

Abstract
The Apc1638N/+ mouse has a chain-terminating mutation in one allele of the adenomatous polyposis coli (Apc) gene that is similar to most mutations observed in the human familial adenomatous polyposis syndrome. Aberrant crypt foci (ACF), the earliest identified neoplastic lesions in the colon, are morphologically abnormal structures that are identifiedmicroscopically in the grossly normal colonic mucosas of rodents treated with colon carcinogens and of human patients. The colons and cecums of 62 Apc1638N/+ mice were evaluated for the spontaneous occurrence of ACF and tumors. Both male and female mice were killed at different times between 5 and 28 weeks of age. Wild-type littermates, ie, Apc+/+ mice, at 22 to 26 weeks of age served as controls. ACF were identified in 97% of the Apc1638N/+ mice starting at 5 weeks of age and not in any wild-type littermates. Although the number of ACF increased with age (P < 0.0001), the average number of crypts per focus of the ACF did not increase significantly. In addition, wild-type Apc protein was detected by immunohistochemistry in all 22 ACF evaluated. Together these data suggest that heterozygous loss of Apc may be sufficient to initiate ACF in these mice and that these mice may be suitable models to study the interaction of environmental factors with an inherited mutation of the Apc gene that is associated with colon cancer.
Inherited mutations in the adenomatous polyposis coli (APC) gene, located on human chromosome 5q21, are responsible for the human familial adenomatous polyposis (FAP) syndrome. 1,2 FAP patients develop hundreds to thousands of benign tumors in the colon, some of which will eventually progress to carcinoma if left in the gastrointestinal tract. Although FAP accounts for only 1% of colon cancer in the United States, the importance of the APC gene in colon cancer goes beyond tumors that develop in this inherited disease. Greater than 80% of all colon tumors, including very small lesions, have mutations that develop somatically in the APC gene, indicating that mutations at this locus are likely involved at an early stage in a high proportion of human colon cancer. 1,2 Mouse models with germline mutations of the homologous mouse Apc gene 3-6 provide tools to study the function of the Apc gene in vivo. These models include the Min (ApcMin/+)mouse, with a random mutation that resulted from treatment with the mutagen ethylnitrosourea, 3,4 and the Apc1638N 5 and ApcΔ716 6 mice, with targeted mutations of the Apc locus that inactivate the gene. These mutations are chain-terminating mutations, similar to most mutations in the human FAP syndrome. In these three mouse models, like human FAP, tumors develop spontaneously throughout the gastrointestinal tract; but the tumors are more numerous in the small intestines of mice, in contrast to the prevalence of tumors in the colons of humans. In each of these models, the inactivation of both alleles of Apc in the germline of the mouse is lethal at an early stage of embryogenesis. 3,5,6 However, the kinetics and extent of tumor development in the different models are different: the Min mouse and the ApcΔ716 mouse develop tumors much more rapidly and in far higher numbers than the Apc1638N/+ mouse, but they rarely if ever develop carcinomas; the Apc1638N/+ mice develop carcinomas as well as desmoid tumors similar to those seen in some FAP patients. 3,5-9 The longer life span and the tumor characteristics make the Apc1638N/+ mouse particularly amenable to the study of tumor pathogenesis from premalignant biomarkers through invasive, metastatic tumors; and longer term dietary studies, which may modulate genetically initiated tumorigenesis.
In the present study we report the presence of aberrant crypt foci (ACF) in the flat colonic mucosa of untreated Apc1638N/+ mice. These microscopically identified, morphologically abnormal structures are seen in rodents treated with colon-specific chemical carcinogens but not in untreated rodents. 10-12 ACF have been identified in the grossly normal colonic mucosa of patients: their frequency is 100-fold higher in patients with colon cancer and is 1000-fold higher in FAP patients than in those without colon cancer. 13-15 ACF in humans are reported to be monoclonal. 16 However, ACF are rare or absent in the Min mouse 17-20 and the ApcΔ716 mouse 21 models. The data presented here, including the retained expression of wild-type Apc protein in ACF in the Apc1638N/+ mouse, suggest that heterozygous loss of Apc may be sufficient to initiate ACF in the colons of these mice and that these mice may be good in vivo models to study the interaction of environmental factors with the inheritance of a single mutant copy of the Apc gene.
