

Abstract
Mice to which human prions efficiently transmit in short incubation periods are valuable not only as research tools of human prions but also as reliable diagnostic tools. We recently produced a line of knock-in mouse expressing a unique human-mouse chimeric PrP (Ki-ChM mouse), which has mouse-specific residues practically only at the C-terminal part after posttranslational modification, and here we attempted transmission of various human prions to assess the susceptibility profile of the mouse. Susceptibility varied considerably depending on prions inoculated: highly susceptible to MM1 and MV1 types of sporadic Creutzfeldt-Jakob disease (CJD), developing disease within ∼150 days, familial CJD with M232R mutation, and dura graft-associated CJD (dCJD) without amyloid plaque; less susceptible to MM2-type sporadic CJD and variant CJD, with some mice lacking any sign of transmission; and totally resistant to VV2 type sporadic CJD and dCJD with amyloid plaque. The rather short incubation time achieved by Ki-ChM mice suggests new approaches to produce mice that develop prion disease with very short incubation periods. We compared the characteristic susceptibility profile of Ki-ChM with those of other precedent transgenic mice and discussed, including the prospects in developing genetically engineered mice susceptible to human prions.
Prion diseases are a group of neurodegenerative diseases characterized by the accumulation of abnormal isoform of host-encoded prion protein (PrP). 1 They have sporadic, infectious, and inherited etiologies and are clinicopathologically diverse. For example, typical Creutzfeldt-Jakob disease (CJD) is characterized by subacutely progressive dementia deteriorating to akinetic mutism within a few months after the onset and, neuropathologically, by diffuse brain atrophy, astrocytic gliosis, and synaptic-type deposits of pathogenic isoform of PrP (PrPSc); Gerstmann-Straeussler-Sheinker syndrome (GSS) is an inherited disease associated with more slowly progressive ataxia and multicentric PrP amyloid plaques; fatal familial insomnia is another inherited disease characterized by slowly progressive ataxia and behavior changes and atrophy of thalamic nuclei and inferior olivery nucleus; a subtype of sporadic CJD, without any mutation in PrP gene, clinicopathologically resembles fatal familial insomnia are often referred to as sporadic fatal insomnia. 2-4 According to the protein-only hypothesis, all of the properties of prions are enciphered in the conformation of PrPSc conformations, and the great diversity of prion diseases reflects the diversity of PrPSc conformation. 1,5 The heterogeneity in physicochemical properties of PrPSc among prions from different prion diseases suggests the existence of considerable diversity in PrPSc conformation. With human prions, the sizes of the protease-resistant core of PrPSc after limited proteolysis (PrPres) on sodium dodecyl sulfate-polyacrylamide gel electrophoresis and the relative ratio of three glycoforms of PrPres are often varied and used for the classification of human prion diseases. 6-8 Not only the physicochemical properties, the transmission properties represented by the incubation periods, occurrence of clinical disease, neuropathology, and distribution of PrPSc deposits on experimental transmission are different between prion strains and also help to distinguish prion strains. 1
Experimental transmission of human prions to animals still plays the major role in the investigations of human prion diseases, because the most important feature of prion, infectivity, cannot be fully reproduced or even evaluated precisely in vitro. However, the long intervals between the inoculation of prions into the animal and the development of illness have made transmission studies so time consuming as to hamper efficient investigation of prion diseases and make screening for prions by bioassay impractical. So, attempts to produce mice with short incubation periods have been incessantly made, and several lines of transgenic (Tg) mice have been developed. 8-13 By transmitting different kinds of human prion diseases to such susceptible mice, comparative study of human prion strains could be efficiently performed and much about human prions has been revealed: for example, the importance of PrPSc conformation for the prion properties, 2,5 and the origin of variant CJD being bovine spongiform encephalopathy. 8,9 On the other hand, as with the case of chimeric-PrP-expressing Tg mice transmission studies, which indicated the existence of the third component involved in the prion replication, 11 developing such mice itself may give insight into the molecular mechanism of prion propagation.
We recently produced a line of knock-in mice expressing a unique human-mouse chimeric PrP (ChM) that has a rather long central human insert and the same length of C-terminal mouse sequence with the chimeric MHu2M PrP; in other words, it has mouse sequence practically only toward the end of the C-terminal part, from codon 215 to codon 231, after posttranslational modification, 14,15 and herein we transmitted different human prion diseases to the mice to test its versatility as a research and diagnostic tool. They proved to be highly susceptible to some types of human prion diseases and developed clinical disease within rather shorter incubation periods, ∼150 days, compared with the precedent Tg mice, while less susceptible or totally resistant to other types of prion diseases. The results of the present study with the novel genetically modified mouse confirm the importance of the PrPSc conformation in propagation of prions, and at the same time provide practical implications for developing mice with shorter incubation times and constructing systematic bioassay systems.
Materials and Methods
Human Prion Diseases Inoculated
The frontal cerebral cortical tissues were isolated at autopsy, on informed consent for research use, from patients with sporadic, inherited, and iatrogenic (dura-graft-associated CJD; dCJD) prion diseases and variant CJD (vCJD). Parts of the tissues were immediately deep-frozen for biochemical study and the other parts were formalin-fixed for pathological assay. The formalin-fixed tissue blocks were then immersed in 90% formic acid for 1 hour at room temperature to attenuate the infectivity. 16 The diagnosis of prion disease was confirmed by identifying characteristic neuropathological findings such as spongiform change, gliosis, and PrP deposition of formalin-fixed brain sections, 4,17 and by identifying PrPres extracted from the brain tissues on Western immunoblots as described. 16 Typing of PrPres was performed based on the classification proposed by Parchi and colleagues: 3,7 for sporadic CJD cases, PrPres of ∼21 kd was designated as PrPres type 1, and PrPres of ∼19 kd as PrPres type 2 (Figure 1A) ▶ . PrP genotyping of patients was performed by polymerase chain reaction-direct sequencing using genomic DNA purified from brain tissues or peripheral blood leukocytes. 18
Figure 1.
