

Abstract
The endothelial surface of atherosclerotic lesions of ApoE knockout mice was interrogated by in vivo biopanning with a phage-displayed constrained peptidyl library. Through repeated biopanning, 103 peptidyl sequences were identified, many are homologous to known proteins. The sequence CAPGPSKSC contains motifs that are shared by 9.7% of selected peptides. On phage or as a synthetic peptide, this constrained peptide selectively bound to atherosclerotic lesion surfaces of ApoE knockout mice in vivo and of human atherosclerotic lesions ex vivo. A cell-surface protein of ∼82 kd recognized by this peptide was affinity-purified and determined by mass spectrometry analysis as glucose-regulated protein 78 (Grp78), indicating the surprising presence of this endoplasmic reticulum chaperone on the endothelial cell surface of atherosclerotic lesions. Peptides that mimicked binding functions of their homologues were demonstrated with three peptides homologous to tissue inhibitor of metalloproteinase-2 (TIMP-2), ie, CNHRYMQMC, CNQRHQMSC, and CNNRSDGMC. Phage carrying CNHRYMQMC bound to atherosclerotic lesion endothelium of ApoE knockout mice in vivo. The three peptides bound to endothelial cells in a dose-dependent manner and were inhibited by TIMP-2 protein. These peptides provide a set of probes to interrogate the cell surface repertoire associated with atherogenesis and thrombotic complications.
Atherosclerosis is a major contributor to the pathogenesis of myocardial and cerebral artery thrombosis as well as to unstable angina. Desquamation of the endothelium after injury was once considered a contributing mechanism in atherogenesis; whereas, it is now accepted that atheroma form in the absence of endothelial cell detachment. 1-3 Whereas the endothelium is physiologically nonadherent for leukocytes, modifications of gene expression patterns appear to contribute to leukocyte localization and transmigration into forming atherogenic lesions. The local endothelium may oxidize or otherwise modify lipoproteins as they translocate into the intima where the accumulation of oxidized low-density lipoprotein (LDL) contributes to the local atherogenic process.
Molecular changes on the endothelial surface are considered important contributors to the initiation, progression, and thrombotic complications of atherosclerosis. 4-6 However, the profile of the endothelial surface protein display during atherogenesis is quite limited. Although adhesive cell-surface glycoproteins, such as selectin 7 and vascular cell adhesion molecule-1 (VCAM-1) 8 appear to play a role in atherogenesis, systematic exploration of the affected endothelial surfaces has been hampered by limitations in technology appropriate to the task. This challenge has been advanced by methods such as random combinatorial search technologies. We here have used in vivo selection from constrained peptidyl libraries for identification of peptide probes that selectively bind to atherosclerotic lesions induced in ApoE knockout mice. The cellular events in this model are thought to mimic, at least in part, those of others in the hypercholesterolemic animal models and perhaps the human disease. 9,10 This in vivo biopanning of atherosclerotic lesion endothelium has used a phage-displayed constrained peptidyl library and is similar to technology pioneered by Pasqualini and Ruoslahti. 11 Selected peptides identified from the lesion-associated phage were analyzed for homing to atherosclerotic lesions in vivo and binding to endothelial cells ex vivo. Grp78 was identified as the endothelial surface target of one selected peptide. In another example, three selected peptides homologous to TIMP-2 recapitulated the binding to lesions and were in turn inhibited by TIMP-2 protein.
Materials and Methods
Reagents
The PHD phage-displayed constrained peptide library was from New England Biolabs (Beverly, MA). TIMP-2 protein was from Roche (Indianapolis, IN). Human umbilical vein endothelial cells and EGM (endothelial growth medium) were from Clonetics, Walkersville, MD. Cells were cultivated in EGM with 5% fetal bovine serum in flasks coated with 0.2% gelatin. The bEND.3 murine endothelial cells (American Type Culture Collection, Rockville, MD) were cultured in Dulbecco’s minimum essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and 2 mmol/L of l-glutamine.
Animals
The parental stocks of C57BL/6J ApoE gene-inactivated (knockout) mice were obtained from Jackson Laboratories, Bar Harbor, ME. These and wild-type C57BL/6J mice were bred in the rodent breeding facility of The Scripps Research Institute and fed ad libitum standard chow diet (no. 5015; Harlan Tekland, Madison, WI). To produce the atherosclerotic ApoE model the mice were fed atherogenic diet no. TD 88051 containing 15.8% (w/w) fat, 1.25% (w/w) cholesterol, and 0.5% (w/w) sodium cholate. All studies were reviewed and approved by the Institutional Animal Care and Use Committee and conducted in the Institutional facilities accredited by Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC), with an assurance from the Public Health Service, registered with the United States Department of Agriculture and in compliance with regulations.
In Vivo Panning of Phage-Displayed Peptide Library
In vivo biopanning was performed in atherosclerotic ApoE knockout mice after 12 weeks on the high-fat diet. All animals had grossly visible atherosclerotic lesions of the aortic arch and the branch point of renal arteries. Thirty minutes before injection of the phage library, 1012 pfu of irradiated helper phage were infused via the tail vein and allowed to circulate for 30 minutes to block nonspecific phage binding and saturate the reticuloendothelial system. Then, 1011 pfu of viable peptide library phage were injected and allowed to circulate for 30 minutes. The mouse was perfused via the heart at arterial pressure with physiological saline. The aorta was removed, washed, and opened to expose lesions at the arch and the branch point of renal arteries. Lesions were dissected free and bound phage were eluted with elution buffer (0.1 mol/L glycine, pH 2.2) and neutralized with 0.1 vol of Tris buffer (2 mol/L Tris-HCl, pH 8). The eluted phages were amplified by Escherichia coli in medium on plates, and titered. Basic phage protocols used are from Phage Display of Peptides and Proteins, A Laboratory Manual (Brian K. Kay, Jill Winter, and John McCafferty, Academic Press). Up to 104 phage were recovered from the first round of selection. Three additional rounds of selection were performed and the phage recovered from the fourth round were plated and individual clones were selected for nucleotide sequence analysis.