Materials and Methods
Mice
To breed genetically modified mice, each breeding cage consisted of one male and two female Apc1638N/+ mice. When offspring were ∼3 weeks old, DNA from tail clips was genotyped at the Apc locus using a polymerase chain reaction assay as previously described. 5 Apc1638N/+ and wild-type mice were separated and were maintained ad libitum on the control-purified AIN 76A diet (Harlan Teklad, Madison, WI) until the mice were killed. A total of 62 Apc1638N/+ mice were evaluated in these studies. Two additional Apc1638N/+ mice died, as described below, and could not be evaluated. Twelve of the wild-type littermates between 22 and 26 weeks old served as controls. Our first (preliminary) experiments were performed with 15 Apc1638N/+ mice between the ages of 5 and 26 weeks of age. For the second study, 13 mice were killed at 9 weeks and 12 mice were killed at 22 to 26 weeks of age. In the last experiment, three groups of eight Apc1638N/+ mice were maintained on the AIN 76A diet for 9 weeks, ie, until 12 weeks of age. One of these groups was killed at 12 weeks of age. The second group was maintained on the AIN 76A diet for an additional 16 weeks, ie, killed at 28 weeks of age, but two mice in this group died before 28 weeks of age. The third group of eight mice at 12 weeks of age was transferred to a Western-style diet 22 for an additional 16 weeks and killed at 28 weeks of age.
Pathology
Animals were lightly anesthetized; and 2 to 3 ml of 0.2% methylene blue (Chroma-Gesellschaft Schmid & Co., distributed by Roboz Surgical Instrument Co., Washington, DC) in phosphate-buffered saline (PBS) (0.01 mol/L phosphate, pH 7.4, 0.137 mol/L NaCl) was instilled into the colon through the anal canal. After 3 to 5 minutes, animals were killed by cervical dislocation; the colon and cecum were removed, cleaned in cold 0.9% NaCl solution, and fixed flat for 1 hour in 10% neutral buffered formalin (Fisher Scientific, Pittsburgh, PA) at 4°C. The colon was divided into two equal pieces with the segment closest to the cecum termed “proximal colon” and the remaining half termed “distal colon.” ACF were scored with a light microscope under ×40 or ×100 magnification as described previously. 10,11 Several ACF with four or more crypts were marked with permanent ink (Davidson Marking System; Bradley Products Inc., Bloomington, MN) and embedded in paraffin. Small intestines were fixed in 95% ethanol and evaluated under a dissecting microscope at ×30 magnification. Tumors and areas of unusual morphology were embedded in paraffin. Histological sections of small intestinal lesions and colonic ACF were stained with hematoxylin and eosin (H&E) for morphological evaluation.
Immunohistochemistry
Paraffin-embedded sections of ACF were evaluated for the expression of full-length Apc protein with rabbit polyclonal antibodies specific for the COOH terminal portion of the Apc protein (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). As a control, an adjacent section was incubated with antibody that had been preincubated with a 10-fold excess peptide that was used to prepare the antibody (Santa Cruz Biotechnology, Inc.) or with normal rabbit serum. The immunohistochemical procedures were similar to those described previously 23 and included antigen retrieval in 0.1 mol/L Tris buffer, pH 8.6, for 3 minutes after reaching full pressure in a pressure cooker. The primary antibody, diluted 1:500 in blocking solution (10% normal goat serum in PBS), was followed by biotinylated goat anti-rabbit IgG antibodies (Vector Laboratories, Burlingame, CA) diluted 1:400. The signal was detected with tyramide signal amplification (TSA Biotin System; NEN Life Science Products, Boston, MA) followed by streptavidin biotinylated horseradish peroxidase complex (Amersham, Arlington Heights, IL) diluted 1:100 and the chromogenic substrate 3,3′-diaminobenzidine (Sigma Chemicals, St. Louis, MO) as used previously. 23 Most of the 22 ACF were also evaluated for the expression of β-catenin as detailed previously. 24 The mouse monoclonal anti-β-catenin antibody (IgG1; Transduction Laboratories, Lexington, KY) was diluted 1:1000 in blocking solution.
