

Abstract
In glaucoma, the increased release of glutamate is the major cause of retinal ganglion cell death. Cannabinoids have been demonstrated to protect neuron cultures from glutamate-induced death. In this study, we test the hypothesis that glutamate causes apoptosis of retinal neurons via the excessive formation of peroxynitrite, and that the neuroprotective effect of the psychotropic Δ9-tetrahydroxycannabinol (THC) or nonpsychotropic cannabidiol (CBD) is via the attenuation of this formation. Excitotoxicity of the retina was induced by intravitreal injection of N-methyl-d-aspartate (NMDA) in rats, which also received 4-hydroxy-2,2,6,6-tetramethylpiperidine-n-oxyl (TEMPOL,a superoxide dismutase-mimetic), N-ω-nitro-l-arginine methyl ester (L-NAME, a nitric oxide synthase inhibitor), THC, or CBD. Retinal neuron loss was determined by TDT-mediated dUTP nick-end labeling assay, inner retinal thickness, and quantification of the mRNAs of ganglion cell markers. NMDA induced a dose- and time-dependent accumulation of nitrite/nitrate, lipid peroxidation, and nitrotyrosine (foot print of peroxynitrite), and a dose-dependent apoptosis and loss of inner retinal neurons. Treatment with L-NAME or TEMPOL protected retinal neurons and confirmed the involvement of peroxynitrite in retinal neurotoxicity. The neuroprotection by THC and CBD was because of attenuation of peroxynitrite. The effect of THC was in part mediated by the cannabinoid receptor CB1. These results suggest the potential use of CBD as a novel topical therapy for the treatment of glaucoma.
In glaucoma, reduction in intraocular pressure is frequently insufficient to prevent progression of visual field loss. Rather, glutamate-induced neurotoxicity plays an important role in glaucoma. 1 Glutamate, an important neurotransmitter released from photoreceptor and bipolar cells, is present in high concentration in retinal ganglion cells. 2 However, when excess glutamate is released into the surrounding medium because of compression or vascular occlusion, it is thought to activate a toxic response in adjacent cells. Excitotoxicity is mediated by overstimulation of the N-methyl-d-aspartate (NMDA) and non-NMDA receptors, a mechanism that has been demonstrated in the retina. 3 Overstimulation of these receptors leads to excessive levels of intracellular calcium. 4 This in turn leads to activation of nitric oxide synthase and excess accumulation of superoxides and nitric oxide (NO), causing lipid peroxidation, mitochondrial dysfunction, DNA damage, and the eventual cell death. 5,6 Elevated glutamate levels have been found in the vitreous of glaucomatous patients and monkeys with glaucoma. 7 Similarly, high glutamine, suggesting high extracellular glutamate, has been found in the retinal Müller cells of glaucomatous monkey eyes. 8 The reaction product of superoxides and NO, peroxynitrite (ONOO−), was recently identified in the retinal ischemia-reperfusion injury. 6 The causal role of ONOO− in the glutamate-induced retinal excitotoxicity in glaucoma, however, has not been determined.
In vitro and in vivo studies have demonstrated that ischemia-induced neurotoxicity can be reduced by antioxidants 9,10 or by inhibitors of nitric oxide synthase. 11 TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidine-n-oxyl),a superoxide dismutase mimetic that permeates biological membranes 12 and scavenges superoxides and hydroxy radicals, 13 for example, has been shown to be an effective neuroprotectant in a gerbil model of cerebral ischemia. 14 A competitive inhibitor of nitric oxide synthase, N-ω-nitro-l-arginine methyl ester (L-NAME), has also been demonstrated as a retinal neuroprotectant in a rat model of NMDA-induced retinal neurotoxicity. 15 The neuroprotective effects of these molecules may be because of their ability to reduce the formation of reactive oxygen species, NO, or ONOO− in these animals. However, this mechanism has not been studied.
Cannabinoid components of marijuana, such as (−)Δ9-tetrahydrocannabinol (THC), or the synthetic cannabinoid WIN55,212-2, have been shown to prevent glutamate- or NMDA-induced neurotoxicity in isolated neurons 16 or in the brain 17,18 via activation of the cannabinoid receptor subtype CB1. The major obstacles to the therapeutic utilization of cannabinoids are their psychotropic effects, which are also mediated by actions on CB1 receptors. However, the nonpsychotropic component of marijuana, cannabidiol (CBD), and the synthetic nonpsychotropic cannabinoid, HU-211, as well as THC have been demonstrated as potent antioxidants and/or NMDA receptor antagonists that protect neuron cultures from glutamate-induced death 19 or from oxidative stress. 20 Immediate administration of HU-211 in a calibrated crush injury of adult rat optic nerve reduces injury-induced metabolic and electrophysiological deficit. 21 These findings have prompted us to study the mechanism of glutamate-induced neurotoxicity in the retina and to investigate the actions of cannabinoids in preventing the neurotoxicity. In this report, we show that formation of superoxide (measured as lipid peroxidation), NO (measured as nitrite/nitrate), and ONOO− (measured as nitrotyrosine) have a key role in NMDA-induced neurotoxicity in the rat retina. Further, we demonstrated that THC and CBD are neuroprotective against NMDA-induced retinal injury and that their protective actions are in part because of an effect in reducing formation of lipid peroxides, nitrite/nitrate, and nitrotyrosine.
Materials and Methods
Animals
Adult male albino Sprague-Dawley rats (∼250 g, 50 to 55 days old) from Harlan (Indianapolis, IN) were used in this study. Animals were maintained in clear plastic cages and subjected to standard 12-hour light/12-hour dark light cycles. Light levels at the bottom of cages were controlled at ≤1.5 foot candles (16.1 lux) to avoid the possibility of light damage in the retina. Animals were used for experiments only after a period of acclimation of at least 5 days in the animal room at regulated temperature (22 to 24°C).
Administration of NMDA, TEMPOL, L-NAME, THC, and CBD
The procedure to induce excitotoxic cell loss in rat retinas was essentially the same as previously described 3,22-25 with the following modifications. The volume of intravitreal injections was originally set at 5 μl. 22,25 To avoid solution loss from the injection site, this volume was reduced to 2 μl. To ensure the proper delivery and even distribution of the intravitreally injected compounds, all solutions for intravitreal injection contained 5 μg/ml of Fast Green FCF (Sigma, St. Louis, MO). Animals anesthetized by intraperitoneal injection of ketamine and xylazine (80 mg/kg and 12 mg/kg, respectively) as previously described 23-25 were given a single intravitreal injection of one of the following solutions. NMDA (Sigma) solution in 0.1 mol/L of phosphate-buffered saline (PBS) was neutralized with 0.1 mol/L of NaOH. TEMPOL (Sigma) and L-NAME (Sigma) were also dissolved in PBS. Injections of NMDA alone, or in combination with TEMPOL or L-NAME, were made with 30-gauge needles mounted to 10-μl Hamilton syringes. For controls, the contralateral eyes were injected with 2 μl of either PBS or neutralized N-methyl-l-aspartate (NMLA, Sigma). The tip of the needle was inserted under the guidance of a Leica Wild M650 dissecting microscope (Leica, Heerbrugg, Switzerland) through the dorsal limbus of the eye. Injections were performed slowly throughout a period of 2 minutes. The volume of the injected solution apparently did not cause significant pressure-induced retinal damage, because both PBS- and NMLA-injected control eyes showed normal retinal morphology with no apparent apoptosis within 7 days. Neomycin/polymyxin B/bacitracin ophthalmic ointment was applied to the injected eyes and animals were allowed to recover. Rats were killed at various times and the eyes were enucleated and processed for further analyses. In parallel studies, rats injected with NMDA were given an intravenous injection of THC, SR141716A (National Institute of Drug Abuse, Research Triangle Park, NC) or CBD (Sigma) through one of the lateral tail veins. These compounds were prepared for injection solutions by slowly adding 100 μl of the stock solutions in absolute alcohol into rapidly stirred 0.9 ml of 25% bovine serum albumin in PBS under argon. Animals were given an intravenous injection of 200 μl of 0.5 μg/μl or 2.5 μg/μl (0.4 or 2 mg/kg of body weight) of THC, 1 or 2 mg/kg of SR141716A, or 2 mg/kg of CBD immediately before the intravitreal injection of NMDA, NMLA, or PBS. For animals to be analyzed at day 7 after NMDA treatment and after the initial injection of THC, a booster injection of THC at thesame doses was given at the third day. For control, the same volumes of vehicle, including alcohol, bovine serum albumin, and PBS were used for the intravenous injection. Experiments were conducted in a blind manner as to the treatment conditions.
