

Abstract
Ebola virus (EBOV) infection causes a severe and often fatal hemorrhagic disease in humans and nonhuman primates. Whether infection of endothelial cells is central to the pathogenesis of EBOV hemorrhagic fever (HF) remains unknown. To clarify the role of endothelial cells in EBOV HF, we examined tissues of 21 EBOV-infected cynomolgus monkeys throughout time, and also evaluated EBOV infection of primary human umbilical vein endothelial cells and primary human lung-derived microvascular endothelial cells in vitro. Results showed that endothelial cells were not early cellular targets of EBOV in vivo, as viral replication was not consistently observed until day 5 after infection, a full day after the onset of disseminated intravascular coagulation. Moreover, the endothelium remained relatively intact even at terminal stages of disease. Although human umbilical vein endothelial cells and human lung-derived microvascular endothelial cells were highly permissive to EBOV replication, significant cytopathic effects were not observed. Analysis of host cell gene response at 24 to 144 hours after infection showed some evidence of endothelial cell activation, but changes were unremarkable considering the extent of viral replication. Together, these data suggest that coagulation abnormalities associated with EBOV HF are not the direct result of EBOV-induced cytolysis of endothelial cells, and are likely triggered by immune-mediated mechanisms.
Ebola hemorrhagic fever (HF) is characterized by hypotension, generalized fluid distribution problems, lymphopenia, coagulative disorders, and hemorrhages. Disseminated intravascular coagulation (DIC) is a prominent manifestation of Ebola virus (EBOV) infection. DIC is a syndrome characterized by coagulation abnormalities including systemic intravascular activation of coagulation leading to widespread deposition of fibrin in the circulation, which contributes to the multiple organ failure and high mortality rates characteristic of EBOV infections. The mechanisms by which EBOV causes multisystemic disease are only partially understood. Although viral replication in target tissues is clearly a factor, various strategies that restrict replication of EBOV have, at best, only delayed onset of lethal disease. 1-3 Recent studies suggested that the EBOV glycoprotein (GP) is the main determinant of vascular cell injury and consequently direct EBOV replication-induced structural damage of endothelial cells triggers the hemorrhagic diathesis. 4,5 This hypothesis has not been rigorously tested in vitro or in vivo.
Viral infections can exert changes in the vascular endothelium in a variety of ways, such as inducing endothelial cell activation indirectly by infecting and activating leukocytes and triggering the synthesis and local production of proinflammatory soluble factors or by directly inducing changes in endothelial cell expression of cytokines, chemokines, and cellular adhesion molecules in the absence of immune mediators (as a direct result of virus infection). Mediators released from activated endothelial cells that can modulate vascular tone, thrombosis, and/or inflammation include nitric oxide, prostacyclin, interferons (IFNs), interleukin (IL)-1, IL-6, and chemokines such as IL-8. 6,7 Few studies have evaluated the host gene response of endothelial cells infected with EBOV; Harcourt and colleagues reported that EBOV inhibits the induction of genes by double-stranded RNA in endothelial cells. 8
Despite more than 1000 known fatal cases of EBOV infection, little has been learned from necropsies regarding the role of the endothelium in disease pathogenesis because only a very limited number of tissues from a handful of cases in 1976 and 1996 have been examined. 9-11 EBOV infection of endothelial cells in postmortem tissues was demonstrated in one of these studies. 11 By comparison, EBOV infection of endothelial cells has been well documented in nonhuman primates. 12-16 However, with few exceptions, these investigations examined monkeys killed when moribund and shed little light on the pathogenesis of EBOV infection during times before death.
The specific goals of this study were: 1) to determine the course of EBOV infection and detectable cytopathic disruption of endothelium in nonhuman primates, specifically, to determine whether or not EBOV infection of endothelial cells is an early event or a terminal event in the disease course relative to the first signs of any disruption/degeneration of the endothelium and/or evidence of DIC; and 2) to determine whether or not EBOV infection directly induces the activation and/or cytolysis of human endothelial cells in vitro. The importance of this study is that it cannot be concluded that EBOV-induced damage to the endothelium is exclusively because of immune-mediated mechanisms without first examining the direct effects of EBOV infection on endothelial cell integrity and vascular permeability. It is possible that EBOV mediates disruption of endothelia via an indirect route rather than only by direct infection of endothelial cells.
Materials and Methods
Animals and Inoculations
Healthy, filovirus-seronegative, adult male cynomolgus (Macaca fascicularis) macaques (n = 21, 4 to 6.5 kg) were used for these studies. The complete clinical and histopathological details of these animals are described in a companion paper. 17 Briefly, animals were inoculated intramuscularly with 1 ml of virus stock that contained 1000 plaque forming units (PFUs) of EBOV-Zaire (1995 isolate). Scheduled necropsies were performed at 1 (n = 3), 2 (n = 3), 3 (n = 4), 4 (n = 4), 5 (n = 4), and 6 (n = 3) days after infection.
Serum Albumin and Total Protein
Concentrations of serum albumin and total protein were measured using a Piccolo Point-Of-Care Blood Analyzer (Abaxis, Sunnyvale, CA).
Histology
Formalin-fixed tissues for histology and in situ hybridization were processed and embedded in paraffin according to conventional methods. 18 Histology sections were cut at 5 to 6 μm on a rotary microtome, mounted on glass slides, and stained with hematoxylin and eosin. Replicate sections of all tissues were mounted on positively charged glass slides (Superfrost Plus; Fisher Scientific, Pittsburgh, PA) and immunohistochemically stained for detection of viral antigen by an immunoperoxidase (IPO) method according to kit procedures (Envision System; DAKO Corp., Carpinteria, CA), or by a fluorescence-based method.
Immunohistochemistry
Immunoenzymatic Methodology
Sections were deparaffinized and rehydrated through a series of graded ethanols, pretreated with DAKO Ready to Use Proteinase K (DAKO) for 6 minutes at room temperature, and blocked with DAKO’s Serum-Free Protein Block (DAKO) for 20 minutes before exposure to antibody. Tissue sections were incubated with primary antibody overnight at 4°C using an anti-EBOV rabbit polyclonal (kindly provided by Cindy Rossi) (1:500) or an equal mixture of mouse monoclonal antibodies to EBOV GP and VP40 (1:5000). 14 An alkaline phosphatase-labeled polymer (DAKO Envision System, alkaline phosphatase) was added for 30 minutes and color development was achieved by exposing tissue to the substrate 6-bromo-2-hydroxyl-3-naphtholic acid (Histomark Red; Kirkegaard and Perry, Gaithersburg, MD) for 50 minutes in the dark. Sections were counterstained with hematoxylin. Negative controls included replicate sections exposed to anti-Marburg virus antibodies and unexposed cynomolgus monkey tissue; archived EBOV-infected cynomolgus tissue served as positive controls.
Immunofluorescence Methodology
Tissue sections were deparaffinized, rehydrated, and incubated in 20 μg/ml of proteinase K for 30 minutes at room temperature. Sections were subsequently rinsed, placed in normal goat serum for 20 minutes, and transferred to a mixture of the anti-EBOV antibodies as described above for 30 minutes at room temperature. After incubation, sections were rinsed and stained with goat anti-mouse Alexa 594 (Molecular Probes, Eugene OR), incubated with a pan-T cell marker, CD3 (DAKO), for 30 minutes at room temperature, rinsed, and incubated in goat anti-mouse Alexa 488 (Molecular Probes).