Results
ACF are putative premalignant lesions that are identified in whole-mount preparations of colons from rodents treated with chemical carcinogens 10,12 and humans at risk for colon tumor development. 12-15 Because tumors in the gastrointestinal tract form spontaneously in the Apc1638N/+ mice, 5,7 the first question posed was: do ACF form spontaneously in these mice? Our first (preliminary) studies were performed with 15 Apc1638N/+ mice between 5 and 26 weeks of age. ACF were identified in the cecum as well as the proximal and distal colons of these mice (Figure 1 ▶ ; A to C). In our youngest mice, 5 to 6 weeks old, there was a total of 5 and 15 ACF with an average of 3.8 ± 3.0 crypts per focus in the two female mice, but no ACF in the single male mouse. This male mouse was the only Apc1638N/+ mouse of these 15 mice in which ACF were not found. In subsequent studies with 47 additional Apc1638N/+ mice (22 males and 25 females) described below, there was only 1 additional mouse, a 12-week-old male mouse, in which ACF were not found. Careful examination of the entire colons of wild-type littermates, ie, Apc+/+ mice, revealed no ACF in 12 mice at 22 to 26 weeks of age. Therefore, the appearance of ACF is limited to mice carrying the mutant Apc1638N allele and occurs as early as 5 weeks of age.
Figure 1.
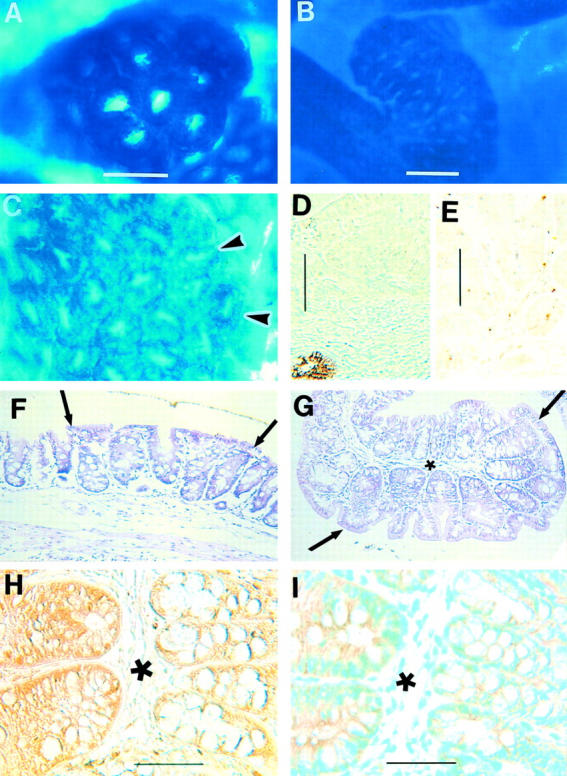
ACF that develop spontaneously in Apc1638N/+ mice. Immunohistochemical staining with diaminobenzidine substrate and methyl green counterstain demonstrates the expression of Apc and β-catenin proteins in paraffin-embedded tissue. A: Methylene blue-stained ACF in unembedded cecum. Note a few normal crypts in the bottom right corner; most normal crypts are out of focus because ACF are slightly raised. B: Methylene blue-stained ACF on a ridge in unembedded proximal colon. C: Methylene blue-stained unembedded cecum with an ACF to the left of the arrowheads (same magnification as A). D: Human colon cancer with a single adjacent normal crypt reveals Apc protein in the normal crypt but not in the cancer (negative control). E: Tumor found in the small intestine of Apc1638N/+ mouse shows lack of expression of Apc protein. The golden dots are from yellow ink that was applied to the tumor before embedding (negative control). F: H&E-stained section of the same cecal ACF as seen in C located between the arrows with yellow ink at the top. Note the enlarged crypts between the arrows (same magnification as A and C). G: H&E-stained section of an ACF in the proximal colon located between the arrows. Note the enlarged crypts and relative loss of goblet cells in the ACF (located below and to the right of the asterisk; same magnification as A). H: Nearby section of the same ACF as seen in G (asterisks denote same location in the two sections) demonstrates Apc protein in both the ACF (located to the left of the asterisk) and the normal adjacent mucosa (located to the right of the asterisk). I: Nearby section of the same ACF as seen in G and H (asterisks denote same location in all three sections) demonstrates normal membranous expression of β-catenin in both the ACF and normal crypts. Scale bars: 150 μm (A); 300 μm (B); 100 μm (D, E); 50 μm (H and I).