Measurement of Nitrite and Nitrate
NO production was determined by measuring the levels of nitrite and nitrate, the stable oxidized products of NO, in the supernatant of PBS homogenate of rat retinas by modified Greiss reagent assay. 26 Briefly, retinas in cold PBS mixed with acid/base-washed, baked Ottawa sand were homogenized in 1.5-ml Eppendorf tubes with Mini-Beadbeater (BioSpec Products, Minebea Co., Ltd.,Bangkok, Thailand) for 20 seconds at 4°C. The homogenate of 210 μl was incubated with nitrate reductase (10 mU) and NADPH (12.5 mmol/L) for 30 minutes at 37°C. The total nitrite in each sample was determined by addition of 200 mU of l-glutamate dehydrogenase, 100 mmol/L NH4Cl, and freshly prepared 4 mmol/L of α-ketoglutarate. Enzyme reduction was monitored by including an NO3− standard. The mixture was incubated at 37°C for 10 minutes followed by addition of 250 μl of Greiss reagent and incubation for another 5 minutes. The absorbance at λ543 nm was recorded versus the blank. Concentrations of total nitrite were calculated from a standard curve constructed using NaNO2/NaNO3 standards. Protein levels were measured by the Bradford method (Bio-Rad, Hercules, CA) and nitrite/nitrate level was expressed as nmol per mg of protein.
Measurement of Lipid Peroxidation
Lipid peroxide concentration was determined by a method that measures the amount of thiobarbituric acid reactivity by the amount of malondialdehyde (MDA) formed during acid hydrolysis of the lipid peroxide compound. 27 Retina homogenate was prepared at a ratio of 1 g wet tissue to 9 ml of 1.15% KCl. The reaction mixture contained 0.1 ml of retina homogenate, 0.1 ml of 8.1% sodium dodecyl sulfate, 0.75 ml of 20% acetic acid solution (buffered to pH 3.5), and 0.75 ml of 0.8% thiobarbituric acid. The mixture was then incubated at 95°C for 1 hour. After cooling, 0.5 ml of distilled water followed by 2.5 ml of the mixture of n-butanol and pyridine (15:1, v/v) was added and the final mixture was shaken vigorously. After centrifugation (4000 rpm, 10 minutes) absorbance of the solvent layer was measured at λ532 nm. Tetraethoxypropane was used as external standard. Lipid peroxide level was expressed in terms of nmol of MDA per mg of protein.
Measurement of Retinal Nitrotyrosine by Immunochemical Slot-Blot Analysis
Peroxynitrite (ONOO−) is a short-lived molecule at physiological pH, but it has been shown to nitrate protein tyrosine residues. Therefore, slot-blot analysis of nitrotyrosine immunoreactivity was used to assay ONOO− formation. 14,28,29 Retinas were homogenized in RIPA lysis buffer (20 mmol/L Tris, pH 7.4, 2.5 mmol/L ethylenediaminetetraacetic acid, pH 8, 1% Triton X-100, 1% deoxycholate, 1% sodium dodecyl sulfate, 50 mmol/L sodium fluoride, 10 mmol/L sodium pyrophosphate, 1 mmol/L of phenylmethyl sulfonyl fluoride). Duplicate protein samples were immobilized onto polyvinylidene difluoride membrane using a slot-blot microfiltration unit. After blocking with 5% nonfat milk, the polyvinylidene difluoride membrane was reacted with a monoclonal anti-nitrotyrosine antibody (Cayman Chemical Co., Ann Arbor, MI) followed by peroxidase-labeled goat anti-mouse IgG and enhanced chemiluminescence. Relative levels of nitrotyrosine immunoreactivity were determined by densitometry and comparison with a standard curve generated from the ONOO−-modified bovine serum albumin (Cayman Chemical Co.).
Measurement of Retinal Nitrotyrosine by Immunohistochemistry (Immunofluorescence)
The distribution of nitrotyrosine in retinal sections was analyzed using immunohistochemistry. 30 OCT-frozen eye sections (8 μm) perpendicular to the retinal surface were analyzed at several adjacent locations along the vertical meridian within 1 to 2 mm of the optic disk according to Hughes. 31 The eye sections were reacted with a polyclonal rabbit anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY), followed by Oregon Green-conjugated goat anti-rabbit antibody (Molecular Probes, Eugene, OR). On completion of the immunostaining, coverslips were applied using Vectashield mounting medium for fluorescence (Vector Laboratories, Burlingame, CA) with or without 1 μg/ml of propidium iodide (Sigma). Data were analyzed using fluorescence microscopy with controlled, constant exposure and Ultra-View morphometric software to quantify intensity of immunostaining.
Terminal dUTP Nick-End Labeling (TUNEL) Assay for in Situ Detection of DNA Fragmentation
TUNEL was performed using the ApopTAG in situ apoptosis detection kit-fluorescein (Intergen, Purchase, NY), following the manufacturer’s directions. Eyes from at least three animals of each group were analyzed. OCT-frozen eye sections (8 μm) perpendicular to the retinal surface were analyzed at three to four adjacent locations along the vertical meridian within 1 to 2 mm of the optic disk. 31 Each assay included a number of controls treated identically to the experimental tissue. For negative-stain control, deionized water was substituted for the TdT enzyme in the reaction buffer. A positive-stain control was generated by incubating a specimen 15 minutes with 1 μl of nuclease enzyme mixed with 50 μl of nuclease buffer (ApoTACs kit; R&D Systems, Minneapolis, MN). On completion of the TUNEL assay, coverslips were applied using Vectashield mounting medium for fluorescence (Vector Laboratories) containing 1 μg/ml of propidium iodide. Tissues were viewed by epifluorescence using standard fluorescein excitation and emission filters. Each section was systematically scanned for positive green fluorescent cells indicating apoptosis. To distinguish between structures that autofluoresced versus those that were TUNEL-positive, all slides were examined first with the rhodamine (red) filter, then with the fluorescein isothiocyanate (green) filter. Autofluorescent structures were visible under both filters, whereas TUNEL-positive cells were detectable using only the green filter. Positively labeled cells were counted in the retinal ganglion cell layer and the inner nuclear layer separately under a fluorescence microscope with ×400 magnification. Images were obtained using a Zeiss Axioplan 2 fluorescent microscope (Carl Zeiss Inc., Germany) equipped with a Spot Camera and Spot Software version 2.2 (Diagnostic Instruments, Inc., Sterling Heights, MI).