For localization of EBOV antigens in endothelial cells, double stains using a marker for endothelial cells and a pool of anti-EBOV antibodies were used. Briefly, tissue sections were pretreated with proteinase K (20 μg/ml) for 30 minutes at room temperature and incubated in normal goat serum for 20 minutes. Sections were then incubated in a rabbit polyclonal antibody for von Willebrand factor (DAKO) for 30 minutes at room temperature. After incubation, sections were placed in goat anti-rabbit Alexa 488 (Molecular Probes) for 30 minutes at room temperature, rinsed, and incubated in a mixture of the anti-EBOV antibodies described above. Sections were rinsed and incubated in goat anti-mouse Alexa 594 (Molecular Probes) for 30 minutes at room temperature. After rinsing in phosphate-buffered saline (PBS), sections were mounted in an aqueous mounting medium containing 4′,6′-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA) and examined with a Nikon E600 fluorescence microscope (Nikon Instech Co., Ltd., Kanagawa, Japan).
In Situ Hybridization
EBOV GP and VP40 RNA were localized in tissues using digoxigenin-labeled DNA probes. Probe constructs were plasmids (pCR2.1; Invitrogen, San Diego, CA) containing complementary DNA sequences for EBOV GP or VP40. Probes were labeled by nick translation with digoxigenin-11-UTP (Boehringer Mannheim, Indianapolis, IN) following the manufacturer’s recommendations. Before hybridization, tissue sections were incubated with 40 μg/ml of nuclease-free proteinase K (Boehringer Mannheim) in Tris-buffered saline, pH 7.6, for 30 minutes at 37°C. For hybridization, probes were denatured at 95°C for 5 minutes, placed on ice, and then applied to tissue sections and incubated overnight at 42°C. After hybridization, sections were washed in buffer and incubated in alkaline phosphatase-conjugated, anti-digoxigenin antibody (Boehringer Mannheim), diluted 1:600, for 1 hour at 37°C. Sections were washed and the color was developed with 5-bromo-4-chloro-3-indolyl phosphate (NBT/BCIP; Life Technologies, Gaithersburg, MD) as the substrate and nitro blue tetrazolium salt (NBT) as the chromagen for 1 hour at 37°C. Sections were counterstained with nuclear fast red (Vector Laboratories). Tissue sections incubated in the pCR2.1 plasmid lacking the EBOV gene inserts served as negative controls.
Cells and Viruses
Normal primary human umbilical vein endothelial cells (HUVECs) and primary human lung-derived microvascular endothelial cells (HMVEC-Ls) were obtained from Clonetics, Inc. (San Diego, CA), and maintained in endothelial cell growth medium (Clonetics), which was supplemented with human recombinant epidermal growth factor, hydrocortisone, fetal bovine serum, gentamicin, and bovine brain extract (Clonetics). HUVECs and HMVEC-Ls were maintained according to the supplier’s recommendations. Viability was estimated by the trypan-blue exclusion procedure and was greater than 99% in all preparations. Experimental infection conditions consisted of 2.5 × 105 HUVECs or HMVEC-Ls in segregated cultures. Cells were infected at a multiplicity of infection of 1.0 with EBOV that was originally obtained from a fatally infected human from the former Zaire in 1995. 2 After adsorption of virus for 1 hour at 37°C, cultures were washed twice with PBS, refed with fresh endothelial cell growth medium, and incubated for various times at 37°C. Additional cultures of HUVECs and HMVEC-Ls were challenged with an equivalent dose of inactivated EBOV (ie, virus inactivated by exposure to 6 million rads from a 60Co source). Culture medium from Vero cells represented a virus-free stock in all mock infection experiments. Virus titration by plaque assay on Vero cells was performed as previously described. 19
Electron Microscopy
HUVECs and HMVEC-Ls were processed by conventional methods for transmission electron microscopy as previously described for cultured cells. 20 Also, portions of liver; spleen; lung; kidney; adrenal gland; skin; and mesenteric, axillary, and inguinal lymph nodes from EBOV-infected monkeys were immersion-fixed in 4% paraformaldehyde plus 1% glutaraldehyde in 0.1 mol/L Millonig’s phosphate buffer (pH 7.4) and processed for transmission electron microscopy as previously described. 14,15 Briefly, tissues and cultured cells were postfixed in 1% osmium tetroxide in 0.1 mol/L Millonig’s phosphate buffer, rinsed, stained with 0.5% uranyl acetate in ethanol, dehydrated in graded ethanol and propylene oxide, and embedded in Poly/Bed 812 resin (Polysciences, Warrington, PA). Areas to be examined by transmission electron microscopy were selected from 1-μm sections stained with toluidine blue. Ultrathin sections were cut, placed on 200-mesh copper electron microscopy grids, stained with uranyl acetate and lead citrate, and examined using a JEOL 1200 EX transmission electron microscope (JEOL Ltd., Peabody, MA) at 80 kV.
HUVECs and HMVEC-Ls were also processed by conventional methods for scanning electron microscopy. Briefly, cells grown on coverslips were treated with 4% paraformaldehyde and 1% glutaraldehyde in 0.1 mol/L Millonig’s phosphate buffer (pH 7.4) for 2 hours. The specimens were postfixed in 1% osmium tetroxide, rinsed, dehydrated in ethanol, dried by the critical point method in liquid carbon dioxide, and mounted on aluminum scanning electron microscopy stubs with silver paint. Samples were viewed in a Hitachi S-4500 scanning electron microscope (Nissei Sangyo America Ltd., Gaithersburg, MD).
Cytokine/Chemokine and Prostacyclin Production
Cytokine/chemokine levels in HUVEC or HMVEC-L cultures were assayed using commercially available enzyme-linked immunosorbent assay kits according to manufacturer’s directions. Cytokines/chemokines assayed included IFN-α, IL-6, and RANTES (BioSource International, Inc., Camarillo, CA); and IL-8 and IFN-β (R&D Systems, Minneapolis, MN). Prostacyclin levels were determined by measuring its stable metabolite 6-keto-prostaglandin F1α by enzyme-linked immunosorbent assay (R&D Systems).
RNase Protection Assays
Total RNA was extracted from infected/treated HUVECs and HMVEC-Ls by using Trizol (Life Technologies, Inc.) and was stored at −70°C until RNA isolations could be performed. Probe sets were obtained from Pharmingen, La Jolla, CA. The Multiprobe RNase Protection Assay was performed according to manufacturer’s directions (Pharmingen) with minor modifications. 17
Results
Effects of EBOV Infection on Levels of Total Serum Proteins and Albumin
We evaluated levels of total serum proteins and albumin in EBOV-infected monkeys as an indicator of the impairment of endothelial barrier integrity and increased endothelial permeability to macromolecules. Total serum proteins did not fluctuate during the course of EBOV infection however decreases in serum albumin levels were seen by day 4 indicating a loss of only small molecular weight proteins (Figure 1) ▶ .
Figure 1.

Total serum protein and albumin values after infection of cynomolgus monkeys with EBOV-Zaire. Note that day 0 values represent controls that were obtained before the animals were challenged with EBOV.
Evaluation of Endothelial Cell Infection and Cytopathology in Vivo
We evaluated tissues longitudinally collected from 21 EBOV-infected cynomolgus monkeys by immunohistochemistry, in situ hybridization, and electron microscopy to determine whether EBOV infection of endothelial cells is an early event or a terminal event in the disease course relative the first signs of any morphological disruption of the endothelium or evidence of DIC. The principal immunohistochemical and in situ hybridization findings are presented in Table 1 ▶ . Endothelial cell immunoreactivity was not detected in any tissue of any animal until day 4, and even by this time was only an infrequent observation. At day 4, EBOV antigen was very rarely seen in endotheliallike cells lining hepatic and adrenal cortical sinusoids (noted in three of four animals). Also, single endothelial cells lining high endothelial venules in the tonsil contained EBOV antigen in one monkey.
