Belaaouaj A, Diaz L, Senior R, Werb Z: α-1-proteinase inhibitor is the critical substrate for gelatinase B in experimental bullous pemphigoid. Cell 2000, 102:647-665 [DOI] [PubMed] [Google Scholar]
- 21.MacIvor D, Shapiro S, Pham C, Belaaouaj A, Abraham S, Ley T: Normal neutrophil function in cathepsin G-deficient mice. Blood 1999, 94:4282-4293 [PubMed] [Google Scholar]
- 22.Huber A, Weiss S: Disruption of the subendothelial basement membrane during neutrophil diapedesis in an in vitro construct of a blood vessel wall. J Clin Invest 1989, 83:1122-1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackarel A, Cottell D, Russell K, FitzGerald M, O’Connor C: Migration of neutrophils across human pulmonary endothelial cells is not blocked by matrix metalloproteinase or serine protease inhibitors. Am J Respir Cell Mol Biol 1999, 20:1209-1219 [DOI] [PubMed] [Google Scholar]
- 24.Loike J, Sodeik B, Cao L, Leucona S, Weitz J, Detmers P, Wright S, Silverstein S: CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the A α chain of fibrinogen. Proc Natl Acad Sci USA 1991, 88:1044-1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Owen CA, Campbell MA, Boukedes SS, Stockley RA, Campbell EJ: A discrete subpopulation of human monocytes expresses a neutrophil-like proinflammatory (P) phenotype. Am J Physiol 1994, 267:775-785 [DOI] [PubMed] [Google Scholar]
- 26.Hunninghake GW, Davidson JM, Rennard S, Szapiel S, Gadek JE, Crystal RG: Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysema. Science 1981, 212:925-927 [DOI] [PubMed] [Google Scholar]