Morphometry of Inner Retinal Thickness Measurements
Eyes from each group at day 7 after NMDA or vehicle treatment with or without THC were analyzed. OCT-frozen eye sections (8 μm) perpendicular to the retinal surface were measured at two adjacent locations along the vertical meridian within 1 to 2 mm of the optic disk. Ultra-View morphometric software was used to measure the thickness of the inner retina (distance between the inner limiting membrane and the inner border of outer nuclear layer). Readings from three to four sections from each eye were averaged to obtain the mean value for that eye.
Comparative Quantitative Reverse Transcriptase-Polymerase Chain Reactions (RT-PCR)
The levels of Thy-1, neurofilament light (NF-L; MW 70 to 80 kd) and internal standard, 18S ribosomal RNA (rRNA), from total RNAs present in the retinas were determined with a comparative quantitative RT-PCR technique (Ambion Inc., Austin TX). Thy-1 antigen is primarily associated with retinal ganglion cells. 32 NF-L is primarily associated with ganglion cell axons. 33 Therefore, determining Thy-1 and/or NF-L mRNA levels relative to the level of an internal standard such as 18S rRNA is one way of assaying for ganglion cell number. We determined the effect of NMDA and THC on Thy-1 and NF-L mRNA. Briefly, total RNA was extracted from sand-homogenized retinas using total RNA purification reagent, RNAwiz (Ambion), which is based on the use of detergent, chaotropic salts, and phenolic extraction to denature the RNase and solubilize the total RNA, and first-strand cDNA synthesis with random hexamers performed on 2 μg of DNase-treated RNA (Life Technologies/Invitrogen, Carlsbad, CA). The individual cDNA species were amplified in a reaction containing a cDNA aliquot, PCR buffer, and MgCl2. Reactions were initiated by incubating at 95°C for 15 minutes (Qiagen, Valencia, CA). PCR (94°C, 40 seconds; 58°C, 30 seconds; 72°C, 30 seconds) was performed for a suitable number of cycles to ensure that amplification was within exponential phase (cycle numbers: Thy-1, 25 or 26; NF-L, 24), followed by a final extension at 72°C for 7 minutes. The abundant internal standard, 18S rRNA, was amplified to the levels comparable to Thy-1 or NF-L by using primer and competimer mixtures (Ambion) at various ratios. Interexperimental variations were avoided by performing all amplifications in a single run. Oligonucleotides primers used were 5′-CGCTTTATCAAGGTCCTTACTC-3′ and 5′-GCGTTTTGAGATATTTGAAGGT-3′ (sense and anti-sense for Thy-1, to amplify a 344-bp fragment) and 5′-ATGCTCAGATCTCCGTGGAGATG-3′ and 5′-GCTTCGCACTCATTCTCCAGTT-3′ (sense and anti-sense for NF-L, to amplify a 365-bp fragment). PCR reaction products were separated on 2% agarose gels using ethidium bromide for visualization. The relative abundance of each PCR product was determined by quantitative analysis of photographs of gels on a densitometer.
Data Analysis and Statistics
All data are reported as mean ± SE. Statistical analysis used analysis of variance to determine whether there was a difference between NMDA-, NMLA-, or PBS-injected control eyes in the presence or absence of TEMPOL, L-NAME, THC, or CBD. Fisher’s posthoc least significant difference test was used for multiple comparisons. P < 0.05 or P < 0.01 (as indicated) was considered significant.
Results
NMDA Increases Peroxynitrite Formation in the Retina in a Dose- and Time-Dependent Manner
Peroxynitrite (ONOO−), the product of superoxide and NO, has been implicated in retinal ischemia-reperfusion injury. To explore the role of ONOO− in retinal cell death induced by glutamate neurotoxicity, we first determined the formation of ONOO− in the rat retina after intravitreal injection of NMDA. Because ONOO− has been shown to nitrate protein tyrosine residues, ONOO− formation in the retina was evaluated by slot-blot analysis of nitrotyrosine immunoreactivity. As shown in Figure 1, A and B ▶ , intravitreal injection of NMDA produced a dose- and time-dependent increase in nitrotyrosine formation, which peaked at 18 hours after the injection.
Figure 1.

Intravitreal NMDA treatment induces time- and dose-dependent ONOO− formation in the retina. NMDA was injected into the vitreous of the rat eyes and the formation of nitrotyrosine, lipid peroxidation, and nitrite/nitrate in the rat retina was determined. A: Dose-dependent increases in nitrotyrosine formation determined by immunochemical slot-blot analysis at 18 hours after NMDA (80 or 200 nmol/eye) injection. B: Time-dependent increases in nitrotyrosine formation determined by immunochemical slot-blot analysis after NMDA (200 nmol/eye) injection. C: Time-dependent increases in MDA formation after NMDA (200 nmol/eye) injection. D: Time-dependent increases in nitrite/nitrate formation after NMDA (200 nmol/eye) injection. All values represent the mean ± SE of a total of six animals for each time point in two experiments. *, Significant at P < 0.05.
To confirm that the above NMDA-induced increases in tyrosine nitration are the result of ONOO− formation, we next analyzed the formation of superoxide anion and NO in the NMDA-treated retinas. Superoxide formation was determined by measuring lipid peroxidation using an assay for MDA formation. As shown in Figure 1C ▶ , intravitreal injection of NMDA (200 nmol/eye) produced a time-dependent increase in MDA formation that paralleled the increase in tyrosine nitration, reaching a maximal level at 18 hours after the injection. Formation of NO was evaluated by measuring the stable NO end products, nitrite and nitrate, which also peaked at 18 hours after the injection (Figure 1D) ▶ . In the above studies, intravitreal injection of PBS or NMLA at 200 nmol/eye in the contralateral eyes had no effect on the formation of nitrotyrosine, MDA, or nitrite/nitrate as compared with uninjected eyes (data not shown). These results suggest that NMDA treatment increases ONOO− formation in the rat retina in a dose- and time-dependent manner.
NMDA-Induced Neurotoxicity Is Prevented by Inhibiting Peroxynitrite Formation
We next evaluated the role of ONOO− formation in NMDA-induced neurotoxicity. As shown in Figure 2A ▶ and the quantitative analysis in Figure 2B ▶ , intravitreal NMDA treatment (80 or 200 nmol/eye) produced a dose-dependent increase in the number of TUNEL-positive retinal cells, which were located almost exclusively in the inner nuclear layer (INL) and ganglion cell layer (GCL). This neurotoxicity was almost completely eliminated by the superoxide scavenger TEMPOL (0.4 mg/eye) or the nitric oxide synthase inhibitor L-NAME (0.1 mg/eye) co-injected with NMDA. As shown in Figure 2C ▶ and the quantitative analysis in 2D, the NMDA-induced neurotoxicity involved significant tyrosine nitration within the retinal pigment epithelium (RPE), inner segment (IS), and inner nuclear layers (INL) with strongest immunoreactivity within the ganglion cell layer (GCL). This tyrosine nitration was almost completely eliminated by TEMPOL or L-NAME. TEMPOL or L-NAME also significantly inhibited the NMDA-induced MDA formation (Figure 2E) ▶ , whereas L-NAME, but not TEMPOL, significantly inhibited the NMDA-induced nitrite/nitrate formation (Figure 2F) ▶ , demonstrating that TEMPOL is not an inhibitor for nitric oxide synthase. The L-NAME or TEMPOL treatment had no detectable effect on the untreated control eyes (data not shown). These results suggest that increased ONOO− formation plays an important role in glutamate-induced retinal neurotoxicity.