Table 1.
Immunohistochemical and in Situ Hybridization Findings from Endothelial Cells of Cynomolgus Monkeys Infected with Ebola Virus
| Animal number | Days P.I. | HEV | Sin | Spl | Lam prop | Ren cap | Alv cap | Heart | Brach plex | Brain |
|---|---|---|---|---|---|---|---|---|---|---|
| CQ9877 | 1 | − | − | − | − | − | − | − | − | − |
| CQ9890 | 1 | − | − | − | − | − | − | − | − | − |
| 0717CQ | 1 | − | − | − | − | − | − | − | − | − |
| CQ9846 | 2 | − | − | − | − | − | − | − | − | − |
| 0331CQ | 2 | − | − | − | − | − | − | − | − | − |
| CQ9028 | 2 | − | − | − | − | − | − | − | − | − |
| 32Q | 3 | − | − | − | − | − | − | − | − | − |
| CQ8667 | 3 | − | − | − | − | − | − | − | − | − |
| CQ9093 | 3 | − | − | − | − | − | − | − | − | − |
| CQ8681 | 3 | − | − | − | − | − | − | − | − | − |
| 28-427 | 4 | − | − | − | − | − | − | − | − | − |
| CQ9887 | 4 | − | + | − | − | − | − | − | − | − |
| CQ9108 | 4 | − | + | − | − | − | − | − | − | − |
| CQ9095 | 4 | + | + | − | − | − | − | − | − | − |
| 28-221 | 5 | ++ | ++ | + | + | + | − | − | + | − |
| 0359CQ | 5 | + | ++ | + | + | + | − | − | − | − |
| 0323CQ | 5 | ++ | ++ | + | + | + | + | − | + | − |
| 48-143 | 5 | ++ | ++ | + | + | + | + | + | + | + |
| CQ9878 | 6 | ++ | ++ | + | + | + | + | + | + | + |
| 28-332 | 6 | ++ | ++ | + | ++ | + | + | ++ | + | ++ |
| CQ9112 | 6 | ++ | ++ | + | ++ | + | + | ++ | + | ++ |
+, EBOV RNA- and antigen-positive cells were rarely detected; ++, EBOV-positive cells were occasionally detected; +++, EBOV-positive cells were frequently detected; and −, no EBOV-positive cells were detected. Note: the whole field from each of two sections of each tissue was examined in its entirety.
P.I., postinfection; HEV, high endothelial venules of lymphoid tissues; Sin, hepatic and adrenal cortical sinusoids; Spl, endothelial cells in spleen; Lam prop, lamina propria of tongue, nares, lip, larynx, and intestines; Ren cap, renal capillaries; Alv cap, alveolar capillaries; Heart, endothelial cells in heart; Brach plex, endothelial cells of the brachial plexus; Brain, endothelial cells in capillaries and venules in the brain.
By day 5, EBOV RNA- and antigen-positive endothelial cells had increased in number and distribution (Table 1) ▶ but were still eclipsed in magnitude by the overwhelming numbers of immunoreactive monocytes, macrophages, and dendritic cells (Figure 2A) ▶ . Immunostaining for von Willebrand factor, as a marker for endothelial cells, and EBOV antigens, confirmed that endothelial cells were an infrequent target of EBOV infection (Figure 2; B to D) ▶ , and also demonstrated the structural intactness of the endothelium. When observed, immunoreactive endothelial cells were most apparent lining high endothelial venules in lymph nodes (four of four), tonsils (four of four), gut-associated lymphoid tissue (four of four), and brachial plexus (three of four) (Figure 2E) ▶ . At day 6, an increase in the number of EBOV-positive endothelial cells was noted with a distribution paralleling that shown at day 5 (Table 1) ▶ .
Figure 2.
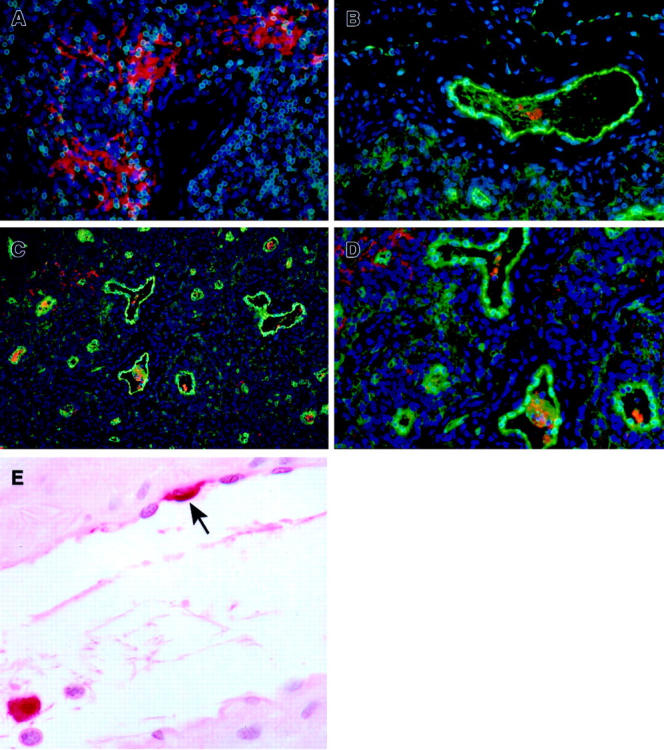
Immunohistochemistry of inguinal lymph nodes of EBOV-Zaire-infected cynomolgus monkeys for cell markers and EBOV at day 5. A: Abundance of EBOV-positive mononuclear cells (red) and extracellular EBOV antigen in extravascular area. Note absence of EBOV-positive endothelial cells. B–D: Double labeling for an endothelial cell marker (von Willebrand factor) (green) and EBOV antigens (red). Note: D is a higher magnification of C. E: Venule, brachial plexus. Rare EBOV-positive endothelial cell (arrow) and typical EBOV-positive monocyte (within lumen). A–D, Immunofluorescence method with 4′,6′-diamidino-2-phenylindole counterstain; E, alkaline phosphatase method. Original magnifications: ×20 (C); ×40 (A, B, D, E).
Ultrastructural changes in endothelia were not detected until day 4 when nonviral tubuloreticular inclusions (TRI), identical to TRI previously described in moribund EBOV-infected monkeys, 13 were observed in occasional endothelial cells in lymphoid tissues (three of four). Infrequent fields in lymphoid tissues also contained endothelial cells that showed morphological evidence of activation (Figure 3, A and B) ▶ , eg, abnormal adhesion to platelets, vacuolization of cytoplasm, increased numbers of intracytoplasmic granules, increased presence of cytoplasmic projections and membrane blebbing, and increased cell thickness.
Figure 3.