The next experiments were designed to characterize the ACF that developed in these Apc1638N/+ mice. In the second study, there was a 3.8-fold increase (P < 0.0001; Figure 2 ▶ ) in the number of ACF at 22 to 26 weeks (in seven females, five males) compared to the number at 9 weeks of age (in seven females, six males). In a follow-up study, this increase was 2.8-fold (P < 0.0001; Figure 3 ▶ ) at 28 weeks (in three females, three males) compared with the number at 12 weeks of age (in four females, four males). However, while the number of ACF increases dramatically with age (Figure 4) ▶ , the number of crypts per ACF does not, ie, the number of ACF with three or more crypts per ACF appears similar at 9 weeks and at 22 to 26 weeks of age (Figure 2) ▶ . At 28 weeks of age, the category of ACF with 15 or more crypts per focus included some ACF with more than 20 crypts, eg, with 23, 30, 33, 60, and 100 crypts. At the earlier age of 9 or 12 weeks, the ACF in this category (with 15 or more crypts per focus) in contrast had less than 20 crypts per focus. The distribution of the ACF was not significantly different among the cecum, proximal colon, and distal colon at 9 (data not shown), 12, or 28 weeks of age; but there was a tendency toward fewer ACF in the distal colon and more in the proximal colon and cecum of these Apc1638N/+ mice (Figure 3) ▶ . There were no detectable differences between the ACF seen in male and female Apc1638N/+ mice. No tumors were identified in the colons of any of our Apc1638N/+ mice by 28 weeks of age, the last time point considered.
Figure 2.

The numbers of ACF per Apc1638N/+ mouse with different numbers of crypts per focus at 9 weeks and 22 to 26 weeks of age. Although the number of ACF per mouse with one or two crypts per focus increases with age, the number of ACF with three or more crypts per focus remains similar at the two time periods.
Figure 3.

The distribution of ACF per Apc1638N/+ mouse in the cecum, proximal colon, and distal colon. All mice, starting at 3 weeks of age, were fed the control AIN 76A diet for 9 weeks and then eight mice were killed. The remaining two groups of eight mice were fed either the control AIN 76A diet or a Western-style diet high in fat and low in calcium and vitamin D 22 for an additional 16 weeks. Two mice on the control diet were found dead before 28 weeks of age.
Figure 4.

The total numbers of ACF per Apc1638N/+ mouse that developed spontaneously in mice maintained on control AIN 76A diet and killed at different ages. The numbers of mice per group were 3 for 5 to 6 weeks of age (first study); 13 for 9 weeks (second study); 8 for 12 weeks (last study); 18 for 22 to 26 weeks (6 from first study and 12 from second study); and 6 for 28 weeks (last study).
H&E-stained sections of 22 of the larger ACF, most comprised of more than six crypts per focus, from Apc1638N/+ mice showed crypts that often were dilated, enlarged, and slightly elevated above the adjacent mucosa (Figure 1, F and G) ▶ . Many ACF have nuclear atypia and loss of mucin-containing cells (Figure 1, F and G) ▶ . The six ACF evaluated from Apc1638N/+ mice killed at 9 to 12 weeks of age varied in size from 4 crypts to greater than 15 crypts, and all displayed minor atypia but not dysplasia. Ten of the 16 ACF from Apc1638N/+ mice killed at 22 to 28 weeks of age varied in size from 6 crypts to 30 crypts and also displayed only atypia. The other six ACF from mice killed at 22 to 28 weeks of age with 12 to 100 crypts showed mild to moderate degrees of dysplasia. When sections of these 22 ACF were immunostained with an antibody to the COOH terminal region of the Apc protein, the colonic epithelial cells within all 22 ACF retained their expression of Apc protein similar to that in the adjacent, normal crypts (Figure 1H) ▶ . Included among the ACF evaluated were ACF with 33, 60, and 100 crypts per focus. Seven human colon cancers and two tumors from the small intestines of Apc1638N/+ mice were stained by the same procedure as negative controls; all tumors, as expected, revealed a marked loss or absence of Apc protein expression (Figure 1, D and E) ▶ indicating the antibody did not cross-react with other proteins in these experiments. Adjacent sections of ACF failed to show the same pattern of staining when incubated with antibody that had been preincubated with a 10-fold excess peptide that was used to prepare the antibody (Santa Cruz Biotechnology, Inc.) or with normal rabbit serum. As a further indication that Wnt signaling was not grossly aberrant, all of the ACF immunostained for the demonstration of β-catenin had only normal membranous expression of β-catenin (Figure 1I) ▶ .