Analysis of Peptide Sequences
The peptide sequences were analyzed with ClustalW 12 software from the European Molecular Biology Laboratory to identify amino acid motifs that are shared among multiple peptides. The peptide sequences were also searched by online databases using the BLAST program accessible at National Center for Biotechnology Information (http://www.ncbi.nim.nih.gov/BLAST). Many of the peptide sequences appeared in known proteins.
Statistical Analysis
The distribution of the account of success X in n independent trials with probability P of success of each trial is called the binomial distribution B (n, p), and can be approximated with equation: P (X = k) = {n!/k! (n − k)!} pk (1 − p)n−k. Where X is the number of times a given peptide motif occurred in n independent sequencing trials, p is the probability of this given motif occur on each trial.
Peptides
Peptides ACAPGPSKSCGGSYK−biotin, ACNHRYMQMCGGSYK−biotin, ACNQRHQMSCGGSYK−biotin, and ACVNRSDGMCGGSYK−biotin were synthesized by the Scripps Peptide Core facility using Fmoc chemistry. A N-terminal alanine (underlined) that is encoded by the phage is uniformly added to the N-terminus of the peptides. An extension of GGSYK was added to the C-terminus of the synthetic peptides (underlined). The tyrosine permitted iodination and the lysine was added for biotinylation. Biotin was attached through the side chain amino group of the C-terminal while the peptides were attached to the beads. The peptides were deblocked, cleaved, and HPLC-purified. The purified peptides were characterized by mass spectrometry. The molecular weight of the peptides exactly matched the predicted mass.
The role of intact disulfide constraints was assessed by reduction and reoxidization of the peptides. Formation of intermolecular disulfide bond in Cys-containing peptide. To peptide at 2.5 mg/ml, 2-mercaptoethenol was added to 0.05 mol/L and held for 4 hours at room temperature. The solution was then lyophilized to remove 2-mercaptoethenol. The reduced peptides were analyzed and divided into two aliquots, one of which was air-oxidized at a concentration of 0.1 mmol/L in folding buffer (2 mol/L GuHCl, 0.2 mol/L Tris-HCl, pH 8.5) while being stirred vigorously with air overnight. The progress of intermolecular disulfide formation was monitored by the Ellman’s reaction. The reoxidized and folded peptides and the reduced control peptides were dialyzed against phosphate-buffered saline (PBS). For in vivo experiments the peptides were dialyzed against sterile physiological saline and sterile-filtered.
Immunohistochemical Analysis
Immunohistochemical analysis for binding was performed on frozen 5-μm sections of ApoE mouse atherosclerotic aortic valve and aorta on poly-l-lysine-coated slides. Biotinylated peptide binding to aortic lesions was detected by fluorescein isothiocyanate (FITC)-conjugated streptavidin (Vector, Burlingame, CA). For endothelial identification, biotinylated mouse CD-31-specific rat monoclonal antibody (Pharmingen, La Jolla, CA) was used, and was detected with either FITC- or Texas red-conjugated streptavidin. Phage staining was performed with rabbit anti-phage antibody followed by a fluorescein-conjugated goat anti-rabbit antibody. The sections of the aorta were analyzed with fluorescent microscopy, and sections of the aortic valves were analyzed using laser-scanning confocal microscopy.
En Face Analysis of Atherosclerotic Lesions
To analyze phage and peptide association with atherosclerotic lesions of the entire ApoE mouse aorta, the targeting reagent, phage carrying a homing peptide sequence or the synthetic peptide, was injected via tail vein. After 30 minutes of circulation, the mice were perfused with sterile physiological saline through a cannula in the left ventricle, and the perfusate was drained from the inferior vena cava, then the vasculature was perfusion-fixed with 4% paraformaldehyde. The aortas were dissected free of other tissues, longitudinally opened, and pinned flat. Biotinylated anti-phage (Sigma, St. Louis, MO) antibody, at 1:500 dilution, was applied for 60 minutes, washed, and was followed by 1:500 dilution of streptavidin-peroxidase (Vector). After three washes with phosphate-buffered saline (PBS) the reaction was developed with diaminobenzidine (DAB) substrate. For human tissue, the affected vessel tissues were dissected and pinned flat and then incubated with biotinylated peptides (10 μg/ml) overnight at 4°C. The tissues were washed three times and streptavidin-peroxidase conjugate was applied for 30 minutes, washed, DAB substrate was added, and reactions were visualized by the colored DAB product (Vector).
Peptide Ligand Blot
Membrane fractions of the murine endothelial cell line (bEND.3) were prepared by Triton X-114 extraction. 12 Cell pellets (5 × 106 cells) were lysed in 1 ml of buffer (1% Triton X-114, 0.1 mol/L Tris, 10 mmol/L EDTA, 2000 U/ml aprotinin, 100 μmol/L phenylmethyl sulfonyl fluoride) on ice for 15 minutes. After centrifugation at 16,110 × g in an Eppendorf centrifuge at 4°C, the supernatant was incubated for 5 minutes at 37°C for phase separation and centrifuged for 2 minutes at room temperature. The lower membrane phase was recovered and re-extracted with 1% Triton X-114 solution. The membrane phase was precipitated with acetone at −20°C and centrifuged for 10 minutes at 16,110 × g The membrane pellet was dried and resuspended in sodium dodecyl sulfate (SDS) sample buffer for SDS-polyacrylamide gel electrophoresis (PAGE) analysis using gradient Tris-glycine gel (8 to 16%). After electrophoresis, the proteins were transferred from gel to nitrocellulose membranes, and blocked with nonfat milk. The biotinylated peptide at 10 μg/ml was used as probe and was incubated with membrane overnight. The blot was washed three times with PBS, incubated with streptavidin-peroxidase for 15 minutes, and developed with DAB substrate (Vector).