Figure 2.

NMDA-induced neurotoxicity is prevented by inhibiting ONOO− formation. NMDA (80 or 200 nmol/eye)-induced apoptosis, formation of nitrotyrosine, lipid peroxidation, and nitrite/nitrate in the rat retina as affected by intravitreal L-NAME (0.1 mg/eye) or TEMPOL (0.4 mg/eye) were determined at 18 hours after NMDA injection with or without L-NAME or TEMPOL. A: Representative retinal distribution of apoptotic nuclei in cryosections determined by TUNEL assay after two doses of NMDA injection and with or without L-NAME or TEMPOL. OCT-frozen eye sections (8 μm) perpendicular to the retinal surface were analyzed at two adjacent locations along the vertical meridian within 1 to 2 mm of the optic disk. Arrows indicate TUNEL-positive cells. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. B: Quantitative analysis of the dose effect of NMDA and the effect of L-NAME and TEMPOL on the NMDA-induced apoptosis in the retinal cryosections determined by TUNEL assay. Values represent the mean ± SE of a total of four animals for each dose in two experiments. *, Significant at P < 0.01. C: Representative retinal distribution of nitrotyrosine residues in cryosections determined by immunohistochemistry after NMDA injection with or without L-NAME or TEMPOL. OCT-frozen eye sections (8 μm) perpendicular to the retina surface were analyzed as described above. RPE, retinal pigment epithelium; IS, inner segment. D: Quantitative analysis of the effect of L-NAME and TEMPOL on the NMDA-induced relative optical density of nitrotyrosine in the retinal cryosections determined by immunohistochemistry. Values represent the mean ± SE of a total of four animals for each dose in two experiments. *, Significant at P < 0.01. E: Effect of L-NAME and TEMPOL on the NMDA-induced MDA formation in the retina. Values represent the mean ± SE of four animals per treatment group. *, Significant at P < 0.05. F: Effect of L-NAME and TEMPOL on the NMDA-induced nitrite/nitrate accumulation in the retina. Values represent the mean ± SE of four animals per treatment group. *, Significant at P < 0.05.
NMDA-Induced Retinal Neurotoxicity Is Prevented by THC
To evaluate the efficacy of systemic THC in preventing the NMDA’s neurotoxic effects, we first determined its effects on apoptosis as shown by TUNEL assay. Rats were given an intravenous injection of THC suspension immediately before the intravitreal injection of NMDA and the retinas analyzed at 18 hours after injection. The typical result from one of three independent TUNEL assays is shown in Figure 3A ▶ and the results quantified in Figure 3B ▶ . As shown, THC (0.4 and 2 mg/kg) produced a dose-dependent reduction in the number of TUNEL-positive cells in the NMDA-treated retinas (80 nmol/eye). In eyes treated with higher dose of NMDA (200 nmol/eye), THC at 0.4 mg/kg or 2 mg/kg also showed dose-dependent reduction in the apoptotic cell number (data not shown).
Figure 3.

NMDA-induced retinal neurotoxicity is prevented by THC. Rats were given an intravenous injection of THC suspension immediately before the intravitreal injection of NMDA. The effect of systemic THC (0.4 or 2 mg/kg) on the NMDA (80 nmol/eye)-induced retinal neurotoxicity is determined by TUNEL analysis at 18 hours after NMDA injection and by measurement of inner retinal thickness and Thy-1 and NF-L mRNA at day 7 of NMDA injection. A: Representative retinal distribution of NMDA-induced TUNEL-positive cells in retinal sections after injection of THC at two levels. OCT-frozen eye sections perpendicular to the retinal surface were analyzed as described before. B: Dose-dependent effect of THC on the NMDA-induced TUNEL-positive cells. Values represent the mean ± SE of a total of three animals for each group in three experiments. *, Significant at P < 0.01. C: Representative H&E-stained retinal cryosections (8 μm) showing the relative inner retinal thickness. OCT-frozen eye sections perpendicular to the retinal surface were measured at two adjacent locations as described before. The thickness of the inner retina (double-headed arrow) was measured by the distance between the inner limiting membrane and the inner border of outer nuclear layer. D: Relative inner retinal thickness analysis to show the loss of inner retinal thickness and protection by THC. Readings from three to four sections from each eye were averaged to obtain the mean value for that eye. All values represent the mean ± SE of a total of four animals for each group in two experiments. *, Significant at P < 0.05. E: Representative negative image of ethidium bromide-stained agarose (2%) gel indicating the level of 18S rRNA relative to Thy-1 or NF-L message in the retina total RNA. F: NMDA-induced reduction of Thy-1 and NF-L mRNA and dose-dependent protection by THC. Values represent the mean ± SE of four experiments. *, Significant at P < 0.05.
This cytoprotective effect of THC was confirmed by analysis of inner retinal thickness on day 7 after NMDA treatment. In this experiment, after the initial injection of THC and NMDA on day 1, a booster intravenous injection of THC at the same dose was given on day 3. Typical retinal images are shown in Figure 3C ▶ and the quantitative analysis is summarized in Figure 3D ▶ . As shown, the NMDA treatment caused a significant decrease in thickness of the inner retina, which was prevented by the THC treatment. Both doses of THC (0.4 and 2 mg/kg) were effective in preventing the cell loss and essentially restored inner retinal thickness to the normal level. Systemic THC injection in PBS-injected eyes had no effect on inner retinal thickness (data not shown).
We next evaluated the treatment effects on retinal ganglion cell loss, by using Thy-1 antigen as a marker for retinal ganglion cells and NF-L as a marker for ganglion cell axons. 31,32 At day 7 after THC and NMDA injections, we measured Thy-1 and NF-L mRNA levels in the retina using comparative quantitative RT-PCR to determine whether changes in the number of ganglion cells vary under NMDA and THC treatments. Figure 3E ▶ shows an ethidium bromide-stained gel of representative samples from a typical experiment. Figure 3F ▶ summarizes the results of four experiments. As shown, intravitreal injection of NMDA caused a significant loss of both Thy-1 and NF-L mRNA at day 7. THC offered a dose-dependent protection against this loss. THC in PBS-injected eyes had no effect on Thy-1 or NF-L mRNA (data not shown).