Because the ACF appeared to increase in number, but not in pathology or the number of crypts per focus, as a function of time, we tested whether a Western-style diet (high in fat and low in calcium and vitamin D 22 ), would influence ACF progression. Three groups of eight Apc1638N/+ mice were placed on the control purified AIN 76A diet at 3 weeks of age. After 9 weeks on the control diet (12 weeks of age), one group was killed and found to have13.9 ± 6.7 (mean ± SD) ACF per mouse. The second group, kept on the control AIN 76A diet for an additional 16 weeks (28 weeks of age), had 38.5 ± 3.3 ACF per mouse or a 2.8-fold increase over the number seen at 12 weeks of age. The third group of mice, transferred to the Western-style diet 22 at 12 weeks of age and kept on the diet for 16 weeks, had 45.6 ± 19 ACF per mouse (Figure 3) ▶ . The average number of crypts per ACF at 28 weeks of age was not increased significantly by the Western-style diet; it was 1.34 ± 0.06 on the AIN 76A diet and 1.47 ± 0.29 crypts per focus when switched to the Western-style diet. The overall number of tumors in the small intestines of these mice appeared to increase from 1.8 ± 1.3 tumors per mouse on the AIN 76A diet to 2.1 ± 1.4 tumors per mouse on the Western-style, but this was not significant. However, only mice switched to the Western-style diet showed progression of some tumors (3) to carcinomas.
Discussion
We describe the spontaneous appearance and distribution of ACF in the colons of Apc1638N/+ mice that inherit a mutation in the Apc gene. The significant increase in the number of ACF but not in the number of crypts per ACF, from 9 to 28 weeks of age, suggests that the inherited Apc mutation may be more involved in the initiation, than in the progression of ACF. Because the largest ACF with between 20 and 100 crypts per focus were seen in the 28-week-old mice but not in the younger mice, it seems that some ACF progress very slowly. With the rapid increase in the numbers of ACF with age, the large numbers of new, small ACF appear to dilute out the larger ACF that are slowly increasing in size. An earlier study with 24 similar mice, in which only two colonic tumors (adenomas) were found in 12- and 13-month-old mice, 7 affirms the long latency between birth and colon tumors in Apc1638N/+ mice. Histological sections of the ACF from Apc1638N/+ mice demonstrate morphologies that range from near normal or minor atypia to dysplastic with the loss of goblet cells and the loss of mucin production as has been reported for ACF in rodents and humans. 12,25 The importance of mucin in the development of colonic tumors was recently demonstrated with the Muc2−/− mouse. 26
The ACF in Apc1638N/+ mice appear similar to ACF in rodents treated with colon-specific chemical carcinogens 10-12 and humans at risk for the development of colonic tumors, 13,14 except the ACF in Apc1638N/+ mice have a more proximal distribution that includes the cecum. Rodents given carcinogens develop tumors and ACF primarily in the distal colon; 12 ACF are not found more proximally in the cecum of these animals. The Apc1638N/+ mice develop tumors primarily in the small intestine with smaller numbers in the cecum and colon; 5,7 ACF develop in the cecum as well as the colon of these mice. These similar patterns of distribution of ACF and tumors further support the role of ACF in colon tumorigenesis.