Peptide-Binding Assay
Analysis of synthetic peptide binding to cells was performed in 96-well plates. Cells (105) were plated into each well, incubated overnight, and then briefly fixed with cold ethanol for 10 seconds. Serial concentrations of peptide in 100 μl of PBS were added and incubated at room temperature with gentle agitation for 4 hours. The wells were washed three times with PBS and the reaction developed using streptavidin-peroxidase substrate (Vector). The reactions were quantitated at 405 nm in a plate reader (Molecular Devices, Sunnyvale, CA). In the inhibition assay, TIMP-2 protein was included with the peptide in the initial incubation.
Membrane Solubilization
Enriched membrane fractions of bEND.3 cells were prepared by sequential extraction with Triton X-100 (TX-100) and Triton X-114 (TX-114). Cell pellets (1 × 107 cells) were lysed in 1 ml of buffer (0.1% TX-100 + 1:50 Complete protease inhibitor cocktail; Roche, Mannheim, Germany) for 1 hour at room temperature. After centrifugation at 16,000 × g for 10 minutes at room temperature, the supernatant was precipitated with 90% acetone and the precipitate extracted with TX-114 (1% TX-114, 0.1 mol/L Tris, 10 mm EDTA, 1:50 protease inhibitor cocktail) for 30 minutes at 4°C. After centrifugation at 16,000 × g, 10 minutes, 4°C, the supernatant was incubated at 37°C for 5 minutes and centrifuged at 16,000 × g, 10 minutes, 37°C for phase separation. The supernatant phase was precipitated with acetone and combined with the acetone-washed membrane phase. The pooled precipitates were then resuspended in 1× Dulbecco’s PBS and purified by affinity chromatography.
Affinity Chromatography
ImmunoPure Monomeric Avidin kit was obtained from Pierce (Rockford, IL). ACAPGPSKSCGGSYK−biotin was coupled to the column according to the manufacturer’s protocol. The solubilized membrane fraction (described above) was passed over the column and protein bound to the peptide was eluted with excess biotin. The eluate was electrophoresed under reducing conditions on SDS-PAGE gels and proteins were visualized with Coomassie Brilliant Blue (Sigma, St. Louis, MO). The affinity-purified protein bands were excised and submitted to the Scripps Center for Mass Spectrometry (La Jolla, CA) for analysis.
Mass Spectrometry
Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry was performed on the trypsin-digested samples using a PerSeptive Biosystems Voyager-STR mass spectrometer with delayed extraction. Samples were irradiated by nitrogen laser (Laser Science Inc.) operated at 337 nm and with a variable attenuator with focus on the sample target. Ions produced were accelerated with a deflection voltage of 30,000 V, and were differentiated according to m/z using a time-of-flight reflectron mass analyzer. Protein was identified from the set of peptide masses using the Mascot search engine and databases.
Western Blot
Cell membrane preparations and controls were subjected to SDS-PAGE using 8 to 16% Tris-glycine gels (Invitrogen, Carlsbad, CA). The proteins were transferred onto polyvinylidene difluoride membranes and blocked with 20 mmol/L Tris, 140 mmol/L NaCl, 0.1% Tween 20, 10% milk (TBS-T/milk). The probes were goat polyclonal antibodies against the N-terminal and C-terminal Grp78 (Santa Cruz Biotechnology, Santa Cruz, CA) and were incubated with target for 1 hour at room temperature. The blots were washed three times with TBS-T and incubated with horseradish peroxidase-conjugated bovine anti-goat IgG (Santa Cruz Biotechnology) for 1 hour at room temperature. The blots were washed three times with TBS-T (20 mmol/L Tris, 140 mmol/L NaCl, 0.1% Tween 20) and developed with ECL reagent (Sigma).
Immunoprecipitation
TX-100/TX-114-enriched bEND.3 membrane extracts were incubated with 4 μg of N-terminal Grp78 antibody, C-terminal Grp78 antibody, or a control antibody for 2 hours at 4°C followed by 1 hour of incubation with protein G-Sepharose beads. The supernatant was removed and beads were then centrifuged for 2 minutes at 2000 rpm, 4°C, and washed three times with the final TX-114 extraction buffer (1% TX-114, 0.1 mol/L Tris, 10 mm EDTA, 1:50 protease inhibitor cocktail) and once with PBS. The precipitated proteins were then analyzed by ligand blot using biotinylated CAPGPSKSC as described above.
Flow Cytometry
Cells were washed with PBS and detached with Enzyme-Free Cell Dissociation Buffer (Life Technologies, Gaithersburg, MD). All subsequent steps were performed on ice. Cells were then pelleted in serum-free growth medium, washed, and resuspended in PBS containing 1:100 dilution of primary antibody for 30 minutes. Cells were then washed and resuspended in PBS containing 1:100 dilution of FITC-labeled secondary antibody for an additional 30 minutes. Unbound secondary was removed by additional washing and cells were analyzed on a FACScan flow cytometer (Becton Dickinson, Mountain View, CA).