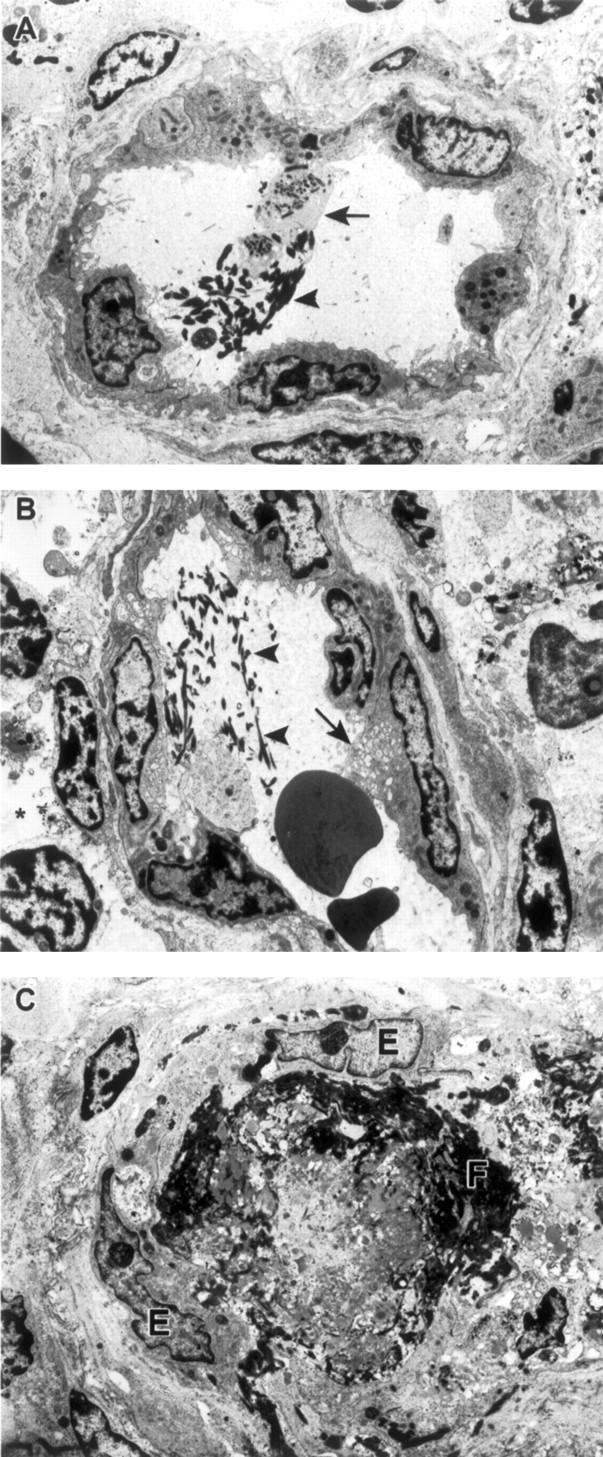
Ultrastuctural appearance of vessels in lymph nodes of EBOV-Zaire-infected cynomolgus monkeys. A: Intravascular fibrin deposits (arrowhead) and adherence of platelets (arrow) to vascular endothelium at day 4. B: Intravascular fibrin deposits (arrowheads), vacuolization of endothelial cell cytoplasm (arrow), and evidence of mild edema (asterisk) at day 4. C: Obstruction of vessel lumen with fibrin and fibrinocellar debris (F). Despite observed coagulopathy, endothelial cells (E) show no evidence of EBOV infection. Original magnifications, ×6500 (A–C).
Evidence of EBOV replication in endothelial cells was not detected by electron microscopy until day 5 when rare endothelial cells lining hepatic sinusoids in two of four monkeys contained characteristic EBOV intracytoplasmic inclusions. 20 Nonetheless evidence suggestive of DIC, as demonstrated by intravascular fibrin deposits and adherence of platelets to vascular endothelium was detected as early as day 4 (three of four) (Figure 3A) ▶ in lymphoid tissues and in hepatic sinusoids; perivascular edema was also evident in occasional foci of lymphoid tissues at day 4. At day 5, extensive intravascular and extravascular deposits of fibrin and fibrinocellular debris were detected in all lymphoid tissues (Figure 3C) ▶ and in hepatic sinusoids (four of four). Foci of endothelial cells in lymphoid tissues showing morphological evidence of activation were more numerous than at day 4. Extravasation of red blood cells was sporadically detected in areas where vessels were obstructed by fibrin thrombi; perivascular edema was evident in these foci and small numbers of macrophages showing evidence of erythrophagocytosis were noted. Also at day 5, higher numbers of endothelial cells containing nonviral TRI were detected in lymphoid tissues. It is worthy to note that these nonviral TRI were not seen in the EBOV-infected endothelial cells in vitro nor was there any association between EBOV inclusions and nonviral TRI in vivo; nonviral TRI were detected in cells with EBOV inclusions and in cells with no morphological evidence of EBOV infection. By day 6, EBOV inclusions were occasionally seen in hepatic sinusoidal endothelial lining cells (three of three) and in high endothelial venules of lymph nodes (three of three). In addition to ultrastructural findings described for day 5, partial detachment of endothelial cells from basement membrane and disruption of basement membrane were infrequent findings in lymphoid tissues at day 6 (three of three). Frank disruption of endothelial integrity was occasionally noted in hepatic sinusoids of all three animals. No morphological evidence of apoptosis was detected in endothelial cells of any tissues of any animal examined at any time point.
Susceptibility of Primary Human Endothelial Cells to EBOV
In vitro experiments were performed to confirm in vivo findings and to further evaluate a potential late-stage contribution of endothelial cell infection to EBOV pathogenesis. Although in vivo observations suggested that endothelial cell infection was a relatively late event in EBOV HF, and not a direct contributor to the hemorrhagic diathesis, late-stage endothelial cell infection may induce other host cell changes that play important roles in the disease and/or outcome of the infection.
Vascular endothelia are remarkably heterogeneous in terms of morphology, marker expression, and function. To determine the susceptibility of different types of endothelial cells to EBOV, cultures of a macrovascular endothelial cell, HUVECs, and a microvascular endothelial cell, HMVEC-Ls, were inoculated with EBOV and analyzed for production of infectious virus and also evaluated for virus-induced cytopathic effects. Viral infectivity titration showed production of ∼6 log10 PFU/ml of infectious EBOV in both HUVECs and HMVEC-Ls by day 6 after infection (Figure 4) ▶ . At day 4, few ultrastructural differences were noted between EBOV-infected cells and mock-infected control cultures with the exception of the characteristic EBOV inclusion material and budding virions, which were striking features. Surprisingly, we detected little cytopathic effect by inverted phase microscopy at day 4 (not shown). By day 6, mild cytopathic effects were observed including swelling of some EBOV-infected cells with detachment infrequently detected in some areas (Figure 5; A to D) ▶ . Ultrastructural evaluation at day 6 showed that nearly all cells in the EBOV-infected cultures contained characteristic EBOV inclusion material and/or budding virions (Figure 5; E to H) ▶ ; occasional EBOV-infected cells showed morphological evidence of necrosis (Figure 5G) ▶ . Ultrastructural evidence of apoptosis was not detected in any of the cultures examined.
Figure 4.

EBOV replication in HUVECs and HMVEC-Ls by infectivity titration.
Figure 5.