The immunohistochemical studies with antibodies to the COOH terminal region of the Apc protein indicate that at least some full-length Apc protein is made by most cells of the ACF. The presence of an intact Apc/β-catenin pathway is further demonstrated by the normal membranous expression of β-catenin and the lack of cytoplasmic or nuclear expression in these ACF. These results suggest that a wild-type allele of Apc is retained by the ACF and that loss of the wild-type copy of the Apc gene is not necessary for the formation of ACF in these mice. A similar expression of wild-type Apc protein was reported for 71% of 58 human adenomas. 27 Together these studies suggest that altered phenotypes (ACF and adenomas) can occur with heterozygous inactivation of the Apc allele and before inactivation of both Apc alleles. However, the loss of the second allele and hence the loss of Apc protein in a small number of cells, which may be sufficient to disrupt the architecture of the crypt, 28 cannot be ruled out. It has been reported that small decreases in expression of the APC gene, and not only complete loss, can be sufficient to influence tumor development. 29
It is interesting that both the ApcMin/+ and ApcΔ716 mice, which also inherit a mutant Apc allele, rarely exhibit classical ACF. 17-21 It is possible that the more rapid and extensive tumor development in the ApcMin/+ and ApcΔ716 mouse models obscures the appearance of ACF as a distinct stage in the progression of tumorigenesis. However, the ApcMin/+ mouse also has been compared directly to the ApcMin/+ mouse in which the Msh2 gene, involved in DNA mismatch repair, has been homozygously inactivated. In the ApcMin/+Msh2−/− mice, tumorigenesis is even more rapid, yet ACF become abundant. 19 Also, although the Apc1638N mutation is a chain-terminating mutation similar to that found in ApcΔ716 mice and human tumors, a truncated protein is not observed in the Apc1638N/+ mice. 5 Therefore, it appears that the Apc1638N/+ mouse expresses a distinctly different phenotype than the ApcMin/+ and ApcΔ716 mice in the premalignant flat mucosa. Although Sorenson and colleagues 30 reported the spontaneous development of ACF in 37-week-old, control, untreated Apc1638N/+ mice; they did not indicate if ACF occurred earlier or whether they were present in the cecum where a large proportion were observed in this study.
The Western-style diet 22 did not significantly promote the progression of ACF and tumors in either the small or large intestine of these mice, although carcinomas were only seen in mice switched to the Western-style diet. This is likely because of the small number of animals, six in the control group and eight in the group on the Western-style diet, and the large variations in the number of ACF and/or tumors per mouse seen in each group killed at 28 weeks of age. Also, greater differences may have been observed if the animals had been kept on the different diets longer and/or if the mice had been allowed to live longer. In studies by Yang and colleagues 31,32 with similar mice, differences in the number of tumors and other pathological lesions were observed between the two diets; but that study used many more mice per dietary group and maintained the mice for a much longer period. In future studies of dietary or other environmental factors with theApc1638N/+ mouse, it will be important to evaluate ACF and tumors in the large intestine as well as tumors in the small intestine. These two areas of the gastrointestinal tract differ in their environment and function; tumorigenesis in these two areas may respond differently to environmental changes.
In summary, ACF develop spontaneously in the Apc1638N/+ mouse as early as 5 weeks of age and do not appear to require the loss of the wild-type Apc allele. Those data and the increase in the number of ACF, but not the number of crypts per ACF, with age suggest that the inheritance of a single mutant Apc1638N allele is an initiating but not a promoting event of colon tumorigenesis in these mice. These studies provide further evidence of the suitability of the Apc1638N/+ mouse as a model to study the in vivo interaction of environmental factors with the inheritance of a mutant Apc allele.
Acknowledgments
We thank Leon Hudson, Heather Pilch, and Karen Stiffler for technical assistance.
Footnotes
Address reprint requests to Theresa P. Pretlow, Ph.D., Institute of Pathology, CWRU, 2085 Adelbert Rd., Cleveland, OH 44106. E-mail: tpp3@po.cwru.edu.
Supported in part by the Public Health Service (grants CA66725, CA87559, CA57179, P30-43703, and P30-13330 from the National Cancer Institute), the American Institute for Cancer Research (grant 95B025), and the American Cancer Society (RPG-95-022-03-CN).