Results
In Vivo Phage Library Interrogation
Four rounds of progressive in vivo panning and selection of the atheroma-bound members of the phage-displayed peptidyl library was performed in which recovery at each round was only from the atherosclerotic lesions. There were 103 individual plaque surface-binding peptidyl sequences identified. They were analyzed with ClustalW software to identify amino acid motifs that are shared by different peptides. The peptidyl sequence data set was prioritized according to frequency of occurrence that is 1) exact, 2) similar, 3) homologous to known proteins. The peptides with greater than 50% homology were aligned and the shared motifs were searched in the online National Center for Biotechnology Information database using BLAST. The unmatched seven amino acid peptide sequences were also searched. Identified common peptide motifs enriched through repeated biopanning are given in Table 1 ▶ . The probability of these shared motifs occurring as a random event is exceedingly small. Some sequences bore significant homology to sequences of known proteins as exemplified by three peptides in Table 2 ▶ with close similarity to tissue inhibitor of metalloproteinase-2 (TIMP-2). The considerable number of peptidyl sequences found to associate with plaque surfaces suggests differential expression of multiple membrane targets on these endothelial cells, indicative of a positional gene expression or molecular processing different from endothelium elsewhere in the vascular tree.
Table 1.
Examples of Identified Shared Peptide Motifs and Homologous Proteins
| Peptide motifs and their homologous protein sequence | Frequency* | P2† | Protein containing the motif | Protein description | Accession number |
|---|---|---|---|---|---|
| 46 GPSKSCG51 | 9.7% | <0.00000001 | Chymase I | Serine protease, convertase | AB00823 |
| CAPGPSKSCG‡ | for angiotensin II generation | ||||
| CTPYPSKSCG | |||||
| CSQYPSRSCG | |||||
| CLMTPSKRCG | |||||
| 140 CEEGWRLKCG150 | 4.9% | <0.000001 | Novel protein | Interferon-induced protein with | BC003804 |
| CQEPTRLKCG | tetratricopeptide repeats | ||||
| CKEPTRAHCG | |||||
| 40 PPPGTRH46 | 3.6% | <0.0001 | Mouse-expressed | Unknown | AA960387 |
| CPHPGTRHC§ | protein | ||||
| CTNTGHRHC | |||||
| 499 FFSRSC504 | 3.5% | <0.00001 | Ricken mouse | Unknown | AAH21390 |
| CEHFFSRSC¶ | cDNA | ||||
| CPGKISRSC | 1300017J01 gene | ||||
| CSQYPSRSC | |||||
| 85 LNNSQAH91 | 2.6% | <0.0001 | Hypothetical protein | Unknown | XP_091658 |
| CLNNSQAHC∥ | |||||
| CPNNKSASC | |||||
| 139 ACLKMRLE146 | 2.6% | ND** | Sec23 homology | Homologous to yeast-secreted | I60247 |
| ACTKMRLEC | protein 23 | ||||
| 321 ACMNQ325 | 2.6% | <0.0001 | AMP deaminase 3 | Generating inosine | NM_009667 |
| ACMNQRVQNC | monophosphate, regulate | ||||
| ACMNQTPDLC | neutrophil activity | ||||
| ACTNQFLQQC | |||||
| 145 NQRH-QMSC152 | 1.8% | ND | TIMP-2 | Tissue inhibitor of | 042146 |
| NQRH-QMSC | metalloproteinase 2 | ||||
| NHRYMQM-C |
*Frequency represents the prevalence of each peptide motif with >55% identity in the peptide data set as calculated based on Clustal W analysis.
†P value is the probability of the shared peptide motif occurring as a random event.
‡Four peptides with this sequence were selected.
§Three peptides with this sequence were selected.
para; and
∥, Two peptides with this sequence were selected.
**Not done.
Table 2.
Examples of Candidate Proteins Mimicked by Multiple Selected Peptides
| Tissue metalloproteinase inhibitor 2 homologous peptides |
| 193 CIKRSDGSC201 |
| CNNRSDGMC |
| 145 NQRYQMGC152 |
| NQRHQMSC |
| 145 NQRY-QMGC152 |
| NHRYMQM-C |
| Tissue metalloproteinase inhibitor 1 homologous peptides |
| 160 CKLQSGTHC168 |
| CKLKSGSLC |
| 197 CTWQSLRS203 |
| CDWQTQRS |
| Megalin (low-density lipoprotein receptor related protein-2) homologous peptides |
| 3116 CLDSSISRC3124 |
| CLDSKVSRC |
| 3964 CNLGDNRTC3972 |
| CLLGEPRTC |
CAPGPSKSC Phage Bound Selectively to Atherosclerotic Lesions
Phage bearing the CAPGPSKSC peptide, the sequence with shared motif and the highest repeat frequency, was analyzed further for in vivo homing to atherosclerotic lesions. Phage carrying this sequence showed preferential binding to atherosclerotic lesions compared to the control wild-type phage (Figure 1A) ▶ . When the same number of control wild-type phage lacking this peptidyl insert were injected into ApoE knockout mice with lesions, there was no demonstrable binding of phage to the atherosclerotic lesions (Figure 1B) ▶ . Phage binding was not detectable on aortic endothelium when this phage was injected in normal BALB/c mice (Figure 1C) ▶ , nor was phage binding evident in young ApoE knockout mice on a normal chow diet and with only slight preatherosclerotic streaking (Figure 1D) ▶ . These observations support the role of the fused peptidyl element CAPGPSKSC in mediation of binding and recognition of a target molecule that is expressed by plaque endothelium during atherogenesis in this model. Further, there is a differential pattern observed in which the peptide appears to bind more to the endothelium at the periphery of the lesions and less to the central aspect of lesions. This suggests that the endothelium at the periphery expresses the target at greater density and that these areas of lesion formation differ to some degree in the phenotype of the overlying endothelium.
Figure 1.