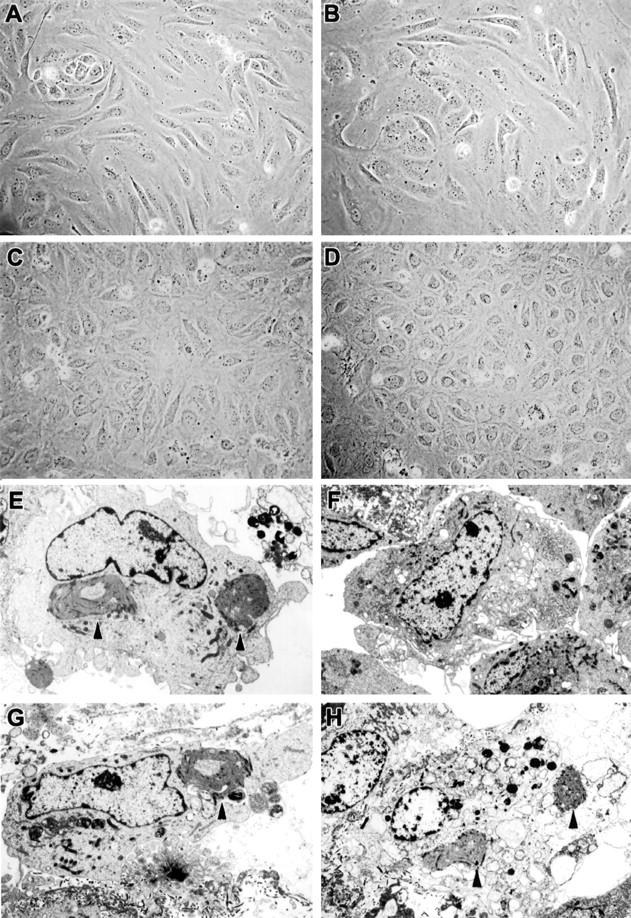
Cytopathology of EBOV-infected HUVECs and HMVEC-Ls at day 6. Inverted phase microscopy of mock-infected (A) and EBOV-infected (B) HUVECs, and mock-infected (C) and EBOV-infected (D) HMVEC-Ls. Note that there are relatively minor differences in cytopathology. E–H: Ultrastructural morphology. E: EBOV-infected HUVECs with typical EBOV intracytoplasmic inclusion (arrowheads). F: Mock-infected HMVEC-Ls. G: EBOV-infected HMVEC-Ls with typical EBOV intracytoplasmic inclusion (arrowhead). H: EBOV-infected HMVEC-Ls with typical EBOV intracytoplasmic inclusions (arrowheads) and morphological evidence of necrosis. Note comparable nuclear morphology in E, F, and G, whereas nuclei in H show degenerative necrotic change as evidenced by pale-staining and random clumping of chromatin. Original magnifications: ×4000 (E); ×3000 (F–H).
Transcriptional Responses of Endothelial Cells to EBOV Infection
As a first step to determine whether EBOV infection induces activation of endothelial cells, we examined mRNA levels of selected genes and compared transcriptional responses of EBOV-infected cells to mock-infected endothelial cell controls and to cultures inoculated with EBOV inactivated by exposure to γ rays. Analysis of total RNA from EBOV-infected HUVECs revealed increased mRNA transcripts for cyclooxygenase (COX)-2 (Figure 6A) ▶ , inducible nitric oxide synthase (iNOS) (not shown), IL-6 (Figure 6C) ▶ , IL-8 (not shown), ICAM-1 (not shown), and monocyte chemotactic protein-1 (MCP-1) (Figure 6C) ▶ by 1 hour. At 24 hours, increased levels of mRNA transcripts for IFN-β and regulated on activation normal T cell-expressed and secreted (RANTES) (Figure 6C) ▶ were detected in EBOV-infected HUVECs, while increased transcripts for cellular inhibitor of apoptosis protein (cIAP)-2 (not shown), growth-related oncogene (Gro)-α, IL-1α (Figure 6C) ▶ , I-309 (not shown), and macrophage inflammatory protein (MIP)-3β (not shown) were observed by 96 hours. Although mRNA levels for these transcripts were sustained through the course of the infection, transient increases were noted for other mRNA transcripts. Specifically, transcripts for VCAM-1 were increased from 1 hour through 24 hours and then returned to baseline levels; transcripts for tissue factor and lipoxygenase were elevated at 1 hour only; transcripts for IFN-γ-inducible protein 10 (IP-10) and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) were increased only at 24 hours (not shown). A transient increase in transcripts for COX-1 was detected at 1 hour; transcripts returned to baseline levels by 24 hours, and were substantially decreased by day 6 (Figure 6A) ▶ .
Figure 6.
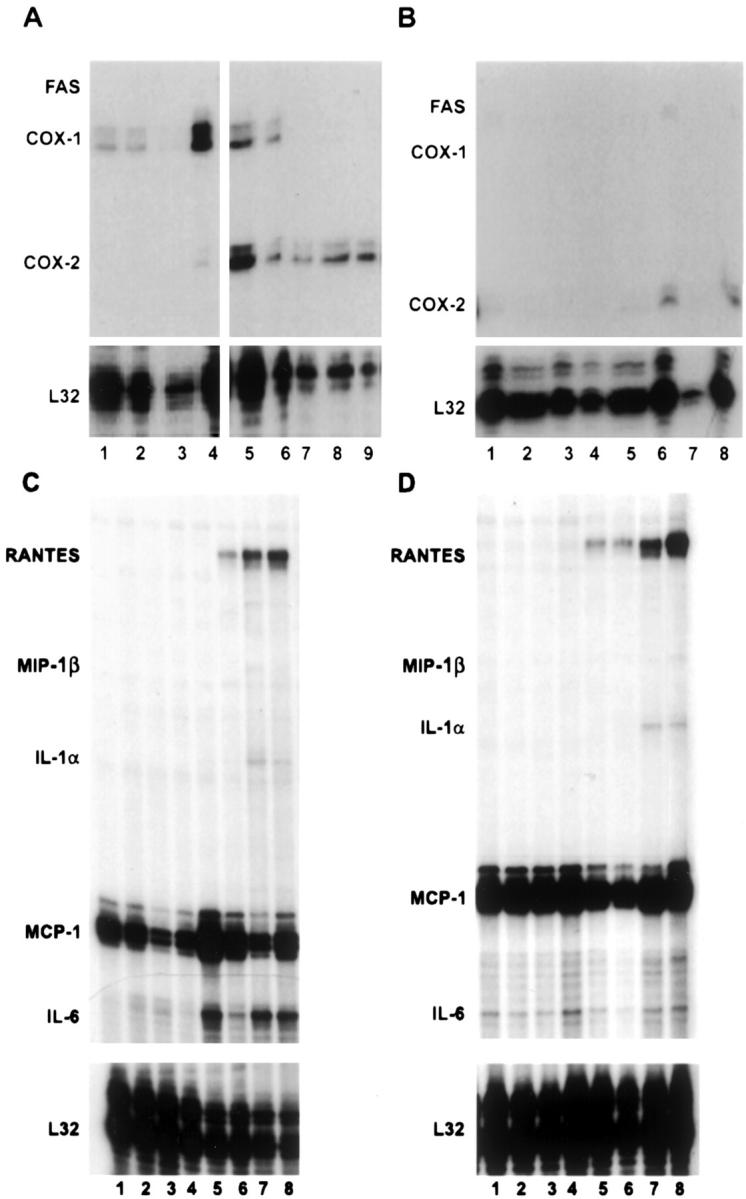
Analysis of mRNA production in EBOV-infected endothelial cell cultures. Representative RNase protection assays are shown. A: COX-1, COX-2, iNOS, Fas; comparison of mock-infected HUVECs at 1 hour (lane 1), 24 hours (lane 2), 96 hours, (lane 3), and 144 hours (lane 4) with EBOV-infected HUVECs at 1 hour (lane 5), 24 hours (lane 6), 96 hours (lane 7), and 144 hours (lane 8), and HUVECs treated with inactivated EBOV at 144 hours (lane 9). B: COX-1, COX-2, iNOS, Fas; comparison of mock-infected HMVEC-Ls at 1 hour (lane 1), 24 hours (lane 2), 96 hours (lane 3), and 144 hours (lane 4) with EBOV-infected HMVEC-Ls at 1 hour (lane 5), 24 hours (lane 6), 96 hours (lane 7), and 144 hours (lane 8). C: Cytokines/chemokines; comparison of mock-infected HUVECs at 1 hour (lane 1), 24 hours (lane 2), 96 hours (lane 3), and 144 hours (lane 4) with EBOV-infected HUVECs at 1 hour (lane 5), 24 hours (lane 6), 96 hours (lane 7), and 144 hours (lane 8). D: Cytokines/chemokines; comparison of mock-infected HMVEC-Ls at 1 hour (lane 1), 24 hours (lane 2), 96 hours (lane 3), and 144 hours (lane 4) with EBOV-infected HMVEC-Ls at 1 hour (lane 5), 24 hours (lane 6), 96 hours (lane 7), and 144 hours (lane 8).