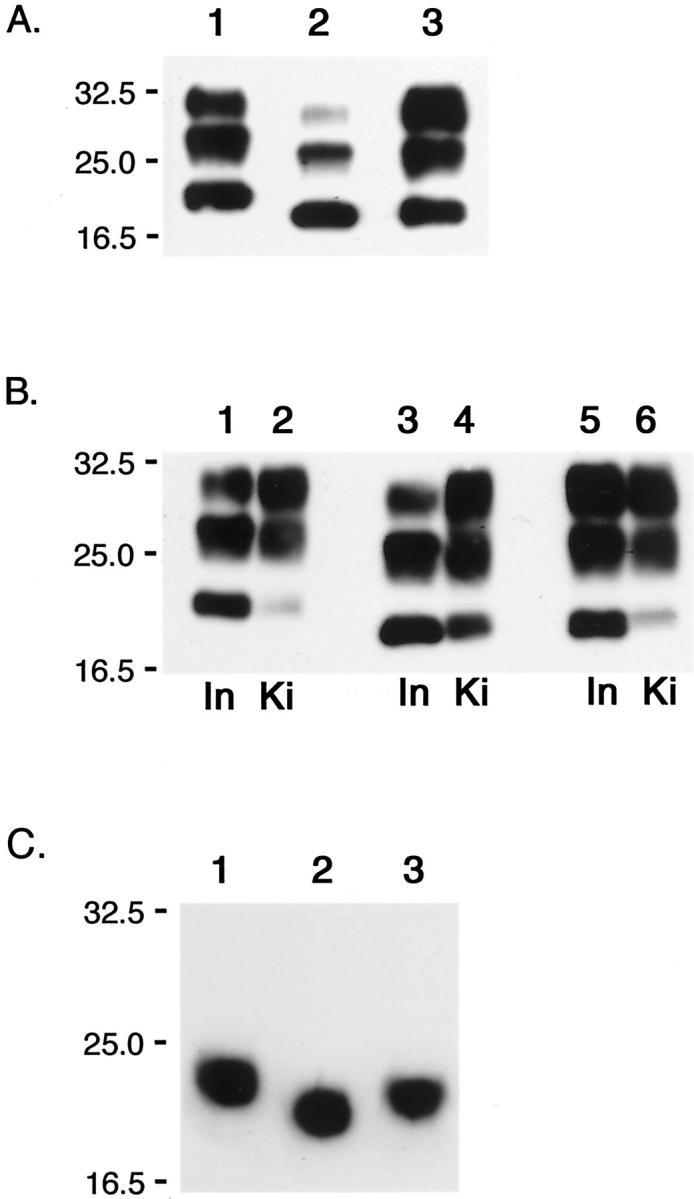
PrPres from patients with prion diseases and prion-inoculated Ki-ChM mice A: PrPres type 1 extracted from MM1 sCJD (H-3, lane 1). PrPres type 2 from MM2-T sCJD (NK, lane 2), and vCJD (96/02, lane 3). B: PrPres isolated from Ki-ChM mice inoculated with MM1 sCJD (H-3, lane 2), MM2-T sCJD (NK, lane 4), and vCJD (96/02, lane 6) are shown along with those from the respective inocula (H-3, lane 1; NK, lane 3; 96/02, lane 5). Ki, PrPres isolated from Ki-ChM mice; In, PrPres isolated from inocula. C: Deglycosylated PrPres isolated from Ki-ChM mice inoculated with human prion diseases (the same samples as shown in B): MM1 sCJD, lane 1; MM2-T sCJD, lane 2; vCJD, lane 3.
After characterization of the genotype and PrPres typing, the brain tissues from different prion disease cases were classified as follows: six sCJD cases [two homozygous for methionine at codon 129 (129M) and PrPres type 1 (MM1), one with valine/methionine at codon 129 and PrPres type 1 (VM1), two with the thalamic form of sCJD with methionine/methionine at codon 129 and PrPres type 2 (MM2-T), and one with valine/valine at codon 129 and PrPres type 2 (VV2)], three vCJD cases, four cases of dura-graft-associated CJD (dCJD) [ a nonplaque type case (np-dCJD) and three plaque-type cases (p-dCJD)], 16 and two cases of inherited prion diseases [one of GSS with the P102L mutation (GSS102) and one of CJD with the M232R mutation (CJD232)] (Figure 1A ▶ , Table 1 ▶ ). All of the cases of vCJD, dCJD, and inherited prion diseases were homozygous for methionine at codon 129; as for the another polymorphic codon, codon 219, all of the cases collected were homozygous for glutamate, except for GSS102, which had lysine at codon 219 on the allele without P102L mutation.
Table 1.