Aortas from atherosclerotic ApoE mice injected with 1011 phage either expressing CAPGPDSKSC or not (control phage). Binding of phage to aorta was visualized with biotinylated anti-phage antibody and streptavidin-linked peroxidase activation of DAB. A: The aorta of an atherosclerotic ApoE knockout mouse injected with CAPGPSKSC phage. Phage binding to the lesions is readily visualized (red-brown). B: The aorta from an atherosclerotic ApoE knockout mouse in which lesions are clearly evident. This mouse was infused with control phage and there is no evident association of phage with the lesions. C: The normal aorta from a BALB/c mouse infused with the CAPGPSKSC phage. No association of phage with the aortic surface is observed suggesting the CAPGPSKSC target molecule is absent or not present in detectable quantity. D: The aorta of a young ApoE knockout mouse on standard chow diet and without atherosclerotic lesions when infused with CAPGPSKSC phage, no localization was detectable indicating that the CAPGPSKSC target molecule is not expressed simply as a result of ApoE gene inactivation.
In Vivo Localization of CAPGPSKSC Peptide
To verify that the peptidyl sequence CAPGPSKSC expressed on phage surface protein III alone was responsible for the selective localization to atherosclerotic lesions, a biotinylated peptide containing the core sequence CAPGPSKSC was synthesized, folded, disulfide bonds constituted by oxidization, and compositional validity established by mass spectrometry. The peptide is predicted to be structurally constrained by computational analysis using molecular dynamics (Insight II, Discover) and energy minimization, and also based on the notable presence of two prolines that are highly constraining. The peptide was injected into atherosclerotic ApoE knockout mice via tail vein, and the tissues were fixed by in vivo perfusion and aortas were freed by dissection. The binding of biotinylated peptide to the aorta was visualized with streptavidin-conjugated peroxidase and DAB substrate. The peptide binding replicated the CAPGPSKSC phage binding to the lesions (Figure 2A) ▶ , supporting that the association of CAPGPSKSC phage with atherosclerotic lesions was mediated by the fused peptidyl sequence. Immunohistochemical staining using an anti-CD31 antibody indicate the endothelial lining is primarily intact on lesion surfaces (Figure 2, B and D) ▶ . The association of this peptide to the atherosclerotic lesions was also analyzed on tissue sections. The experiment was replicated in atherosclerotic ApoE mice by infusing biotinylated CAPGPSKSC peptide and sections of the aorta were prepared and detected with streptavidin-conjugated FITC. Peptide binding to the surface of atherosclerotic lesions (Figure 2C) ▶ was quite evident in contrast with the lack of any evident signal elsewhere (Figure 2C) ▶ .
Figure 2.

A: An aorta from an atherosclerotic ApoE knockout mouse and infused with the synthetic biotinylated CAPGPSKSC peptide. Peptide binding to the lesions is visualized (red-brown) and replicates the phage binding. B: A histological section of a typical atherosclerotic lesion of the aorta of an atherosclerotic ApoE knockout mouse. Endothelium overlying the atherosclerotic lesion was identified with anti-CD31 antibody and labeled dark brown (arrows). C: An adjacent section. The binding of this peptide is most intense to the endothelium overlying the atherosclerotic lesion (yellow arrows), but negative on endothelial cells not directly overlying lesions (white arrows). D: A positive control with all endothelial cells recognized by an anti-CD31 antibody. E: A negative control and autofluorescence resulted from elastic tissue in the vessel wall. Original magnifications, ×200.
Peptide CAPGPSKSC Binding to Human Atherosclerotic Lesions
The biotinylated peptide exhibited similar preferential binding ex vivo to resected human arterial atherosclerotic lesions (Figure 3) ▶ consistent with a similar localized expression of the CAPGPSKSC molecular target by human endothelium.
Figure 3.

Image of an atherosclerotic lesion of a human iliac artery segment. The tissue before staining is shown at the left. Binding of biotinylated CAPGPSKSC peptide to the lesion is demonstrated after application to the lumenal surface of the vessel (right). The binding of phage to the lesion was visualized with enzyme-linked streptavidin conversion of DAB substrate to the brownish red product. The association of CAPGPSKSC peptide to the lesion, but not to nonatherosclerotic surface of this and other vessels (not shown), indicates that a human homologue of the target molecule is present. Lesions are outlined.
The Putative Target for CAPGPSKSC
Peptide CAPGPSKSC bound two protein bands, an ∼82-kd band and an ∼120-kd band, in a ligand blot of the lysate of the mouse endothelial cell bEND.3. The ∼82-kd protein was enriched relative to the ∼120-kd protein in the Triton X-114 extracted membrane fraction (Figure 4) ▶ and represents the candidate target for CAPGPSKSC. The size of this protein excludes known adhesion molecules enriched in atherosclerotic lesions, such as E-selectin and VCAM.
Figure 4.

Identification of a target molecule in mouse endothelial cells (bEND.3). The total protein lysate (lane 1) or membrane preparation (lane 2) was separated by SDS-polyacrylamide gel electrophoresis then transferred to nitrocellulose membrane. The latter was probed with biotinylated peptide CAPGPSKSC. Two sharp bands are observed on a Western blot of a whole cell lysate of mouse endothelial cell line (bEND.3). The protein bands bound by the CAPGPSKSC peptide were ∼82 kd and ∼120 kd. The sharpness of the detected bands suggests that these target proteins are not glycosylated. The ∼82-kd protein was also more abundant in cellular fractions enriched for membrane proteins by Triton X-114 extraction, consistent with a membrane localization of this protein.