To evaluate the potential mechanisms underlying the protective effects of systemic THC in preventing NMDA neurotoxicity in the retina, we first determined whether THC treatment attenuated the formation of proteins nitrated on tyrosine, lipid peroxidation, and nitrite/nitrate levels in the NMDA-treated rat retinas. Rats were given an intravenous injection of THC suspension immediately before the intravitreal injection of NMDA and the retinas analyzed at 18 hours after injection. In Figure 4A ▶ , which shows the analysis of immunofluorescence of nitrotyrosine residues in the retina, THC (0.4 and 2 mg/kg) caused a dose-dependent reduction in the NMDA (80 and 200 nmol/eye)-induced immunofluorescence of nitrotyrosine. In Figure 4B ▶ , which shows the analysis of immunochemical slot blot, systemic THC (2 mg/kg) significantly inhibited tyrosine nitration induced by intravitreal NMDA (200 nmol/eye), confirming the immunofluorescence result. In these analyses, THC alone had no effect. As shown in Figure 4C ▶ , THC (2 mg/kg) significantly reduced NMDA-induced MDA formation. Further, THC (2 mg/kg) significantly reduced NMDA (200 nmol/eye)-induced nitrite/nitrate formation (Figure 4D) ▶ . THC alone did not alter MDA formation or nitrite/nitrate levels in the intact eye. Together, these results suggest that THC protects the retinal neurons, as do TEMPOL and L-NAME, and that this effect involves reduction in levels of superoxide anion, NO, and ONOO−.
Figure 4.

THC protects the NMDA-induced retinal neurotoxicity by inhibiting ONOO− formation. Rats were given an intravenous injection of THC suspension immediately before the intravitreal injection of NMDA. Effect of systemic THC on the NMDA-induced formation of nitrotyrosine, lipid peroxidation, and nitrite/nitrate in the retina is shown at 18 hours after NMDA injection. All values represent the mean ± SE of four to five animals for each group. *, Significant at P < 0.05. A: Dose-dependent effect of THC on the NMDA-induced relative optical density of nitrotyrosine in retinal cryosections (8 μm) determined by immunohistochemistry. OCT-frozen eye sections perpendicular to the retinal surface were analyzed as described before. B: Effect of THC on the NMDA-induced nitrotyrosine formation determined by immunochemical slot-blot analysis. C: Effect of THC on the NMDA-induced MDA formation in the retina. D: Effect of THC on the NMDA-induced nitrite/nitrate accumulation in the retina.
The Cannabinoid Receptor CB1 Has a Role in the Neuroprotective Effect of THC
Although THC has been demonstrated in vitro as a potent antioxidant that protects neuron cultures from glutamate-induced death 19 or oxidative stress, 20 THC is a CB1 cannabinoid receptor agonist and is therefore psychoactive. To determine whether the protective effect of THC on the apoptotic effect of NMDA in the retina is mediated via the CB1 receptor, SR141716A, an antagonist/inverse agonist specific to the CB1 receptor subtype was injected along with THC. TUNEL assay was performed at 18 hours after injection. The typical result of the TUNEL assays and the quantitative analysis of four experiments are shown in Figure 5 ▶ . As shown, THC at 2 mg/kg reduced the number of NMDA-induced TUNEL-positive cells in the inner retina, and SR141716A at 1 to 2 mg/kg produced a dose-dependent increase in the number of these cells. However, even at the high level at 2 mg/kg, SR141716A did not completely block the neuroprotective effect of THC. It is noted that the antagonist alone at 2 mg/kg did not increase the NMDA-induced TUNEL-positive cells, suggesting that the observed neuroprotection-blocking effect by SR141716A was not because of its inverse agonist activity. This result suggests that the activation of CB1 receptors contributes significantly, but not exclusively, to the protective effect of THC.
Figure 5.

The cannabinoid receptor CB1 has a role in the neuroprotective effect of THC. SR141716A, an antagonist/inverse agonist specific to the CB1 receptor subtype, was injected along with THC immediately before the intravitreal injection of NMDA. Effects of systemic SR141716A (1 or 2 mg/kg) and THC (2 mg/kg) on the NMDA (200 nmol/eye)-induced retinal neurotoxicity were determined by TUNEL analysis at 18 hours after NMDA injection. OCT-frozen eye sections (8 μm) perpendicular to the retinal surface were analyzed as described before. A: Representative retinal distribution of NMDA-induced TUNEL-positive cells in retinal sections after injection of THC along with SR-141716A. B: Dose-dependent attenuation by SR-141716A on the protective effect of THC on the TUNEL-positive retinal cells induced by NMDA. Values represent the mean ± SE of four experiments. *, Significant at P < 0.05. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
NMDA-Induced Retinal Neurotoxicity Is Prevented by Cannabidiol (CBD)
To confirm that blocking of nitrotyrosine formation is necessary for neuroprotection in the retina, the nonpsychotropic constituent of marijuana, CBD, was injected in rats immediately before the intravitreal injection of NMDA. CBD does not activate the cannabinoid receptors and so is devoid of psychoactive effects. 34 The retinas were analyzed 18 hours later to determine whether CBD treatment attenuated the formation of ONOO− and whether it reduced the apoptotic effect of NMDA in the retina. Figure 6A ▶ shows that CBD (2 mg/kg) significantly attenuated the NMDA (200 nmol/eye)-induced tyrosine nitration in the retina. Figure 6B ▶ shows the typical result of TUNEL assays and the results quantified in Figure 6C ▶ . These figures show that CBD also reduces the NMDA (80 nmol/eye)-induced apoptosis in the retina. Moreover, in eyes treated with higher dose of NMDA (200 nmol/eye), CBD at 2 mg/kg also showed reduction in the number of apoptotic cells (data not shown).
Figure 6.
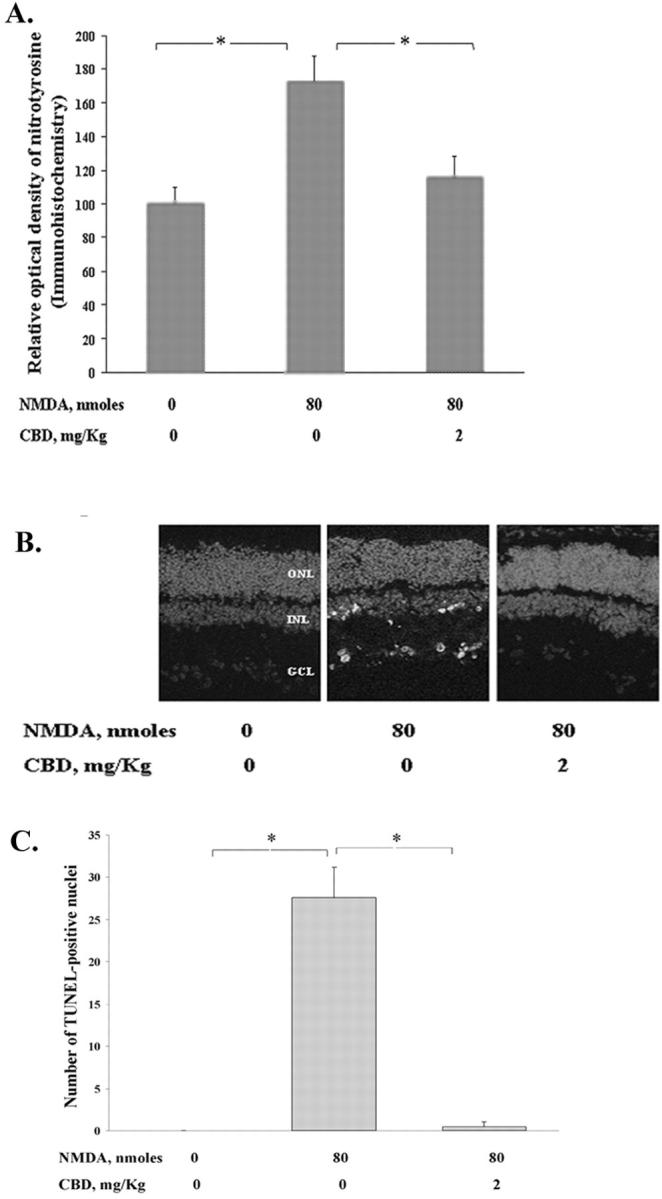
NMDA-induced retinal neurotoxicity is prevented by CBD. Rats were given an intravenous injection of CBD suspension immediately before the intravitreal injection of NMDA. Effect of systemic CBD (2 mg/kg) on the NMDA (80 nmol/eye)-induced formation of nitrotyrosine and retinal apoptosis was determined at 18 hours after NMDA injection. OCT-frozen eye sections (8 μm) perpendicular to the retinal surface were analyzed as described before. A: Effect of CBD on the NMDA-induced relative optical density of nitrotyrosine in retinal cryosections determined by immunohistochemistry. All values represent the mean ± SE of a total of four animals for each group in two experiments. *, Significant at P < 0.05. B: Representative retinal distribution of NMDA-induced TUNEL-positive cells in retinal sections after injection of CBD. C: Quantitative analysis of the effect of CBD on the NMDA-induced apoptosis in the retinal cryosections determined by TUNEL assay. Values represent the mean ± SE of a total of four animals for each dose in two experiments. *, Significant at P < 0.01.