Transmission of Human Prions to Ki-ChM Mice
| Inoculum | Polymorphic codons | Mice examined* | Incubation period (days)† | ||||
|---|---|---|---|---|---|---|---|
| Sacrificed | Alive | ||||||
| 129 | 219 | Sympt. | Pathol. | Pathology (+) | Pathology (−) | ||
| MM1 sCJD | |||||||
| H-3 | M/M | E/E | 6/6 | 6/6 | 151 ± 6.7 | ||
| NR | M/M | E/E | 4/4 | 4/4 | 134 ± 12.0 | ||
| VM1 sCJD | |||||||
| SM | M/V | E/E | 5/5 | 5/5 | 141 ± 5.3 | ||
| MM2-T sCJD | |||||||
| NK | M/M | E/E | 2/6 | 3/6 | 386, 539, 558 | 583, 618, 786 | |
| YN | M/M | E/E | 0/5 | 1/5 | 575 | 391, 659, 674, 778 | |
| VV2 sCJD | |||||||
| AK | V/V | E/E | 0/5 | 0/3 | 636, 684, 772 | >750 | |
| Variant CJD | |||||||
| 96/02 | M/M | E/E | 0/6 | 6/6 | 593, 633, 686 | ||
| 686, 692, 943 | |||||||
| 96/07 | M/M | E/E | 1/5 | 4/5 | 587, 893, 896 | 507 | |
| 910 | |||||||
| 96/45 | M/M | E/E | 1/5 | 3/5 | 563, 790, 852 | 329, 584 | |
| Dura graft-associated CJD | |||||||
| TM/non-plaque | M/M | E/E | 5/5 | 5/5 | 167 ± 24.7 | ||
| TM/plaque | M/M | E/E | 0/6 | 0/6 | 518, 768, 821 | ||
| 821, 835, 840 | |||||||
| KR/plaque | M/M | E/E | 0/7 | 0/6 | 549, 549, 722 | ||
| 829, 836, 885 | |||||||
| NT/plaque | M/M | E/E | 0/5 | 0/3 | 545, 714, 714 | >900 | |
| Inherited diseases | |||||||
| GSS102 (NG) | M/M | E/K‡ | 3/3 | 3/3 | 365 ± 26.6 | ||
| CJD232 (TM) | M/M | E/E | 4/4 | 4/4 | 177 ± 4.9 | ||
M/M, V/V and E/E; methionine, valine, and glutamate homozygosity, respectively. M/V and E/K; methionine/valine and glutamate/lysine heterozygosity, respectively.
*The number of mice with symptomatic or pathological evidence of prion disease divided by the number of mice examined symptomatologically or pathologically: Sympt., symptomatically positive for prion disease; Pathol., pathologically positive. The difference between the numbers of symptomatologically examined mice and pathologically examined ones indicates the number of mice alive.
†The period from inoculation to sacrifice of mice that became terminally ill with (pathology +) or without (pathology −) pathological evidence of prion disease: it is shown as mean ± SD or by individual numbers. For mice alive (alive), the observation period since inoculation is shown.
‡The polymorphism, lysine at codon 219, is on the allele without the pathogenic mutation P102L.
Transmission Experiments
The production of the knock-in mouse expressing human-mouse chimeric PrP (Ki-ChM) used in the current transmission study has been described in detail before. 14,15 Briefly, the chimeric molecule is based on mouse PrP sequence, in which the sequence between SmaI site and BstEII site were replaced by the corresponding human PrP sequence with methionine at codon 129. Consequently, after processing of the N-terminal signal peptide (residues 1 to 22) in the posttranslational modification, the molecule practically has mouse-specific PrP residues only at the C-terminus (Figure 2) ▶ . The relative expression level of chimeric PrP in the brain ofKi-ChM was assessed by Western blot and proved similar to that of endogenous PrP in wild-type mice. A 10% (w/v) homogenate of the brain tissue from frontal cortex of each human prion disease was prepared in sterile phosphate-buffered saline using a disposable glass homogenizer and 20 μl was inoculated. The mice were examined daily after appearance of central nervous system dysfunction and were sacrificed if any sign of distress appeared or the diagnosis of prion disease was made. The criteria for clinical diagnosis of mouse prion disease were as described. 19 At autopsy, the whole brain was cut mid-sagitally into two halves: the right half was immersion-fixed in 10% buffered formalin for histopathological analysis and the left half was immediately frozen for Western immunoblots.
Figure 2.

Comparison of the structures of different human-mouse chimeric PrP and human PrP The diagrams represent states after posttranslational modification (N-glycosylation is omitted). The vertical short lines indicate the mouse-specific residues. Black and white arrowheads indicate methionine and valine at codon 129, respectively.
The Ki-ChM mouse has been shown to be highly susceptible to a sporadic CJD, case H-3, homozygous for methionine at codon 129. 14 Therefore, we performed end-point titration study to determine the infectivity of this prion strain in Ki-ChM mouse. Brain homogenate was serially diluted and inoculated intracerebrally to Ki-ChM mice, and the infectivity titer was calculated by the Behrens-Karber method as described. 14
Neuropathological Examinations of Ki-ChM Mice
Formalin-fixed mouse brains were treated with formic acid to inactivate infectivity, embedded in paraffin, and then cut into 5-μm sections. Deparaffinized sections were pretreated with hydrolytic autoclaving before immunohistochemistry to enhance the immunoreactivity. 20 PrP-N antiserum, 21 rabbit-derived polyclonal antibodies raised against synthetic polypeptides corresponding to the N-terminal residues 25 to 49 of human PrP, was used as the primary antiserum and anti-rabbit immunoglobulins conjugated with peroxidase-conjugated dextran polymer, Envision (DAKO, Glostrup, Denmark), was used as the secondary antibody. Hematoxylin and eosin and Congo red staining was performed with standard techniques. The mice were judged neuropathologically positive when fine-granular PrP-immunopositive deposits, so-called “synaptic-type deposits,” in the gray matter, or plaque-like PrP-positive deposits are recognized.
Western Immunoblotting of Ki-ChM Mouse-Derived PrPres
PrPres was extracted from the mouse brain tissues as described 22 with a little modification. Briefly, brain tissues were homogenized in 50 mmol/L Tris-HCl (pH 8.0), 10 mmol/L NaCl, and 10 mmol/L MgCl2 with glass homogenizers. DNase1 (50 U, Takara, Bio Inc, Otsu, Japan) was added to the homogenate and incubated at 37°C for 60 minutes. Then 30% N-lauroylsarcosine (Sarcosyl) was added to final concentration of 10% and rehomogenized. The homogenate was centrifuged at 22,000 × g for 30 minutes. The supernatant was digested with proteinase-K (20 μg/ml; Wako Chemicals, Osaka, Japan), at 37°C for 60 minutes, and then centrifuged at 453,000 × g for 60 minutes (CP100H model, RP100AT6; Hitachi Koki Co, Tokyo, Japan). The final pellet was resuspended in 1× Laemmli’s sample buffer and boiled. For deglycosylation, PrPres denatured in 1× Laemmli’s sample buffer was treated with N-glycosidase F (PNGase F; New England Biolabs, Beverly, MA) in 100 mmol/L sodium phosphate (pH 7.5)/2% Nonidet P-40. The 3F4 monoclonal antibody 23 was used as the primary antibody. Horseradish peroxidase-conjugated anti-mouse IgG was used as the secondary antibody. Enhanced chemiluminescence (Amersham Biosciences, UK) was used to visualize the blots on Hyperfilm ECL (Amersham Biosciences UK); for analysis of PrPres glycoform ratios images obtained through an imaging device, Versa Doc 5000, were quantified with Quantity One software (Bio-Rad Laboratories, California, USA).