Identification of Grp78 as a Cell-Surface Target for CAPGPSKSC
Affinity chromatography of the TX-100/TX-114-extracted membrane fraction of bEND.3 cells using biotinylated CAPGPSKSC coupled to a monomeric avidin column yielded a protein with a molecular mass of ∼82 kd (Figure 5A) ▶ . A protein band of lower molecular weight was also observed (Figure 5A) ▶ . Both bands were excised and subjected to in-gel tryptic digestion followed by MALDI mass spectrometry analysis (Figure 5B) ▶ . The resulting peptide masses were searched against protein databases using the Mascot search engine. 13 The cryptic peptide map of the purified ∼82-kd protein strongly matched the theoretical peptide map of 78-kd glucose-regulated protein precursor (Grp78), suggesting Grp78 as the putative cell-surface target of the CAPGPSKSC peptide (Figure 5C) ▶ . The cryptic map of the lower band also matched that of the Grp78 (data not shown), indicating it is a proteolytic product of the ∼82-kd protein. The same ∼82-kd band (Figure 5D) ▶ was detected with separate blotting of the TX-100/TX-114-enriched membrane extract probed with either the CAPGPSKSC peptide or antibodies (N- and C-terminal) against Grp78. Finally, identity of GPR78 as the ∼82-kd band recognized by the peptide was further confirmed with Grp78 immunoprecipitated with either N-terminal or C-terminal Grp78 antibodies from the extracts. Ligand blots using the biotinylated CAPGPSKSC peptide were shown to recognize the precipitated Grp78, but were negative in a control precipitation performed with an anti-tissue factor antibody (Figure 5E) ▶ . Result from flow-cytometric analysis of the bEND.3 cells with the goat anti-Grp78 antibody is consistent with cell surface presence of Grp78 proteins in these cells (Figure 5F) ▶ .
Figure 5.
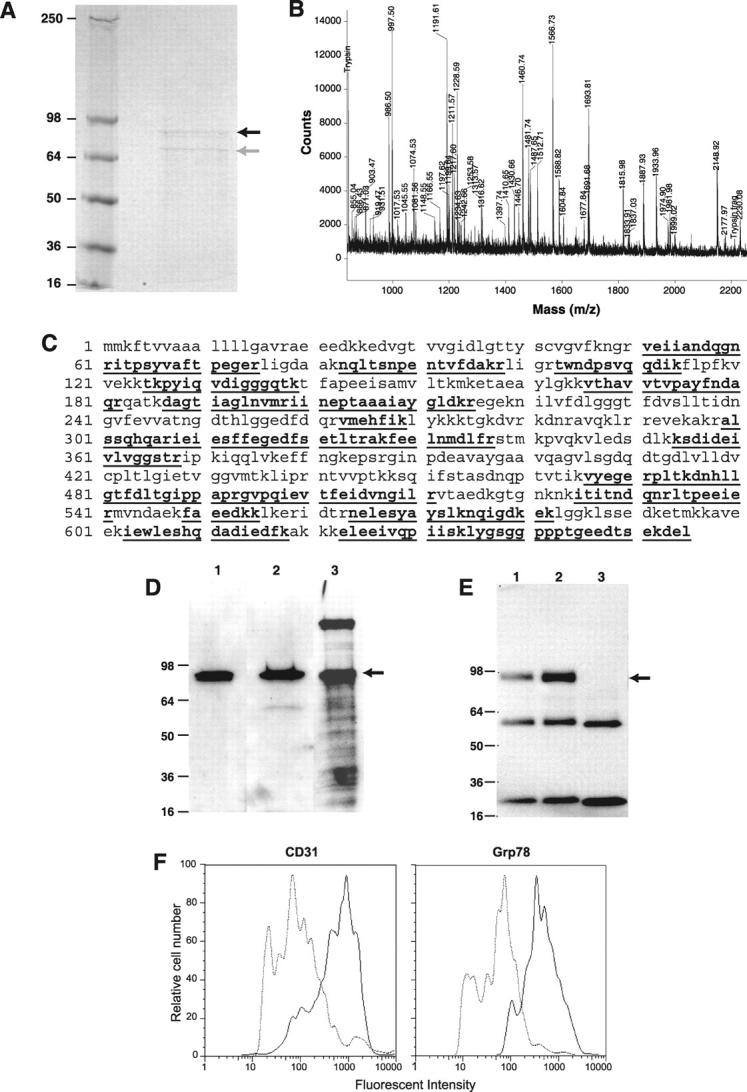
Characterization of Grp78 as a target for CAPGPSKSC peptide. A: The Coomassie-stained gel after protein affinity purification on an immobilized CAPGPSKSC column. The black arrow indicates a protein band of ∼82 kd and the gray arrow indicates a proteolytic product of the ∼82-kd protein. B: The cryptic peptide mixture of the ∼82-kd protein band analyzed by matrix-assisted laser desorption/ionization mass spectrometer. C: The matched cryptic peptide sequences of the ∼82-kd protein band in mouse Grp78 (shown in bold and underlined). D: The Western blotting of the enriched bEND.3 membrane extract with antibodies against 1) the N-terminus of Grp78, 2) the C-terminus of Grp78, and 3) by peptide-ligand blot using biotinylated CAPGPSKSC. E: Immunoprecipitation of the enriched bEND.3 membrane extract with antibodies against 1) the N-terminus of Grp78, 2) the C-terminus of Grp78, 3) a control antibody (anti-tissue factor). Blotting was performed using biotinylated CAPGPSKSC peptide. The lower bands are immunoglobulin fragments in the reaction. F: The flow cytometry analysis of cell surface Grp78 molecules on bENd.3 cells using anti-Grp78 polyclonal antisera. Note a shift of fluorescent intensity in cells stained with anti-Grp78 antisera, similar to cells stained with anti-CD31 antibody.
The TIMP-2 Homologous CNQRHQMSC Sequence Bound to Endothelium
Three TIMP-2 peptidyl homologues were further analyzed to investigate whether peptides from this data set with homology to known proteins mimic the binding activity of their homologous proteins. The CNQRHQMSC phage was injected into ApoE knockout mice, sections of lesion-laden aortic valves were prepared (Figure 6A) ▶ . The CNQRHQMSC phage bound to endothelial cells overlying lesions as demonstrated by FITC-conjugated streptavidin (Figure 6B) ▶ . In contrast, no phage association was detectable on the lesion surface when a control phage was injected (Figure 6C) ▶ .