For EBOV-infected HMVEC-Ls, increased mRNA transcripts for RANTES (Figure 6D) ▶ and TRAIL were detected by 24 hours, while increased levels of transcripts for COX-2 (Figure 6B) ▶ and IL-8 (not shown) were noted at 48 hours and transcripts for IL-1α were elevated by 96 hours (Figure 6D) ▶ ; VCAM-1 transcripts were down-regulated by 2 hours (not shown). Changes in mRNA transcripts for cIAP2, COX-1, growth-related oncogene-α, ICAM-1, IL-6, IFN-β, iNOS, IP-10, I-309, lipoxygenase, MCP-1, and MIP-3β, which were detected in EBOV-infected HUVECs, were not observed in EBOV-infected HMVEC-Ls (not shown). In most cases, it seemed that EBOV replication was required to induce changes in levels of mRNA transcripts for the endothelial cell cultures; the only changes noted when cultures were treated with inactivated EBOV was an increase in COX-2 (Figure 6A) ▶ and MCP-1 (not shown) transcripts detected in HUVECs. Changes in levels of transcripts for IL-1β, IL-4, IL-12, IFN-γ, leukotriene A4, leukotriene B4, MIP-1α, MIP-1β, P-Selectin, transforming growth factor (TGF)-β1, TGF-β2, TGF-β3, TNF-α, TNF-β, bcl-2, caspase 3, caspase 8, endothelial nitric oxide synthase (eNOS), decoy receptor (DCR)-1, DCR2, death receptor (DR)-3, DR4, DR5, Fas, FasL, TNFRp55, TNF receptor type 1-associated death domain, receptor interacting protein, TNF receptor-associated factor (TRAF)-1, TRAF2, TRAF3, TRAF4, testosterone-repressed prostate message-2, cIAP1, neuronal apoptosis inhibitory protein, and X-linked inhibitor of apoptosis protein were not detected in any of the EBOV-infected or mock-infected endothelial cell cultures.
Cytokine/Chemokine and Prostacyclin Expression in EBOV-Infected Endothelial Cells
The potential importance of cytokines/chemokines in EBOV HF was hypothesized in other studies. 21-23 Prostacyclin may also be important in the development of EBOV HF because it has a number of important functions including vasodilation, inhibition of platelet aggregation, and potentiation of the chemotactic effects of other mediators. Levels of cytokines/chemokines and prostacyclin were determined at regular intervals after inoculation of endothelial cell cultures to confirm transcriptional responses and to further investigate the temporal response of endothelial cells to EBOV infection. Analysis of fluids from EBOV-infected HUVECs showed increased levels of IFN-β, IL-6, IL-8, RANTES, and 6-keto-prostaglandin F1α (as a measure of prostacyclin) (Figure 7) ▶ , whereas elevated levels of IL-8, RANTES, and 6-keto-prostaglandin F1α were detected in EBOV-infected HMVEC-Ls (Figure 7) ▶ . Results were recorded as positive if OD values were greater than twice the OD of the mock-infected controls. Increased levels of IL-6 were initially detected at 24 hours in EBOV-infected HUVECs. The accumulated levels rose sharply at day 4 (>12,000 pg/ml) and were maintained through 6 days. Elevated levels of IL-6 were not detected in EBOV-infected HMVEC-Ls. By 48 hours, increased levels of IFN-β, RANTES, and prostacyclin were observed in EBOV-infected HUVECs. Accumulated levels of RANTES markedly increased at day 4 (>16,000 pg/ml) and were sustained through day 6, while levels of IFN-β and prostacyclin continued to increase from 48 hours through day 6. Increased levels of IFN-β were not detected in HMVEC-Ls. Detectable levels of RANTES were first seen at day 4 in EBOV-infected HMVEC-Ls and increased through day 6, whereas accumulated levels of prostacylin were only noted at day 6. Constitutive expression of IL-8 was high (>1000 pg/ml) for HUVECs and HMVEC-Ls (not shown). EBOV infection induced twofold to threefold increases in the levels of IL-8 at days 4 and 6 in these cultures. Increases in accumulated levels of IFN-α were not detected in any of the endothelial cell cultures. Likewise, EBOV rendered noninfectious by treatment with gamma rays failed to induce increased production of cytokines/chemokines or prostacyclin in any of these cultures.
Figure 7.

Analysis of cytokine/chemokine and prostacyclin (measuring its stable metabolite, 6-keto-prostaglandin F1α) accumulation in culture fluids of EBOV-infected HUVECs and HMVEC-Ls by enzyme-linked immunosorbent assay.
Discussion
Dysregulation of endothelial cell functions can cause a wide range of vascular effects that lead to changes in vascular permeability or hemorrhage. 24,25 Vascular damage can be induced by immunological mechanisms and/or by direct infection of the vascular tissue. Several microbial diseases are characterized by severe vascular lesions attributed to direct microbial replication-induced damage to endothelial cells. For example, intracellular replication of Rickettsia rickettsii, the etiological agent of Rocky Mountain spotted fever, directly induces lethal injury to host endothelial cells causing pathophysiological changes including thrombosis, hemorrhage, and vasculitis. 26,27 Another example is Nipah virus infection, in which a systemic vasculitis with extensive thrombosis was attributed to infection, damage, and necrosis of endothelial cells. 28 The etiology of the hemorrhagic diatheses in fatal cases caused by the filoviruses, Marburg virus, and EBOV, was searched for in tissues from initial outbreaks in 1967 and 1976, but no vascular lesions were identified. 10 Nonetheless, there has been much speculation that EBOV replication-induced structural damage of endothelial cells triggers the hemorrhagic diathesis.
In the current study, and consistent with the original histological observations of Murphy 10 in fatal human cases, we have shown that EBOV infection of endothelial cells does not extensively disrupt the architecture of the vascular endothelium in EBOV-infected cynomolgus monkeys. Although EBOV replicated in endothelial cells of these animals, endothelial cell infection was only seen focally at late stages of disease, after the onset of the hemorrhagic abnormalities that characterize EBOV HF. 17 Although we observed ultrastructural evidence of endothelial cell activation and disruption, it is likely that the vasoactive effects on endothelial cells are mediated indirectly as we were unable to associate these changes with the presence of intracytoplasmic EBOV antigens. Additionally, our data showed that although EBOV is capable of replicating in microvascular and macrovascular human endothelial cells in vitro, replication does not directly induce cytopathology.
A notable finding in our EBOV-infected cynomolgus monkey model, was the observation that total serum protein levels did not fluctuate during the course of infection however decreases in serum albumin levels were seen by day 4. A decrease in the level of total proteins would be predicted in cases of severe necrosis and damage to the endothelium versus selective loss of a small molecular weight protein, such as albumin. Because albumin is the serum protein most responsible for maintaining colloid osmotic pressure, excessive loss would result in a reduced plasma osmotic pressure, resulting in edema. A decrease in osmotic pressure may also be attributed to a reduced synthesis of albumin, and it cannot be discounted that EBOV-induced impairment of hepatocyte function contributes to the decreased levels of serum albumin. Previous studies demonstrated that TNF-α can increase albumin permeation in the systemic vasculature, 29 but surprisingly only small increases in circulating levels of TNF-α were detected during the late stages of EBOV-infection in our cynomolgus monkeys. 17 However, previous studies showed a priming effect for TNF-α in the presence of agents such as H2O2. 30 Because substantially increased systemic serum nitrate levels, indicative of increased in vivo nitric oxide production, were observed in these EBOV-infected monkeys, 17 the impact of low concentrations of TNF-α on vascular permeability cannot be discounted.