Results
Susceptibility of Ki-ChM Mice to Human Prions
The Ki-ChM mouse was highly susceptible to prions from patients with MM1-sCJD and MV1-sCJD with all of the mice inoculated succumbing to disease within quite short incubation periods, ∼150 days (Table 1) ▶ . Ki-ChM mice proved to be also highly susceptible to prion diseases of other etiologies; after inoculation of np-dCJD and CJD232, an iatrogenic CJD and a inherited disease respectively, all of the mice developed clinical disease in 160 to 180 days. Although the incubation period for GSS102 was relatively long, ∼360 days, the occurrence of the clinical disease suggests fair susceptibility of the mice to this type of inherited prion disease. On the other hand, the Ki-ChM mouse was much less susceptible to the prions from MM2-T-sCJD and vCJD, with a few mice developing clinical prion disease only after more than 500 days after inoculation. Although the occurrence of clinical disease was rather low, many of the mice inoculated with vCJD prions showed pathological evidence of transmission as described below. However, among the mice inoculated with either MM2-T-sCJD or vCJD, there were individuals that completely lack any sign of transmission. As for VV2-sCJD and p-dCJD, Ki-ChM mice seemed totally resistant to them with none of the mice showing clinical or pathological sign of transmission. We graded the susceptibility of Ki-ChM mouse to a given human prion as either susceptible (when all of the mice inoculated developed clinical illness), less susceptible (when some mice survived without any clinical or pathological signs of prion disease), or resistant (when none of the mice had even pathological sign of prion disease).
End-Point Titration of MM1-sCJD Prion
After the inoculation with serially-diluted H-3, all mice developed clinical disease at dilution rates lower than 105: at the dilution of 10−1, the incubation time was 141 ± 9.2 days; at 10−2, 153 ± 7.3 days; at 10−3, 198 ± 12.3 days; at 10−4, 243 ± 58.5 days (Table 2) ▶ . At dilution rates more than 105, only a fraction of mice inoculated developed illness, but the incubation time exceeded 300 days only toward the end-point: at 10−5, two of five mice inoculated developed illness with incubation time, 269 days; at 10−6, only one succumbed at 348 days. The infectivity titer of the brain tissue of the MM1-sCJD, H-3, was calculated as 7.8 logLD50 U/g.
Table 2.
Transmission of Serially Diluted Brain Homogenate from MM1-sCJD (H-3)
| Dilution rate of the inoculum | Incubation periods* (days ± S.D.) | Occurrence (diseased/inoculated) |
|---|---|---|
| 10−1 | 141 ± 9.2 | 7/7 |
| 10−2 | 153 ± 7.3 | 4/4 |
| 10−3 | 198 ± 12.3 | 5/5 |
| 10−4 | 243 ± 58.5 | 5/5 |
| 10−5 | 269, 269 | 2/5 |
| 10−6 | 348 | 1/5 |
| 10−7 | >377 | 0/6 |
| 10−8 | >377 | 0/6 |
*Incubation periods: Means ± SD days from the inoculation to the development of disease.
Neuropathological Examinations of Ki-ChM Mice
Pathologically, the brains of MM1- and VM1-sCJD-inoculated Ki-ChM exhibited diffuse spongiform changes typical of prion disease; iatrogenic CJD, np-dCJD, and inherited prion diseases, GSS102 and fCJD232, also exhibited similar changes (Figure 3B) ▶ . These involved the cerebral cortex, caudate/putamen, hippocampus, thalamus, substantia nigra, superior colliculus, and to a lesser extent, the hypothalamus and pons. PrP deposition in the affected mouse brains was observed as fine granular synaptic-type deposits in gray matter (Figure 3, A and C) ▶ . The intensity of the immunostaining grossly correlated with the severity of spongiform changes in multiple gray matter areas.
Figure 3.