Figure 6.

Images of phage bound to aortic valves. These aortic valves were from ApoE knockout mice with atherosclerotic lesions. The mice were infused with 1011 pfu of phage carrying the CNQRHQMSC sequence. In A, a H&E-stained section of aortic valve from an atherosclerotic ApoE knockout mouse is shown. Note the aortic valve is thickened by the presence of atherosclerotic lesions. In B, the endothelial cells report red with biotinylated rat anti-mouse CD 31 antibody and the associated phage report green with anti-phage antibody. Co-localization reports yellow. In experiments performed with control phage, no phage association was evident as shown in C. These data indicate that the CNQRHQMSC phage can bind to atherosclerotic lesion-involved endothelial surfaces. Original magnifications, ×100.
TIMP-2 Homologous Peptides Bound to Human Umbilical Vein Endothelial Cells
All three TIMP-2 homologous peptides showed dose-dependent binding to human umbilical vein endothelial cells (Figure 7A) ▶ . The binding of these peptides to cells was competitively inhibited by TIMP-2 protein (Figure 7B) ▶ consistent with the presence of a TIMP-2-binding site on the surface of these cells. The regions of TIMP-2 homologous to the peptides differ from the contact regions of TIMP-2 to known TIMP-2-binding molecules such as TM-MMP as revealed by crystallography. 14 The presence of a TIMP-2-binding site other than a member of the metalloproteinase family has been postulated. 15,16 Bindings of these peptides to cell surfaces are consistent with the presence of a receptor for TIMP-2.
Figure 7.

The three synthetic peptides with homology to TIMP-2 bind to endothelial cells in a dose-dependent manner (top). Binding of peptides (0.5 μmol/L) was inhibited by purified TIMP-2 protein (0.05 μmol/L) (bottom), indicating that the peptides mimicking TIMP-2 binding are consistent with a novel binding site for TIMP-2 protein on the surface of these cells. The scrambled control peptide did not bind.
Discussion
Although it is reasonable to hypothesize differences in gene expression and cell-surface molecular repertoire for endothelial cells associated with the active atherogenic lesion as well as those overlying the formed plaque surface; there is limited evidence for such positional and pathological differences. Most current data addressing the molecular pathobiology of atherosclerosis has been developed from hypothesis-driven experimental progression from identified data sets and has revealed much about the composition of atherosclerotic plaques. In the present experiments we have used a relatively unbiased exploratory approach to search for selective differences in the molecular repertoire of the surface of endothelial cells associated with atherosclerotic lesions, differences that are postulated to result from participation in the atherogenic process. The data were developed by in vivo competitive selection in which association of members of a constrained phage displayed peptidyl library with atherosclerotic lesions rather than the competing nonatherogenic endothelium was used in a progressive selection process and the peptidyl ligand encoded by inserted gene then identified. Selection by lesions in the atherosclerotic ApoE knockout mouse of phage-expressed peptidyl ligands should occur only if their targets were selectively expressed only, or at higher density, by the lesion-associated endothelium in contrast to the far more abundant competing normal vascular endothelium in the mouse. The one imposed bias in these experiments was the size and cyclic constrained structural characteristics of the peptidyl repertoire.
For the current studies we used the high-fat diet fed ApoE knockout mouse as the atherosclerosis model. ApoE is a constituent of cholesterol-rich lipoprotein and mediates binding to the hepatic LDL R-related protein and thereby clearance from the circulation. In its absence ApoE knockout mice display severe hypercholesterolemia with elevated cholesterol forms of VLDL, IDL, and to a lesser degree LDL. 10 Consequently, ApoE knockout mice fed high-fat diets develop early fatty streak lesions within a few months of birth, with lesions that mimic aspects of early human lesions.
The phage displayed cyclic peptide CAPGPSKSC as well as the synthetic peptide preferentially bound lesions of both atherosclerotic ApoE-deficient mice and human specimens, in contrast to ApoE mice fed standard diet and lacking lesions or nonatherosclerotic human blood vessels. The preferential binding of the synthetic CAPGPSKSC peptide to atherosclerotic lesion surfaces in vivo provides direct evidence of the validity of the peptidyl ligand and that the surface protein repertoire differs for the atherosclerotic lesion-associated endothelium. The CAPGPSKSC peptide has some homology to chymase, a serine protease that is responsible for angiotensin II generation (Table 1) ▶ . 17-19 The disulfide constraint coupled with the presence of two prolines in the CAPGPSKSC sequence imparts a predicted rigid ring structure. Peptide-folding experiments also supported that, under oxidizing condition, the peptide quickly circularized and the monomeric loop structure seems to be the favored conformation for this peptide because dimers or higher order aggregates were notably absent. There have been other reports of homing peptides selective to the vasculature of different organs, 20 consistent with hypothesized positional differences in endothelium. 21 Our data further reveals molecular heterogeneity of endothelium overlying atherosclerotic lesions. On the aortic atherosclerotic lesions of ApoE knockout mice binding of the CAPGPSKSC peptide and phage is intensified at the edge of the lesions. The intensity of peptide binding might be correlated with some form of endothelial cell stimulus-driven response. On the lesions of the aortic arch, the binding of the CAPGPSKSC peptide is consistently absent in the center of lesions, which may reflect local endothelial denudation, consistent with peptide recognition of an endothelial cell surface target not exposed on the subendothelial matrix. The CAPGPSKSC peptide binds to human lesions as well, indicative of a conserved target homologue. Human lesions, because of their greater age or other facets of the atherosclerotic process, differ in degree and morphology from the young lesions of ApoE knockout mice. Nevertheless, these observations support that certain aspects are shared between the animal model and human atherosclerosis.