To confirm in vivo findings, primary cultures of endothelial cells were used. In vitro infection of these cells resulted in a noncytolytic infection, with the production of progeny virus and induced cells to secrete chemoattractants, such as the CXC chemokine IL-8, the proinflammatory cytokine IL-6, and transcripts for the proinflammatory protein COX-2. Although very high levels of IFN-α were detected in our EBOV-infected monkeys by day 4, 17 and nonviral TRI that are known to be associated with the production of IFN-α, 31 were observed in endothelial cells in vivo, we failed to detect production of IFN-α in EBOV-infected HUVECs or HMVECs. These observations strongly suggest that other factors triggered by EBOV infection induce endothelial cells to produce IFN-α in vivo and support the hypothesis that endothelial cell disturbances in vivo are primarily unrelated to overt cytopathology induced by EBOV replication. Interestingly, the predominate type I IFN species observed in our in vitro infections was IFN-β. This observation is distinct from previous in vitro work using primate monocytes/macrophages, an early cellular target of EBOV, where IFN-α was the predominant type I IFN species produced. 23 The observation of increased circulating levels of IFN-β at late stages of disease, after the onset of DIC, and the lack of IFN-β produced from EBOV-infected monocytes/macrophages in vitro is consistent with the observation that endothelial cells were late targets of EBOV infection in vivo.
It is worthy to note that EBOV infection did not induce observable levels of apoptosis either directly or indirectly in endothelial cells in vitro or in vivo. Importantly, morphological evidence of apoptosis was not detected by electron microscopy. Relatively strong increases in transcripts for the anti-apoptotic gene cIAP2, and COX-2, which is known to have anti-apoptotic properties, 32,33 were detected in EBOV-infected HUVECs. Proapoptotic transcripts were not seen in any of the EBOV-infected endothelial cell cultures, consistent with the hypothesis that EBOV induces protective modalities in cells that it infects. Furthermore, these observations support the view that development of vascular hemorrhage/leakage is not a direct result of destruction of the endothelium by EBOV replication. These findings are in contrast with results reported for other viral hemorrhagic fevers including dengue 34 and African swine fever, 35 which induce apoptosis in endothelial cells in vitro, but are consistent with Lassa virus. 36
Although endothelial cells were readily infected in vitro, the predilection of EBOV for endothelial cells in vivo was not consistent and developed according to anatomical location. Although this phenomenon may be a function of the remarkably heterogeneous nature of vascular endothelia in terms of morphology, receptor/marker expression, and function, it is also possible that the host factor required for EBOV replication in endothelial cells is located on the basolateral surface and in vivo is somewhat protected from viremia. For example, over 4 log10 PFU/ml of EBOV was detected in circulation of these monkeys at day 4 17 yet the only endothelial cells containing EBOV RNA and antigens were endothelial cells lining hepatic sinusoids. As illustrated by Schnittler and Feldmann 37 in postulating a model for routes of filoviral dissemination, the portal liver sinuses are lined by a discontinuous endothelium that does not rest on a regular basement membrane. This endothelium contains transcellular gaps, allowing virus particles to enter the space of Disse. In addition to providing more immediate access to hepatocytes as proposed, 37 these gaps may also enhance access to the basolateral aspects of the sinusoidal endothelial cells. It is also worthy of note that sinusoidal endothelial lining cells express the receptor DC-SIGNR, which was recently postulated to be a receptor for EBOV. 38 HEVs were also earlier targets of EBOV than other endothelial cells, and it is possible that increased trafficking of EBOV-infected monocytes and dendritic cells through these vessels disseminated virus more readily to HEVs.
Although we have defined endothelial cell transcriptional and protein responses, our findings are limited by their intended focus on the endothelium. These findings exclude responses from immune cells and complex immune cell-endothelial cell interactions that are also likely to contribute to multifactoral causes of EBOV HF. However, in focusing on an important cellular target of EBOV disease, we have addressed key vascular responses and identified many factors that regulate immune cell responses and endothelial cell barrier functions central to EBOV HF. The potential role of endothelial cell responses in EBOV pathogenesis remains speculative, but these results are vital for defining factors that contribute to EBOV HF. Our data imply that EBOV infection primarily affects the function rather than the structure of endothelial cells. We conclude that EBOV-induced coagulopathy results primarily from vascular disruption induced by factors secreted from infected monocytes/macrophages and dendritic cells, whereas direct virus-induced cytolysis of endothelial cells plays a minimal, secondary role in the hemorrhagic diathesis.
Acknowledgments
The authors thank Denise Braun, Joan Geisbert, Lynda Miller, Roswita Moxley, Jeff Brubaker, Steve Moon, Neil Davis, and Larry Ostby for their expert technical assistance; Gabriela Dveksler, Aileen Marty, Rahda Maheshwari, and Chou-Zen Giam for helpful discussions and comments; and Dana Scott for skillful assistance with necropsies.
Footnotes
Address reprint requests to Thomas W. Geisbert, USAMRIID, Attn: MCMR-UIV, 1425 Porter St., Fort Detrick, MD 21702-5011. E-mail: tom.geisbert@amedd.army.mil.
Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the United States Army. Research was conducted in compliance with the Animal Welfare Act and other Federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996. The facility where this research was conducted is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The research described herein was sponsored by the Medical Chemical/Biological Defense Research Program, United States Army Medical Research and Material Command (project no. 02-4-4J-081).