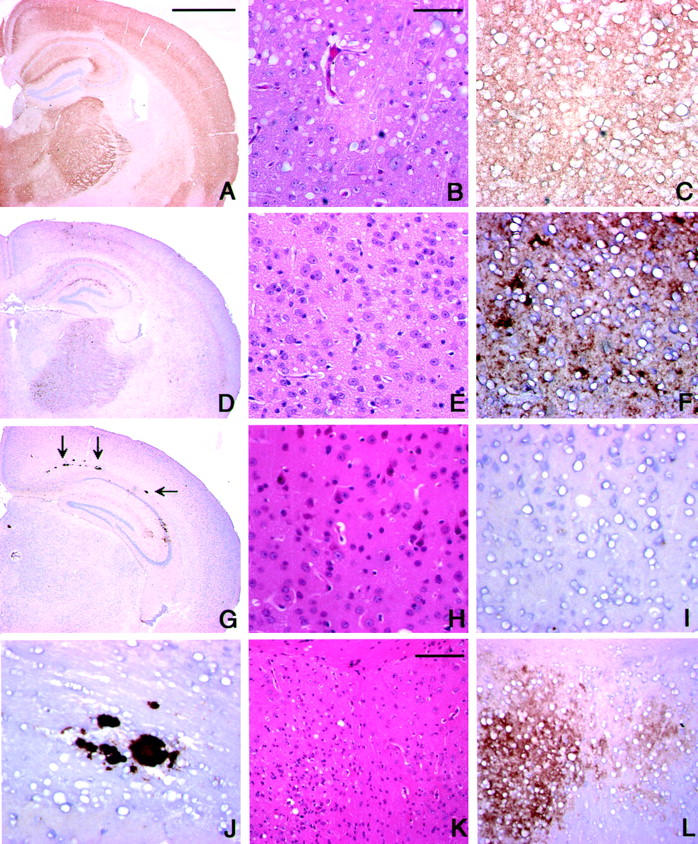
Pathological findings in prion-inoculated Ki-ChM mice A–C: VM1 inoculated; D–F: MM2-T (NK) inoculated; G–L: vCJD (96/02) inoculated. B, E, H, and K are H&E staining and the others are PrP immunostaining. B, C, E, F, H, and I: Cerebral cortex; J: periventricular white matter; K and L: septal area. A and D: Synaptic-type diffuse deposition of PrP is in cerebral cortex, hippocampus, thalamus, and caudate/putamen. G: Arrows indicate plaque-type PrP deposits in the periventricular white matter; synaptic-type deposits are only in parts of hippocampus. J: A magnified view of plaque-type PrP deposits in G. Vacuoles of various size are seen in B and E, but are absent in H. K: Spongiform change with gliosis is evident in the left side of image, while it is much milder in the right side; its intensity parallels the PrP deposition shown in L. Scale bars: 1 mm (A, D, G); 50 mm (B, C, E, F, H, I, J); 100 mm (K, L).
Brains of the three mice inoculated with MM2-T-sCJD showed spongiform and synaptic-type PrP deposition with a distribution similar to those in MM1/VM1-inoculated mice (Figure 3 ▶ ; D to F). Some coarse granular PrP deposits were also observed in gray matter (Figure 3F) ▶ .
The pathology of vCJD-inoculated mice was unique. Spongiform changes and synaptic-type PrP deposition were observed only focally near the inoculation site (hippocampus, thalamus, or septal nuclei) and were much milder than those in MM1/VM1-inoculated mice (Figure 3; G, H, I, K, and L) ▶ . Meanwhile, plaque-type PrP deposits were found in the periventricular white matter and in the gray matter near the inoculation site, mainly thalamus and hippocampus (Figure 3, G and J) ▶ . When stained by Congo red dye, at least some of the plaques exhibited birefringence under polarized light, indicative of amyloid (data not shown).
Western Immunoblotting of Ki-ChM Mouse-Derived PrPres
To confirm the successful transmission of vCJD prions to Ki-ChM mice, PrPres in the brain tissue of vCJD-inoculated mice was examined by Western immunoblot (Figure 1B) ▶ . Although PrPres was present also in the vCJD-inoculated Ki-ChM mouse as MM1- or MM2-T-sCJD-inoculated Ki-ChM mice, the unglycoform of the vCJD-inoculated mouse seemed to migrate slightly slower than that of the inoculated vCJD prion (Figure 1B ▶ , lanes 5 and 6). The molecular mass of deglycosylated PrPres from the vCJD-inoculated Ki-ChM mouse was estimated as ∼20 kd because it took a mean position between those of MM1-inoculated mice (∼21 kd) and MM2-T-inoculated mice (∼19 kd) (Figure 1C) ▶ ; PrPres size was preserved through transmission for MM1- and MM2-T-sCJD but not for vCJD. The shift of PrPres size of vCJD prion through transmission to Ki-ChM mice may reflect the influence of the PrPSc conformation on transmission; a similar phenomenon was observed in transmission studies with Tg mice expressing PrP with 129V. 9 Likewise, the relative ratio of PrPres glycoform was not always preserved through transmission. Although the dominance of di- and mono-glycoform relative to nonglycoform was preserved for vCJD, the glycoform ratios changed for MM1- and MM2-T-sCJD (Figure 1, A and B) ▶ . The ratios of di-, mono-, and unglycoform of PrPres from MM1-sCJD patient (H-3) were 15%, 48%, and 37%, respectively, however, in the PrPres from the recipient Ki-ChM mouse the ratios of di-, mono-, and unglycoform were 51%, 42%, and 7%, and the diglycoform was the most abundant (Figure 1B ▶ , lanes 1 and 2). Similarly, although the ratio of di-, mono-, and unglycoform of PrPres from MM2-T-sCJD (NK) was 8%, 44%, and 48%, respectively, diglycoform of the PrPres from the Ki-ChM accounted for 51% (Figure 1B ▶ , lanes 3 and 4). As a possible explanation for the difference in glycoform ratios between PrPres from inocula and mice, the contribution of the effects of postmortem glycolytic changes cannot be ruled out because as for human samples the antemortem and postmortem conditions were not constant among cases, in contrast to those of mouse samples.