Discussion
The present results demonstrate that oxidative stress as indicated by increases in lipid peroxidation and formation of nitrite and nitrotyrosine plays an important role in the NMDA-induced retinal neurotoxicity. Our finding that L-NAME and TEMPOL attenuate lipid peroxidation and nitrotyrosine formation and prevent NMDA-induced retinal cell death strongly supports a causal role for these reactive oxygen species in NMDA-induced neurotoxicity. Our finding of the neuroprotective effect of TEMPOL without effect on nitrite formation agrees well with a previous study in brain injury. 14 Furthermore, our studies show that systemic THC or CBD also exhibits neuroprotective effects by attenuating nitrotyrosine formation. To the best of our knowledge, this is the first demonstration for TEMPOL, THC, and CBD as retinal neuroprotectants in vivo via attenuating lipid peroxidation and/or nitrotyrosine formation.
Peroxynitrite is difficult to measure because of its instability. Therefore, the presence of ONOO− in pathophysiological processes is commonly demonstrated by the detection of nitrotyrosine. 35 It should be noted that there are other potential sources of tyrosine nitration in addition to ONOO−. 36 However, nitrotyrosine is considered to be a likely indicator for ONOO− under conditions of simultaneous production of NO and superoxide. 28,29,37,38 Our time course study results showing that increases in nitrate/nitrite accumulation and lipid peroxidation parallel nitrotyrosine formation and neurotoxicity strongly implicate ONOO− formation in NMDA neurotoxicity. Although lipid peroxidation, NO, and ONOO− accumulation in clinical glaucoma has not been published, a preliminary study supporting a role of these reactive oxygen species in monkey retinas with experimental glaucoma has been reported. 39 We noticed that there are decreases in the formation of nitrotyrosine, MDA, and nitrates after 18 hours of NMDA injection (Figure 1) ▶ . These decreases are likely because of neuronal cell loss, which mostly occurs at 18 hours of neurotoxicity.
Various doses of THC (0.1, 1, and 10 mg/kg) have been shown to have neuroprotectant function in rat models of cerebral ischemia. 18,40 In the present work, THC at 0.4-mg/kg and 2-mg/kg dose levels produced a dose-dependent decrease in numbers of NMDA-induced TUNEL-positive cells even at high doses of NMDA (200 nmol/eye). 3 To determine the protracted retinal damage and the protective effect of THC, we performed additional experiments at day 7 after NMDA injection. At this time, the net cell loss was evaluated by measuring the inner retinal thickness and by directly determining the expression of ganglion cell-associated genes Thy-1 and NF-L. The results of these analyses are generally in good agreement with the results of TUNEL assay. However, we noticed that the dose-dependent protective effect of THC on Thy-1 and NF-L expressions was not observed in the inner retinal thickness measurements (Figure 3) ▶ . This may be because of the different sensitivities of comparative quantitative RT-PCR compared to the morphological study of inner retina thickness.
Our result shows a dose-dependent, partial blockade of the effect of THC by SR141716A, an antagonist/inverse agonist of the CB1 receptor. This suggests that the neuroprotective effect of THC is because of, at least in part, the activation of CB1 receptor. In support of this possibility, the CB1 receptor is expressed in the inner neurons of the retina, including bipolar, amarcrine, and horizontal cells. 41 Activation of CB1 receptors in these cells could reduce excitotoxicity by inhibiting voltage-sensitive calcium channels 42,43 as well as by enhancing voltage-dependent potassium channel activity (which in turn inhibits excitation). 44 These changes in turn reduce the formation of superoxides and NO. Our result on the effect of THC is similar to the recently reported effect of WIN55,212-2, another cannabinoid receptor agonist, in a murine model. 45
Our result also shows that SR141716A at 2 mg/kg did not completely block the neuroprotective effect of THC. To determine whether the neuroprotective effect of THC is entirely CB1 receptor-dependent, higher SR141716A concentrations than 2 mg/kg should also be tested in our study. However, SR141716A at higher concentrations than required for inhibition of CB1 receptors is known to produce masking actions by indirect effects on other receptor systems. 46,47 Thus, blockade of neuroprotective effect of THC by SR141716A is not absolute proof of CB1 receptor involvement. An alternative procedure to determine the role of CB1 in THC-mediated retinal neuroprotection should be performed in CB1 receptor knockout mice.
Next, we evaluated the effect of CBD, a nonpsychotropic cannabinoid that does not activate the CB1 receptors. 34 Our study shows that CBD is also neuroprotective in vivo via blocking nitrotyrosine formation. This result confirms that blocking of nitrotyrosine formation is necessary for neuroprotection. Our result is supported by previous in vitro analyses, which have shown that both psychotropic and nonpsychotropic cannabinoids that contain phenolic structures, such as THC and CBD, are similarly potent antioxidants that protect neuron cultures from glutamate-induced cell death 19 or oxidative stress. 20 The latter has been shown in cultured neurons isolated from either CB1 receptor knockout mice or wild-type mice. 30
It should be mentioned that CBD is not only an antioxidant per se but it also inhibits the degradation of an endogenous cannabinoid, arachidonoyl ethanolamide or anandamide, 48 which has been shown as a neuroprotectant through multiple mechanisms including noncannabinoid receptor targets. 49-52 In addition to the role as an antioxidant, THC could activate the neuroprotective mitogen-activated protein kinase-signaling pathway. 53 Future studies will be required to determine the exact mechanism of neuroprotection by cannabinoids in glutamate-induced retinal neurotoxicity.
In addition to possessing neuroprotective or retinal neuroprotective activity as demonstrated here and elsewhere, cannabinoids such as THC, WIN55,212-2, endogenous cannabinoid 2-arachidonoylglycerol, as well as nonpsychotropic HU-211 have been demonstrated to induce dose-related reductions in intraocular pressure in human and in animal models. 54-58 This suggests that cannabinoids may offer a multifaceted therapy for glaucoma. The neuroprotective effect of these nonpsychotropic or endogenous cannabinoids is currently under investigation in our laboratory.