References
- 1.Huggins JW: Prospects for treatment of viral hemorrhagic fevers with ribavirin, a broad-spectrum antiviral drug. Rev Infect Dis Suppl 1989, 4:S750-S761 [DOI] [PubMed] [Google Scholar]
- 2.Jahrling PB, Geisbert J, Swearengen JR, Jaax GP, Lewis T, Huggins JW, Schmidt JJ, LeDuc JW, Peters CJ: Passive immunization of Ebola virus-infected cynomolgus monkeys with immunoglobulin from hyperimmune horses. Arch Virol Suppl 1996, 11:135-140 [DOI] [PubMed] [Google Scholar]
- 3.Jahrling PB, Geisbert TW, Geisbert JB, Swearengen JR, Bray M, Jaax NK, Huggins JW, LeDuc JW, Peters CJ: Evaluation of immune globulin and recombinant interferon-α2b for treatment of experimental Ebola virus infections. J Infect Dis Suppl 1999, 179:S224-S234 [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Delgado R, Xu L, Todd RF, Nabel EG, Sanchez A, Nabel GJ: Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science 1998, 279:1034-1037 [DOI] [PubMed] [Google Scholar]
- 5.Yang Z, Duckers HJ, Sullivan NJ, Sanchez A, Nabel EG, Nabel GJ: Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat Med 2000, 6:886-889 [DOI] [PubMed] [Google Scholar]
- 6.Moncada S, Palmer RMJ, Higgs EA: Relationship between prostacyclin and nitric oxide in the thrombotic process. Thromb Res Suppl 1990, XI:3-13 [DOI] [PubMed] [Google Scholar]
- 7.Mantovani A, Bussolino F, Dejana E: Cytokine regulation of endothelial cell function. EMBO J 1992, 6:2591-2599 [DOI] [PubMed] [Google Scholar]
- 8.Harcourt BH, Sanchez A, Offermann MK: Ebola virus inhibits induction of genes by double-stranded RNA in endothelial cells. Virology 1998, 252:179-188 [DOI] [PubMed] [Google Scholar]
- 9.Dietrich M, Schumacher HH, Peters D, Knobloch J: Human pathology of Ebola (Maridi) virus infection in the Sudan. Pattyn SR eds. Ebola Virus Haemorrhagic Fever. 1978:pp 37-42 Elsevier/North-Holland Biomedical Press, New York
- 10.Murphy FA: Pathology of Ebola virus infection. Pattyn SR eds. Ebola Virus Haemorrhagic Fever. 1978:pp 43-60 Elsevier/North-Holland Biomedical Press, New York
- 11.Zaki SR, Goldsmith CS: Pathologic features of filovirus infections in humans. Curr Top Microbiol Immunol 1999, 235:97-116 [DOI] [PubMed] [Google Scholar]
- 12.Baskerville A, Fisher-Hoch SP, Neild GH, Dowsett AB: Ultrastructural pathology of experimental Ebola haemorrhagic fever virus infection. J Pathol 1985, 147:199-209 [DOI] [PubMed] [Google Scholar]
- 13.Geisbert TW, Jahrling PB, Hanes MA, Zack PM: Association of Ebola related Reston virus particles and antigen with tissue lesions of monkeys imported to the United States. J Comp Pathol 1992, 106:137-152 [DOI] [PubMed] [Google Scholar]
- 14.Jaax NK, Davis KJ, Geisbert TW, Vogel P, Jaax GP, Topper M, Jahrling PB: Lethal experimental infection of rhesus monkeys with Ebola-Zaire (Mayinga) virus by the oral and conjunctival route of exposure. Arch Pathol Lab Med 1996, 120:140-155 [PubMed] [Google Scholar]
- 15.Davis KJ, Anderson AO, Geisbert TW, Steele KE, Geisbert JB, Vogel P, Connolly BM, Huggins JW, Jahrling PB, Jaax NK: Pathology of experimental Ebola virus infection in African green monkeys. Arch Pathol Lab Med 1997, 121:805-819 [PubMed] [Google Scholar]
- 16.Ryabchikova EI, Kolesnikova LV, Netesov SV: Animal pathology of filoviral infections. Curr Top Microbiol Immunol 1999, 235:145-173 [DOI] [PubMed] [Google Scholar]
- 17.Geisbert TW, Hensley LE, Larsen T, Young HA, Reed DS, Geisbert JB, Scott DP, Kagan E, Jahrling PB, Davis KJ: Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am J Pathol 2003, 163:2347-2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prophet EB Mills B Arrington JB Sobin LH eds. Laboratory Methods in Histotechnology. 1992:pp 25-59 Armed Forces Institute of Pathology, Washington DC
- 19.Jahrling PB: Filoviruses and arenaviruses. Murray PR Baron EJ Pfaller M Tenover FC Yolken RH eds. Manual of Clinical Microbiology. 1999:pp 1125-1136 ASM Press, Washington DC
- 20.Geisbert TW, Jahrling PB: Differentiation of filoviruses by electron microscopy. Virus Res 1995, 39:129-150 [DOI] [PubMed] [Google Scholar]
- 21.Baize S, Leroy EM, Georges-Courbot M-C, Capron M, Lansoud-Soukate J, Debre P, Fisher-Hoch SP, McCormick JB, Georges AJ: Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat Med 1999, 5:423-426 [DOI] [PubMed] [Google Scholar]
- 22.Villinger F, Rollin PE, Brar SS, Chikkala NF, Winter J, Sundstrom JB, Zaki SR, Swanepoel R, Ansari AA, Peters CJ: Markedly elevated levels of interferon (IFN)-α, IFN-γ, interleukin (IL)-2, IL-10, and tumor necrosis factor-α associated with fatal Ebola virus infection. J Infect Dis 1999, 179(Suppl 1):S188-S191 [DOI] [PubMed] [Google Scholar]
- 23.Hensley LE, Young HA, Jahrling PB, Geisbert TW: Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily. Immunol Lett 2002, 80:169-179 [DOI] [PubMed] [Google Scholar]
- 24.Dvorak HF, Brown LF, Detmar M, Dvorak AM: Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995, 146:1029-1039 [PMC free article] [PubMed] [Google Scholar]
- 25.Kevil CG, Payne DK, Mire E, Alexander JS: Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem 1998, 273:15099-15103 [DOI] [PubMed] [Google Scholar]
- 26.Walker DH: Pathology and pathogenesis of the vasculotropic rickettsioses. Walker DH eds. Biology of Rickettsial Diseases. 1988:pp 115-138 CRC Press, Boca Raton
- 27.Eremeeva ME, Silverman DJ: Rickettsia rickettsii infection of the EA. hy 926 endothelial cell line: morphological response to infection and evidence for oxidative injury. Microbiology 1998, 144:2037-2048 [DOI] [PubMed] [Google Scholar]
- 28.Wong KT, Shieh WJ, Kumar S, Norain K, Abdullah W, Guarner J, Goldsmith CS, Chua KB, Lam SK, Tan CT, Goh KJ, Chong HT, Jusoh R, Rollin PE, Ksiazek TG, Zaki SR: Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am J Pathol 2002, 161:2153-2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Worrall NK, Chang K, LeJeune WS, Misko TP, Sullivan PM, Ferguson TB, Jr, Williamson JR: TNF-alpha causes reversible in vivo systemic vascular barrier dysfunction via NO-dependent and -independent mechanisms. Am J Physiol 1997, 273:H2565-H2574 [DOI] [PubMed] [Google Scholar]
- 30.Feldmann H, Bugany H, Mahner F, Klenk HD, Drenckhahn D, Schnittler H-J: Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J Virol 1996, 70:2208-2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimley PM, Kang YH, Silverman RH, Davis G, Hoofnagle JH: Blood lymphocyte inclusions associated with alpha interferon. Lab Invest 1983, 48:30A-31A [Google Scholar]
- 32.Teruyama K, Abe M, Nakano T, Iwasaka-Yagi C, Takahashi S, Yamada S, Sato Y: Role of transcription factor Ets-1 in the apoptosis of human vascular endothelial cells. J Cell Physiol 2001, 188:243-252 [DOI] [PubMed] [Google Scholar]
- 33.Cao Y, Prescott SM: Many actions of cyclooxygenase-2 in cellular dynamics and in cancer. J Cell Physiol 2002, 190:279-286 [DOI] [PubMed] [Google Scholar]
- 34.Avirutnan P, Malasit P, Seliger B, Bhakdi S, Husmann M: Dengue virus infection of human endothelial cells leads to chemokine production, complement activation, and apoptosis. J Immunol 1998, 161:6338-6346 [PubMed] [Google Scholar]
- 35.Vallee I, Tait SW, Powell PP: African swine fever virus infection of porcine aortic endothelial cells leads to inhibition of inflammatory responses, activation of the thrombotic state, and apoptosis. J Virol 2001, 75:10372-10382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lukashevich IS, Maryankova R, Vladyko AS, Nashkevich N, Koleda S, Djavani M, Horejsh D, Voitenok NN, Salvato MS: Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: different effects on IL-8 and TNF-alpha gene expression. J Med Virol 1999, 59:552-560 [PMC free article] [PubMed] [Google Scholar]
- 37.Schnittler H-J, Feldmann H: Marburg and Ebola hemorrhagic fevers: does the primary course of infection depend on the accessibility of organ-specific macrophages? Clin Infect Dis 1998, 27:404-406 [DOI] [PubMed] [Google Scholar]
- 38.Simmons G, Reeves JD, Grogan CC, Vandenberghe LH, Baribaud F, Whitbeck JC, Burke E, Buchmeier MJ, Soilleux EJ, Riley JL, Doms RW, Bates P, Pohlmann S: DC-SIGN and DC-SIGNR bind Ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 2003, 305:115-123 [DOI] [PubMed] [Google Scholar]