Discussion
Ki-ChM mice showed quite different levels of susceptibility to various human prions: highly susceptible to MM1-sCJD, MV1-sCJD, np-dCJD, GSS102, and CJD232; while much less susceptible to MM2-T-sCJD, vCJD, and totally resistant to VV2-sCJD and p-dCJD. Until today, several lines of transgenic (Tg) mice highly susceptible to different human prions have been reported: Tg mice expressing human PrP with 129M, eg, Tg(HuPrP129M+/+ Prnp0/0), 35 mice: 12 Tg mice expressing human PrP with valine at codon 129 (129V), eg, Tg(HuPrP129V+/+ Prnp0/0), 152 mice; 8,9 and Tg mice expressing human-mouse chimeric MHu2M PrP and its modified forms, eg, Tg(MHu2M)5378/Prnp0/0 mice 10,11 and Tg(MHu2M, M165V,E167Q)22372/Prnp0/0, 13 respectively. Interestingly, the susceptibility profiles to various human prion diseases were different between these Tg mice (Table 3) ▶ . Briefly, Tg mice expressing human PrP with 129V were highly susceptible to all of the human prions examined except for vCJD, although the incubation periods are not very short. 8,9 Tg (HuPrP129M+/+ Prnp0/0) mice were as susceptible to prions with 129M as Tg mice expressing PrP with 129V but seem less susceptible to prions with 129V; as for vCJD, although they hardly developed clinical disease, all of the inoculated mice showed pathological signs of transmission of vCJD. 12 Tg (MHu2M) Prnp0/0 mice were reported to be highly susceptible to some typical CJD with 129M, eg, MM1-sCJD, with short incubation periods, ∼200 days, 10,11 and sporadic fatal insomnia as well, 2 but seem less susceptible to prions with 129V 5,11 and vCJD. 13 Tg mice expressing another type of chimeric PrP, Tg(MHu2M, M165V,E167Q)Prnp0/0, which is distinct from MHu2M PrP in two residues at codons 165 and 167 showed dramatically short incubation periods, ∼110 days, for MM1-sCJD. 13 Compared with those Tg mice, the susceptibility profile of Ki-ChM mouse is characterized by the rather short incubation periods, ∼150 days, for some types of prions including MM1-sCJD. The reason for the rather short incubation periods of Ki-ChM mouse is not obvious. In the above-mentioned Tg mice expressing MHu2M(M165V, E167Q) dramatically short incubation periods for MM1-sCJD were achieved by replacing the two amino acid residues at the C-terminal end of the central human sequence part to mouse residues. 13 Because these residues are postulated to be involved in the interface with the third component in the conversion reaction, protein X, the abbreviated incubation periods could be presumably attributed to the improved interaction with the third component. 24 On the other hand, in the chimeric PrP of Ki-ChM mouse the corresponding two residues remain as human residues, instead this chimeric PrP is different from MHu2M PrP in that the N-terminal part including the octapeptide repeat region is all human sequence after posttranslational modification. The role of the octapeptide repeat region in the conversion of PrPC to PrPSc and its propagation has not been defined. But given the facts that amplified repeats are associated with familial prion diseases, 1 and the experimental fact that Tg mice expressing N-terminus-truncated PrP are relatively resistant to transmission of prions, 25 it seems possible that the octapeptide repeat region plays some role in prion propagation, and N-terminal human sequence has contributed to the abbreviated incubation periods of Ki-ChM mouse. In combination with other mutations that are known to shorten incubation periods, 13 chimeric PrP with all human sequence in the N-terminal part may further shorten the incubation.
Table 3.
Comparison of the Results of the Transmission Studies of Various Human Prions between Different Genetically Modified Mice
| Transgene | ChM | MHu2M | MHu2M (M165V, E167Q) | HuPrP-129M | HuPrP-129V | |||||||||
| Codon 129 | Met | Met | Met | Met | Val | |||||||||
| Tg mice | KiChM | Tg(MHu2M) 5378/Prnp0/0 | Tg(MHu2M, M165V, E167Q) 22372/Prnp0/0 | Tg (HuPrP129M Prnp0/0)−35 | Tg(HuPrP,M129) 440/Prnp0/0 | Tg (HuPrP,V129)152/Prnp0/0 | ||||||||
| Expression level | 1 × Mo | 1 × Hu | 1∼2 × Hu | 2 × Hu | 2 × Hu | 2 × Hu | 4–8 × Hu | |||||||
| References | Present paper | Korth et al 13 | Korth et al 13 | Asante et al 12 | Korth et al 13 | Hill et al 9 | Korth et al 13 | |||||||
| Incubation Times* | ||||||||||||||
| sCJD-MM1 | 134–153 | 180–217 | 106–114 | 223–237* | 155–165 | 210±4 | 254±6 | |||||||
| -MM2 | >650, >680 | 232 ± 5, >580 | 368–556 | |||||||||||
| -MM2-T | 539, 558 | 221 ± 6 | 303 ± 20 | 699 ± 30 | ||||||||||
| -MV1 | 141 ± 5 | 214–215 | 124 ± 3 | 241 ± 1§ | 176 ± 2 | |||||||||
| -MV2 | >640 | >450 | 437 ± 31§ | 307–419 | 209–231 | |||||||||
| -VV2 | >750 | 433−531 | >450 | 354§ | 248–448 | 195–223 | ||||||||
| vCJD | >700 | 563–647 | 335–380 | 340–720 | 228 ± 15 | |||||||||
| Susceptibility¶ | ||||||||||||||
| Susceptible | sCJD-MM1 | sCJD-MM1 | sCJD-MM1 | sCJD-MM1*§ | sCJD-MM1 | sCJD-MM1§ | sCJD-MM1 | |||||||
| -MV1 | -MM2-T | -MV1 | -MV1 | -VV2 | -MM2 | |||||||||
| np-dCJD | vCJD | -MV2 | -MV2 | |||||||||||
| CJD232 | -MM2 | -VV2 | ||||||||||||
| GSS101 | ||||||||||||||
| Less susceptible | sCJD-MM2-T | sCJD-VV2 | sCJD-VV2 | sCJD-MV2*§ | sCJD-MM2-T | vCJD | ||||||||
| vCJD | vCJD | -MM2-T | -VV2§ | -VV2 | ||||||||||
| vCJD | vCJD | |||||||||||||
| Resistant | sCJD-VV2 | sCJD-MM2 | sCJD-MV2 | sCJD-MM2 | ||||||||||
| p-dCJD | -MV2 | |||||||||||||
| -VV2 | ||||||||||||||
| End-point titration for MM1-sCJD‡ | ||||||||||||||
| LogLD50/g | 7.8 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | |||||||
| For sCJD-MM1 | (case,H-3) | |||||||||||||
| PrPres size through the first passage† | ||||||||||||||
| sCJD-MM1 | ∼21 kDa | ∼21 kDa | ∼21 kDa | ∼21 kDa | N.D. | Some, shifted | N.D. | |||||||
| -MM2-T | ∼19 kDa | ∼19 kDa | N.D. | N.D. | N.D. | |||||||||
| vCJD | ∼20 kDa | ∼ 19 kDa | ∼19 kDa | ∼19 kDa | N.D. | Shifted | N.D. | |||||||
N.D.; not described.