References
- 1.Kinzler KW, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 2.Ilyas M, Straub J, Tomlinson IPM, Bodmer WF: Genetic pathways in colorectal and other cancers. Eur J Cancer 1999, 35:335-351 [DOI] [PubMed] [Google Scholar]
- 3.Moser AR, Pitot HC, Dove WF: A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247:322-324 [DOI] [PubMed] [Google Scholar]
- 4.Su L-K, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF: Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256:668-670 [DOI] [PubMed] [Google Scholar]
- 5.Fodde R, Edelmann W, Yang K, Van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM, Kucherlapati R: A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci USA 1994, 91:8969-8973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M: Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA 1995, 92:4482-4486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang K, Edelmann W, Fan K, Lau K, Kolli VR, Fodde R, Khan PM, Kucherlapati R, Lipkin M: A mouse model of human familial adenomatous polyposis. J Exp Zool 1997, 277:245-254 [PubMed] [Google Scholar]
- 8.Smits R, van der Houven van Oordt W, Luz A, Zurcher C, Jagmohan-Changur S, Breukel C, Khan PM, Fodde R: Apc1638N: a mouse model for familial adenomatous polyposis-associated desmoid tumors and cutaneous cysts. Gastroenterology 1998, 114:275-283 [DOI] [PubMed] [Google Scholar]
- 9.Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF, Pitot HC, Halberg RB, Itzkowitz SH, Groden J, Coffey RJ: Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 2003, 124:762-777 [DOI] [PubMed] [Google Scholar]
- 10.Bird RP: Observation and quantification of aberrant crypts in the murine colon treated with a colon carcinogen: preliminary findings. Cancer Lett 1987, 37:147-151 [DOI] [PubMed] [Google Scholar]
- 11.Pretlow TP, O’Riordan MA, Kolman MF, Jurcisek JA: Colonic aberrant crypts in azoxymethane-treated F344 rats have decreased hexosaminidase activity. Am J Pathol 1990, 136:13-16 [PMC free article] [PubMed] [Google Scholar]
- 12.Pretlow TP, Siddiki B, Augenlicht LH, Pretlow TG, Kim YS: Aberrant crypt foci (ACF)—earliest recognized players or innocent bystanders in colon carcinogenesis. Schmiegel W Schölmerich J eds. Colorectal Cancer: Molecular Mechanisms, Premalignant State and Its Prevention, Falk Symposium No. 109 1999:pp 67-82 Kluwer Academic Publishers Lancaster
- 13.Pretlow TP, Barrow BJ, Ashton WS, O’Riordan MA, Pretlow TG, Jurcisek JA, Stellato TA: Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res 1991, 51:1564-1567 [PubMed] [Google Scholar]
- 14.Roncucci L, Stamp D, Medline A, Cullen JB, Bruce WR: Identification and quantification of aberrant crypt foci and microadenomas in the human colon. Hum Pathol 1991, 22:287-294 [DOI] [PubMed] [Google Scholar]
- 15.Takayama T, Katsuki S, Takahashi Y, Ohi M, Nojiri S, Sakamaki S, Kato J, Kogawa K, Miyake H, Niitsu Y: Aberrant crypt foci of the colon as precursors of adenoma and cancer. N Engl J Med 1998, 339:1277-1284 [DOI] [PubMed] [Google Scholar]
- 16.Siu I-M, Robinson DR, Schwartz S, Kung H-J, Pretlow TG, Petersen RB, Pretlow TP: The identification of monoclonality in human aberrant crypt foci. Cancer Res 1999, 59:63-66 [PubMed] [Google Scholar]
- 17.Jacoby RF, Marshall DJ, Newton MA, Novakovic K, Tutsch K, Cole CE, Lubert RA, Kelloff GJ, Verma A, Moser AR, Dove WF: Chemoprevention of spontaneous intestinal adenomas in the ApcMin mouse model by the nonsteroidal anti-inflammatory drug piroxicam. Cancer Res 1996, 56:710-714 [PubMed] [Google Scholar]