In conclusion, our results indicate that lipid peroxidation and ONOO− formation play an important role in NMDA-induced retinal neurotoxicity and cell loss in the retina, and that THC and CBD, by reducing the formation of these compounds, are effective neuroprotectants. The present studies could form the basis for the development of new topical therapies for the treatment of glaucoma.
Acknowledgments
We thank Dr. Deborah Lewis for discussions of the manuscript; Mr. Nabil Yang for his technical assistance; and Dr. Ellen S. Liberman (National Institutes of Health) and Mr. Kevin Gormley (National Institute of Drug Abuse (NIDA), Research Triangle Park, NC) for providing (−)Δ9-tetrahydroxycannabinol.
Footnotes
Address reprint requests to Gregory I. Liou, Ph.D., Department of Ophthalmology, Medical College of Georgia, 1120 15th St., Augusta, GA 30912. E-mail: giliou@mail.mcg.edu.
Supported in part by an unrestricted departmental award from Research to Prevent Blindness Inc., New York, NY (to G. I. L. and R. B. C.), the American Heart Association (to R. W. C.) and the National Institutes of Health (grants EY12078 to G. I. L., EY04618 and EY11766 to R. B. C.).
KG died on Nov. 22, 2001.
References
- 1.Dreyer EB, Lipton SA: New perspectives on glaucoma. JAMA 1999, 281:306-308 [DOI] [PubMed] [Google Scholar]
- 2.Thoreson WB, Witkovsky P: Glutamate receptors and circuits in the vertebrate retina. Prog Ret Eye Res 1999, 18:765-810 [DOI] [PubMed] [Google Scholar]
- 3.Lam TT, Abler AS, Kwong JMK, Tso MOM: NMDA-induced apoptosis in rat retina. Invest Ophthalmol Vis Sci 1999, 40:2391-2397 [PubMed] [Google Scholar]
- 4.Sucher NJ, Leis S, Lipton SA: Calcium channel antagonists attenuate NMDA receptor mediated neurotoxicity of retinal ganglion cells in culture. Brain Res 1991, 551:297-302 [DOI] [PubMed] [Google Scholar]
- 5.Coyle JT, Puttfarcken P: Oxidative stress, glutamate and neurodegenerative disorders. Science 1993, 262:689-693 [DOI] [PubMed] [Google Scholar]
- 6.Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA: Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol 1996, 114:299-305 [DOI] [PubMed] [Google Scholar]
- 7.Carter-Dawson L, Shen F, Harwerth RS, Smith EL, III, Crawford MLJ, Chuang A: Glutamine immunoreactivity in Muller cells of monkey eyes with experimental glaucoma. Exp Eye Res 1998, 66:537-545 [DOI] [PubMed] [Google Scholar]
- 8.Shibuki H, Katai N, Yodoi J, Uchida K, Yoshimura N: Lipid peroxidation and peroxynitrite in retinal ischemia-reperfusion injury. Invest Ophthalmol Vis Sci 2000, 41:3607-3614 [PubMed] [Google Scholar]
- 9.Ciani E, Groneng L, Voltattorni M, Rolseth V, Contestabile A, Paulsen RE: Inhibition of free radical production or free radical scavenging protects from the excitotoxic cell death mediated by glutamate in cultures of cerebellar granule neurons. Brain Res 1996, 728:1-6 [PubMed] [Google Scholar]
- 10.MacGregor DG, Higgins MJ, Jones PA, Maxwell WL, Watson MW, Graham DI, Stone TW: Ascorbate attenuates the systemic kainate-induced neurotoxicity in the rat hippocampus. Brain Res 1996, 727:133-144 [DOI] [PubMed] [Google Scholar]
- 11.Iadecola C, Xu X, Zang F, Hu J, El-Fakahany EE: Prolonged inhibition of brain nitric oxide synthase by short-term systemic administration of nitro-L-arginine methyl ester. Neurochem Res 1994, 19:501-505 [DOI] [PubMed] [Google Scholar]
- 12.Mitchell JB, Samuni A, Krishna MC, DeGraff WG, Ahn MS, Samuni U, Russo A: Biologically active metal-independent superoxide dismutase mimics. Biochemistry 1990, 29:2802-2807 [DOI] [PubMed] [Google Scholar]
- 13.Laight DW, Andrews TJ, Haj-Yehia AJ, Carrier MJ, Anggard EE: Microassay of superoxide anion scavenging activity in vitro. Environ Toxicol Pharmacol 1997, 3:65-68 [DOI] [PubMed] [Google Scholar]
- 14.Cuzzocrea S, McDonald MC, Mazzon E, Siriwardena D, Costantino G, Fulia F, Cucinotta G, Gitto E, Cordaro S, Barberi I, DeSarro A, Caputi AP, Thiemermann C: Effects of TEMPOL, a membrane-permeable radical scavenger, in a gerbil model of brain injury. Brain Res 2000, 875:96-106 [DOI] [PubMed] [Google Scholar]
- 15.Morizane C, Adachi K, Furutani I, Fujita Y, Akaike A, Kashii S, Honda Y: N-Nitro-L-arginine methyl ester protects retinal neurons against NMDA-induced neurotoxicity in vivo. Eur J Pharmacol 1997, 328:45-49 [DOI] [PubMed] [Google Scholar]
- 16.Shen M, Thayer SA: Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol 1998, 54:459-462 [DOI] [PubMed] [Google Scholar]
- 17.Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA: Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 1999, 19:2987-2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van der Stelt M, Veldhuis WB, Bär PR, Veldink GA, Vliegenthart JFG, Nicolay K: Neuroprotection by Δ9-tetrahydrocannabinol, the main active compound in marijuana, against ouabain-induced in vivo excitotoxicity. J Neurosci 2001, 21:6475-6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hampson AJ, Grimald M, Axelrod J, Wink D: Cannabidiol and (−)Δ9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA 1998, 95:8268-8273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marsicano G, Moosmann B, Hermann H, Lutz B, Behlt C: Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem 2002, 80:448-456 [DOI] [PubMed] [Google Scholar]
- 21.Yoles E, Belkin M, Schwartz M: HU-211, a nonpsychotropic cannabinoid, produces short-and long-term neuroprotection after optic nerve axotomy. J Neurotrauma 1996, 13:49-57 [DOI] [PubMed] [Google Scholar]
- 22.Siliprandi R, Canelia R, Carmignoto G, Schiavo N, Zanellato A, Zanoni R, Vantini G: NMDA-induced neurotoxicity in the adult rat retina. Vis Neurosci 1992, 8:567-573 [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Schlamp CL, Nickells RW: Experimental induction of retinal ganglion cell death in adult mice. Invest Ophthalmol Vis Sci 1999, 40:1004-1008 [PubMed] [Google Scholar]
- 24.Kido N, Tanihara H, Honjo M, Inatani M, Tatsuno T, Nakayama C, Honda Y: Neuroprotective effects of brain-derived neurotrophic factor in eyes with NMDA-induced neuronal death. Brain Res 2000, 884:59-67 [DOI] [PubMed] [Google Scholar]
- 25.Inomata Y, Hirata A, Yonemura N, Koga T, Kido N, Tanihara H: Neuroprotective effects of interleukin-6 on NMDA-induced rat retinal damage. Biochem Biophys Res Commun 2003, 302:226-232 [DOI] [PubMed] [Google Scholar]
- 26.Hevel JM, Marletta MA: Nitric-oxide synthase assays. Methods Enzymol 1994, 233:250-258 [DOI] [PubMed] [Google Scholar]
- 27.Ohkawa H, Ohishi N, Yagi K: Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 1979, 95:351-358 [DOI] [PubMed] [Google Scholar]