*Incubation times; the mean (and the standard deviation or standard error of mean if available) of the days from the inoculation to the development of illness. For comparison between different genetically modified mice, results for sporadic CJD cases have been cited.
†PrPres sizes through the first passages; the mobility on SDS-PAGE of the PrPres derived from the mice inoculated with the indicated human prions.
‡End-point titration for sCJD-MM1; this was calculated according to Behrens-Karber’s method based on the results of the transmission studies using serially-diluted brain homogenates.
§Because the classification of human prion diseases are different, their type-1 and type-2 PrPres were interpreted as our PrPres type 1, and their type 3 as our PrPres type 2.
¶Susceptible, all mice inoculated developed clinical prion diseases; less susceptible, some of the mice inoculated lacked either clinical or pathological sign of prion disease; resistant, none of the mice had any sign of prion disease.
Another point to note in the present study is the less susceptibility of Ki-ChM mice to some type of human prion diseases, such as MM2-T-sCJD and vCJD, and the total resistance to VV2-sCJD and p-dCJD. The possibility of smaller amounts of the PrPSc in those inocula than those of efficiently transmitting prions is not plausible; on Western immunoblot analysis for molecular typing of PrPres of the samples, the PrPres of every sample was recognized as clear blots and the difference in the concentration of PrPres between the most abundant and the least was within 10-fold; in addition, in a transmission study challenging Ki-ChM mice with serially diluted brain homogenate from MM1-sCJD (H-3), at a dilution of 10−2, the incubation periods did not make much difference in the incubation times, and even at the dilution of 10−4, the mean incubation time did not exceed 250 days (Table 2) ▶ . Whether a mouse more susceptible to some types of prions becomes unsusceptible all of the more to other types of prions is a problem of practical importance; if so, the versatility of genetically modified mice designed to have short incubation periods may be limited to some specific prions. In the already reported transmission studies with Tg mice expressing other kinds of chimeric PrP, similar tendency was observed, too; they showed shorter incubation periods for MM1-sCJD prions than Tg mice expressing human PrP, however the former were less susceptible to other types of prion diseases than the latter (Table 3) ▶ . 8-13
The importance of the PrPSc conformation in the transmission is demonstrated in the presented results. Among the human prion diseases subjected to transmission to Ki-ChM mice in this study, the five prions, MM1-sCJD, np-dCJD, MM2-T-sCJD, p-dCJD, and vCJD, had the identical primary structure of PrP, however, their transmissibility varied greatly: MM1-sCJD and np-dCJD transmitted quite efficiently, MM2-T-sCJD and vCJD transmitted inefficiently, and p-dCJD did not transmit at all. Given that the PrPres sizes reflect the PrPSc conformation to some degree, 26,27 the efficiently transmitting two prions and the inefficiently transmitting two could have rather different PrPSc conformation because MM1-sCJD and np-dCJD prions have PrPres of 21 kd, whereas MM2-T-sCJD and vCJD have PrPres of 19 kd. This indicates that the PrPSc conformation has more influence on the transmission efficiency of prions than the primary structure, being consistent with recently reported experiments. 28,29 In addition to the identical primary structure, the PrPres of the three prions, MM1-sCJD, np-dCJD, and p-dCJD have the identical PrPres size of 21 kd, being practically indistinguishable from one another; 16 despite that, p-dCJD never transmitted to the Ki-ChM mouse, whereas the other two prions transmitted very efficiently. Such heterogeneity in transmission properties among prions with the identical type of PrPres and identical etiology is also observed in other transmission studies; 11,13 this suggests a considerable heterogeneity among the prions categorized into the same group based on the properties of PrPres and etiology. According to protein-only hypothesis, this may stem from a subtle structural variation of PrPSc that does not manifest in the currently recognized properties of PrPres, but transmission experiments could detect the difference.
Although much remains to be elucidated about the mechanism of prion propagation, it seems true that the infectivity titer of a prion varies among different hosts depending on the host-encoded PrP, for instance a vCJD case, 96/02, exhibited quite different transmissibility to Ki-ChM and Tg(MHu2M, M165V,E167Q)Prnp0/0, and that each of the many human prion strains has its optimal host-encoded PrP, which makes the host animal very susceptible to the prion and develop illness in rather short incubation periods. Assessing the transmissibility profile of a given human prion to a panel of Tg or knock-in mice with different primary structures of PrP, and specifying the optimal host-encoded PrP would facilitate in developing more accurate classification of human prions. On specifying the optimal PrP for a prion, quantitation of the infectivity of the prion for the mouse by end-point titration should be taken into account as well as the length of the incubation periods. The present study sets an example; as for Ki-ChM mouse, the infectivity titer of a case of MM1-sCJD, H-3, which transmitted to this line of mice in ∼150 days, was determined to be 7.8 logLD50 U/g. This parameter is important because such titrated infectivity would not only allow objective comparison between different lineages of mice, but also would guarantee the reliability of the mouse as a bioassay tool for the type of prion. In addition, the accumulation of such systematic data would help develop genetically engineered mice suitable for bioassay for various human prions, and, moreover, advance our understanding of the molecular mechanism of prion propagation.
Footnotes
Address reprint requests to Tetsuyuki Kitamoto, Department of Neurological Science, Tohoku University Graduate School of Medicine, 2-1 Seiryo-machi, Sendai 980-8575, Japan. E-mail: kitamoto@mail.cc.tohoku.ac.jp.