- 18.Paulsen JE, Namork E, Steffensen IL, Eide TJ, Alexander J: Identification and quantification of aberrant crypt foci in the colon of Min mice—a murine model of familial adenomatous polyposis. Scand J Gastroenterol 2000, 35:534-539 [DOI] [PubMed] [Google Scholar]
- 19.Reitmair AH, Cai J-C, Bjerknes M, Redston M, Cheng H, Pind MTL, Hay K, Mitri A, Bapat BV, Mak TW, Gallinger S: MSH2 deficiency contributes to accelerated APC-mediated intestinal tumorigenesis. Cancer Res 1996, 56:2922-2926 [PubMed] [Google Scholar]
- 20.Steffensen IL, Paulsen JE, Eide TJ, Alexander J: 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine increases the numbers of tumors, cystic crypts and aberrant crypt foci in multiple intestinal neoplasia mice. Carcinogenesis 1997, 18:1049-1054 [DOI] [PubMed] [Google Scholar]
- 21.Oshima H, Oshima M, Kobayashi M, Tsutsumi M, Taketo MM: Morphological and molecular processes of polyp formation in Apc Δ716 knockout mice. Cancer Res 1997, 57:1644-1649 [PubMed] [Google Scholar]
- 22.Newmark HL, Lipkin M, Maheshwari N: Colonic hyperplasia and hyperproliferation induced by a nutritional stress diet with four components of Western-style diet. J Natl Cancer Inst 1990, 82:491-496 [DOI] [PubMed] [Google Scholar]
- 23.Hao XP, Pretlow TG, Rao JS, Pretlow TP: Inducible nitric oxide synthase (iNOS) is expressed similarly in multiple aberrant crypt foci and colorectal tumors from the same patients. Cancer Res 2001, 61:419-422 [PubMed] [Google Scholar]
- 24.Hao XP, Pretlow TG, Rao JS, Pretlow TP: β-catenin expression is altered in human colonic aberrant crypt foci. Cancer Res 2001, 61:8085-8088 [PubMed] [Google Scholar]
- 25.Siu I-M, Pretlow TG, Amini SB, Pretlow TP: Identification of dysplasia in human colonic aberrant crypt foci. Am J Pathol 1997, 150:1805-1813 [PMC free article] [PubMed] [Google Scholar]
- 26.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, Kucherlapati R, Lipkin M, Yang K, Augenlicht L: Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 2002, 295:1726-1729 [DOI] [PubMed] [Google Scholar]
- 27.Iwamoto M, Ahnen DJ, Franklin WA, Maltzman TH: Expression of β-catenin and full-length APC protein in normal and neoplastic colonic tissues. Carcinogenesis 2000, 21:1935-1940 [DOI] [PubMed] [Google Scholar]
- 28.Augenlicht L: Adhesion molecules, cellular differentiation, and colonic crypt architecture. Gastroenterology 1994, 107:1894-1898 [DOI] [PubMed] [Google Scholar]
- 29.Yan H, Dobbie Z, Gruber SB, Markowitz S, Romans K, Giardiello FM, Kinzler KW, Vogelstein B: Small changes in expression affect predisposition to tumorigenesis. Nat Genet 2002, 30:25-26 [DOI] [PubMed] [Google Scholar]
- 30.Sorensen IK, Kristiansen E, Mortensen A, van Kranen H, van Kreijl C, Fodde R, Thorgeirsson SS: Short-term carcinogenicity testing of a potent murine intestinal mutagen, 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine (PhIP), in Apc 1638N transgenic mice. Carcinogenesis 1997, 18:777-781 [DOI] [PubMed] [Google Scholar]
- 31.Yang K, Edelmann W, Fan K, Lau K, Leung D, Newmark H, Kucherlapati R, Lipkin M: Dietary modulation of carcinoma development in a mouse model for human familial adenomatous polyposis. Cancer Res 1998, 58:5713-5717 [PubMed] [Google Scholar]
- 32.Yang WC, Mathew J, Velcich A, Edelmann W, Kucherlapati R, Lipkin M, Yang K, Augenlicht LH: Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res 2001, 61:565-569 [PubMed] [Google Scholar]