- 28.Ischiropoulos H, Beers MF, Ohnishi ST, Fisher D, Garner SE, Thom SR: Nitric oxide production and perivascular nitration in brain after carbon monoxide poisoning in the rat. J Clin Invest 1996, 97:2260-2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawa T, Akaike T, Maeda H: Tyrosine nitration by peroxynitrite formed from nitric oxide and superoxide generated by xanthine oxidase. J Biol Chem 2000, 275:32467-32474 [DOI] [PubMed] [Google Scholar]
- 30.El-Remessy AB, Behzadian MA, Abou-Mohamed G, Franklin T, Caldwell RW, Caldwell RB: Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am J Pathol 2003, 162:1995-2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hughes WF: Quantitation of ischemic damage in the rat retina. Exp Eye Res 1991, 53:573-582 [DOI] [PubMed] [Google Scholar]
- 32.Beale R, Osborne NN: Localization of the Thy-1 antigen to the surface of rat retinal ganglion cells. Neurochem Int 1982, 4:587-595 [DOI] [PubMed] [Google Scholar]
- 33.Osborne NN, Wood JPM, Cupido A, Melena J, Chidlow G: Topical flunarizine reduces IOP and protects the retina against ischemia-excitotoxicity. Invest Ophthalmol Vis Sci 2002, 43:1456-1464 [PubMed] [Google Scholar]
- 34.Mansbach RS, Rovetti CC, Winston EN, Lowe JA, III: Effects of the cannabinoid CB1 receptor antagonist SR141716A on the behavior of pigeons and rats. Psychopharmacology (Berl) 1996, 124:315-322 [DOI] [PubMed] [Google Scholar]
- 35.Misko TP, Highkin MK, Veenhuizen AW, Manning PT, Stern MK, Currie MG, Salvemini D: Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J Biol Chem 1998, 273:15646-15653 [DOI] [PubMed] [Google Scholar]
- 36.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, Van Der Vliet A: Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 1998, 391:393-397 [DOI] [PubMed] [Google Scholar]
- 37.Halliwell B, Zhao K, Whiteman M: Nitric oxide and peroxynitrite. The ugly, the uglier and the not so good: a personal view of recent controversies. Free Radic Res 1999, 31:651-669 [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Joseph J, Feix J, Hogg N, Kalyanaraman B: Nitration and oxidation of a hydrophobic tyrosine probe by peroxynitrite in membranes: comparison with nitration and oxidation of tyrosine by peroxynitrite in aqueous solution. Biochemistry 2001, 40:7675-7686 [DOI] [PubMed] [Google Scholar]
- 39.Whitetree AR, Harwerth RS, Smith EL, III, Crawford MLJ, Feldman R, Carter-Dawson L: Nitric oxide and peroxynitrite in monkey retinas with experimental glaucoma. Invest Ophthalmol Vis Sci 2001, 42:S749(Abstract 4010) [Google Scholar]
- 40.Louw DF, Yang FW, Sutherland GR: The effect of δ-9-tetrahydrocannabinol on forebrain ischemia in rat. Brain Res 2000, 857:183-187 [DOI] [PubMed] [Google Scholar]
- 41.Yazulla S, Studholme KM, McIntosh HH, Deutsch DG: Immunocytochemical localization of cannabinoid CB1 receptor and fatty acid amide hydrolase in rat retina. J Comp Neurol 1999, 415:80-90 [DOI] [PubMed] [Google Scholar]
- 42.Pan X, Ikeda SR, Lewis DL: Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol Pharmacol 1996, 49:707-714 [PubMed] [Google Scholar]
- 43.Twitchell W, Brown S, Mackie K: Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 1997, 78:43-50 [DOI] [PubMed] [Google Scholar]
- 44.Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S: Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther 1995, 273:734-743 [PubMed] [Google Scholar]
- 45.Ayoub G, Garcia N, Fischer L, Roach K, Chan R, Nudelman A, Quirkm A, Morris C: Neuroprotection of retinal ganglion cells under glaucomatous conditions. International Cannabinoid Research Society, 12th Annual Symposium on the Cannabinoids, 2002, Abstract 134
- 46.Cheer JF, Cadogan AK, Marsden CA, Fone KC, Kendall DA: Modification of 5-HT2 receptor mediated behaviour in the rat by oleamide and the role of cannabinoid receptors. Neuropharmacology 1999, 38:533-541 [DOI] [PubMed] [Google Scholar]
- 47.Darmani NA, Pandya DK: Involvement of other neurotransmitters in behaviors induced by the cannabinoid CB1 receptor antagonist SR 141716A in naive mice. J Neural Transm 2000, 107:931-945 [DOI] [PubMed] [Google Scholar]
- 48.Bisogno T, Hanus L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I, Moriello AS, Davis JB, Mechoulam R, Di Marzo V: Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol 2001, 134:845-852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Stelt M, Veldhuis WB, van Haaften GW, Fezza F, Bisogno T, Bar PR, Veldink GA, Vliegenthart JF, Di Marzo V, Nicolay K: Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J Neurosci 2001, 21:8765-8771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinor AD, Irvin SM, Greenberg DA: Endocannabinoids protect cerebral cortical neurons from in vitro ischemia in rats. Neurosci Lett 2000, 278:157-160 [DOI] [PubMed] [Google Scholar]
- 51.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED: Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400:452-457 [DOI] [PubMed] [Google Scholar]
- 52.Maingret F, Patel AJ, Lazdunski M, Honore E: The endocannabinoid anandamide is a direct and selective blocker of the background K(+) channel TASK-1. EMBO J 2001, 20:47-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valjent E, Pages C, Rogard M, Besson MJ, Maldonado R, Caboche J: Delta9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopamineric transmission. Eur J Neurosci 2001, 14:342-352 [DOI] [PubMed] [Google Scholar]
- 54.Tiedeman JS, Shields MB, Weber PA, Crow JW, Cocchetto DM, Harris WA, Howes JF: Effect of synthetic cannabinoids on elevated intraocular pressure. Ophthalmology 1981, 88:270-277 [DOI] [PubMed] [Google Scholar]
- 55.Beilin M, Neumann R, Belkin M, Green K, Bar-Ilan A: Pharmacology of the intraocular pressure lowering effect of systemic dexanabinol (HU-211), a non-psychotropic cannabinoid. J Ocul Pharmacol Ther 2000, 16:217-229 [DOI] [PubMed] [Google Scholar]
- 56.Laine K, Jarvinen K, Mechoulam R, Breuer A, Jarvinen T: Comparison of the enzymatic stability and intraocular pressure effects of 2-arachidonylglycerol and noladin ether, a novel putative endocannabinoid. Invest Ophthalmol Vis Sci 2002, 43:3216-3222 [PubMed] [Google Scholar]
- 57.Panikashvill D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E: An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001, 413:527-531 [DOI] [PubMed] [Google Scholar]
- 58.Song ZH, Slowey CA: Involvement of cannabinoid receptors in the intraocular pressure-lowering effects of WIN55212-2. J Pharmacol Exp Ther 2000, 292:136-139 [PubMed] [Google Scholar]