Supported by a grant from the Organization for Pharmaceutical Safety and Research (to S. M. and T. K.); a grant from the Ministry of Health, Labor, and Welfare, Japan (to S. M. and T. K.); and grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (to T. M. and T. K.).
References
- 1.Prusiner SB: The prion diseases. Brain Pathol 1998, 8:499-513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ, Prusiner SB: Prion protein conformation in a patient with sporadic fatal insomnia. N Engl J Med 1999, 340:1630-1638 [DOI] [PubMed] [Google Scholar]
- 3.Parchi P, Giese A, Capellari S, Brown P, Schultz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julein J, Vital C, Ghetti B, Gambetti P, Kretzschmar H: Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999, 46:224-233 [PubMed] [Google Scholar]
- 4.Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, Kretzschmar HA, Lantos PL, Masullo C, Schlote W, Tateishi J, Weller RO: Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol 1995, 5:459-466 [DOI] [PubMed] [Google Scholar]
- 5.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB: Evidence of the conformation of the pathogenic isoform of the prion protein enciphering and propagating prion diversity. Science 1996, 274:2079-2082 [DOI] [PubMed] [Google Scholar]
- 6.Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG: Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA 2000, 97:10168-10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AAF, Trojanowski JQ, Petersen RB, Gambetti P: Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996, 39:767-778 [DOI] [PubMed] [Google Scholar]
- 8.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF: Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996, 383:685-690 [DOI] [PubMed] [Google Scholar]
- 9.Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, Doey L, Lantos P: The same prion strain causes vCJD and BSE. Nature 1997, 389:448-450 [DOI] [PubMed] [Google Scholar]
- 10.Telling GC, Scott M, Hsiao KK, Foster D, Yang S-L, Torchia M, Sidle KCL, Collinge J, DeArmond SJ, Prusiner SB: Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA 1994, 91:9936-9940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen F, DeArmond SJ, Prusiner SB: Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995, 83:79-90 [DOI] [PubMed] [Google Scholar]
- 12.Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, Welch J, Hill AF, Lloyd SE, Wadsworth JDF, Collinge J: BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO 2002, 21:6358-6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, Parchi P, Gambetti P, Will R, Ironside J, Heinrich C, Tremblay P, DeArmond SJ, Prusiner SB: Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci USA 2003, 100:4784-4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitamoto T, Mohri S, Ironside JW, Miyoshi I, Tanaka T, Kitamoto N, Itohara S, Kasai N, Katsuki M, Higuchi J, Muramoto T, Shin R-W: Follicular dendritic cell of the knock-in mouse provides a new bioassay for human prions. Biochem Biophys Res Commun 2002, 294:280-286 [DOI] [PubMed] [Google Scholar]
- 15.Kitamoto T, Nakamura K, Nakao K, Shibuya S, Shin RW, Gondo Y, Katsuki M, Tateishi J: Humanized prion protein knock-in by Cre-induced site-specific recombination in the mouse. Biochem Biophys Res Commun 1996, 222:742-747 [DOI] [PubMed] [Google Scholar]
- 16.Shimizu S, Hoshi K, Muramoto T, Honma M, Ironside JW, Kuzuhara S, Sato T, Yamamoto T, Kitamoto T: Creutzfeldt-Jakob disease with florid-type plaques after cadaveric dura mater grafting. Arch Neurol 1999, 56:357-362 [DOI] [PubMed] [Google Scholar]
- 17.Ironside JW: Pathology of variant Creutzfeldt-Jakob disease. Arch Virol Suppl , 16:143-151 [DOI] [PubMed] [Google Scholar]
- 18.Kitamoto T, Ohta M, Doh-ura K, Hitoshi Y, Terao J, Tateishi J: Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Straussler syndrome. Biochem Biophys Res Commun 1993, 191:709-714 [DOI] [PubMed] [Google Scholar]
- 19.Mohri S, Tateishi J: Host genetic control of incubation periods of Creutzfeldt-Jakob disease. J Gen Virol 1989, 70:1391-1400 [DOI] [PubMed] [Google Scholar]
- 20.Kitamoto T, Shin R-W, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi J: Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol 1992, 140:1285-1294 [PMC free article] [PubMed] [Google Scholar]
- 21.Kitamoto T, Muramoto T, Hilbich C, Beyreuther K, Tateishi J: N-terminal sequence of prion protein is also integrated into kuru plaques in patients with Gerstmann-Straussler syndrome. Brain Res 1991, 545:319-321 [DOI] [PubMed] [Google Scholar]
- 22.Kitamoto T, Mohri S, Tateishi J: Organ distribution of proteinase-resistant prion protein in humans and mice with Creutzfeldt-Jakob disease. J Gen Virol 1989, 70:3371-3379 [DOI] [PubMed] [Google Scholar]
- 23.Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H: Mouse polyclonal and monoclonal antibody to scrapie-associated fibril protein. J Virol 1987, 61:3688-3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB: Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci USA 1997, 94:10069-10074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, von Mering C, Aguzzi A, Weissmann C: Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 2000, 27:399-408 [DOI] [PubMed] [Google Scholar]
- 26.Caughey B, Raymond GJ, Bessen RA: Strain-dependent differences in β-sheet conformations of abnormal prion protein. J Biol Chem 1998, 273:32230-32235 [DOI] [PubMed] [Google Scholar]
- 27.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B: Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995, 375:698-700 [DOI] [PubMed] [Google Scholar]
- 28.Peretz D, Williamson RA, Legname G, Matsunaga Y, Vergala J, Burton DR, DeArmond SJ, Prusiner SB, Scott MR: A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 2002, 34:921-932 [DOI] [PubMed] [Google Scholar]
- 29.Hill AF, Joiner S, Linehan J, Desbuslais M, Lantos PL, Collinge J: Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci USA 2000, 97:10248-10253 [DOI] [PMC free article] [PubMed] [Google Scholar]