The cell membrane target protein of ∼82 kd was identified as Grp78 (glucose-regulated protein precursor 78). Strict conservation of this protein across species supports the activity of CAPGPSKSC peptide recognizing both mouse and human Grp78 protein. Grp78 is a heat-shock protein that was originally characterized as an endoplasmic reticulum chaperone and also known as BiP. 22 Recent work has demonstrated its presence on the surface of cells under certain conditions; and studies indicate that this molecule may play roles in some pathological processes. Grp78 associates with class I major histocompatibility protein on the surface of human umbilical vein endothelial cells in culture. 23 It has recently been implicated in mediating signal transduction when associated as a co-receptor with the protease inhibitor α2-macroglobulin (α2M). 24 This signaling function is greatly enhanced under conditions of certain forms of cellular activation such as unstimulated versus stimulated macrophages, 25 rheumatoid synovial versus normal fibroblasts, 26 and highly metastatic versus nonmetastatic prostate cancer cells. 27 Antibodies against Grp78 also co-precipitate the primary receptor for α2M, the low-density lipoprotein receptor-related protein, a scavenger receptor in the LDL superfamily, members of which are strongly implicated in atherogenesis. 24 Grp78 itself binds to mutated forms of the LDL receptor and is involved in their retention in the endoplasmic reticulum. 28 Recently, Grp78 was identified as a co-receptor for coxsackievirus A9 (CAV-9), and functions in concert with the other known CAV-9 virus receptor αvβ3 integrins. Grp78 interaction with major histocompatibility complex class I molecules is essential for mediating virus internalization. 29
Local oxygen tension is considered to play an important role in the cell biology of angiogenesis. Oxidative stress promotes vascular inflammation and monocyte/macrophage recruitment to the endothelial cell by increasing expression of molecules such as VCAM-1 and monocyte chemoattractant protein-1 (MCP-1). 30,31 In other studies, oxygen donors such as tauroursodeoxycholic acid and hydrogen peroxide were shown to induce the level of Grp78 expression. 32,33 A mechanism by which Grp78 might participate in the pathobiology of atherosclerosis may be through its induction in response to homocysteine. 34 Hyperhomocysteinemia is a common risk factor of cardiovascular disease. Homocysteine induces endoplasmic reticulum stress, resulting in up-regulation of cholesterol biosynthesis. Overexpression of Grp78 has been observed to inhibit homocysteine-induced gene expression. 35 These data, combined with the interaction of Grp78 with the LDL receptor protein 28 and apolipoprotein B, 36 support a potentially significant role for Grp78 in the pathobiology of atherosclerosis. One must consider that the proadhesive and proinflammatory phenotype of the lesional surface endothelium may contribute to the thrombotic complication of atheroma. In this regard, Grp78 was recently shown to inhibit tissue factor procoagulation activity. Conversion of factor X to Xa and factor VII to VIIa was significantly lower on the surface of Grp78-overexpressed cells. 37 These data indicate an anti-coagulative function for Grp78 on the surface of lesion-lining endothelial cells.
Many of the atheroma-binding peptides demonstrated significant homology with known proteins. It is reasonable to speculate that the binding partners of these proteins may be preferentially present on endothelial cells of atherosclerotic hosts at higher density on the lesions. To explore this hypothesis, a set of peptides with homology to TIMP-2 was analyzed. The dose-dependent binding of the TIMP-2 homologous peptides to endothelium and their inhibition by TIMP-2 protein is consistent with the presence of a heretofore-unrecognized TIMP-2 receptor. TIMP-1 and TIMP-2 are multifunctional proteins with diverse actions; both inhibitors exhibit growth factor-like activity and can inhibit angiogenesis. 38,39 Their metalloproteinase inhibitor activity was believed to have therapeutic potential in atherosclerosis. 40 These data suggest that at least some of the peptides mimic the binding of their homologues. A large-scale sampling of this peptide data set may advance identification of the binding partners and thereby the repertoire of the atherosclerotic endothelial surfaces. The rupture-prone vulnerable plaques usually contain a substantial number of macrophages and lymphocytes. 41,42 The matrix-degrading proteases produced by the inflammatory cells are hypothesized to contribute to the weakening of the protective fibrous cap of these plaques. In this respect it is of interest that we identified a considerable number of peptidyl identities consistent with proteases and protease inhibitors.
Despite the potential importance of plaque surface molecules in atherogenesis, most efforts have been focused on proteins overexpressed within the lesions. However, relevant to the local endothelial surfaces, a number of scavenger receptors have been identified on the endothelial surface, molecules that bind to different as well as modified lipoproteins in processes implicated in atherogenesis. 43 The members of this family of membrane proteins are expanding and now include CD36, 44 macrosialin, 45,46 and SRB-1. 47,48 Modified lipoproteins containing various oxidized phospholipids, 49-51 as well as shear stress, 52,53 can elicit expression of adhesion molecules and cytokines implicated in atherogenesis. Recruitment of mononuclear leukocytes to the intima is one of the early events, 1 thus the focus on specific adhesion molecules. 54-56 Several classes of adhesive cell-surface glycoproteins, such as selectin 7 and VCAM-1, 8 are implicated in atherogenesis and enhance attachment and subsequent transmigration of monocytes and lymphocytes, as well as production of cytokines. Here, we have applied a novel biopanning strategy to interrogate the differential molecular profile of the endothelial surface of atherosclerotic lesions. In addition to providing insight into the pathophysiology of atherosclerosis, the peptidyl probes recognizing lesion-surface molecules may be used to develop noninvasive imaging agents for the early detection of atherosclerosis as well as monitoring the progression of this disease.
Acknowledgments
We thank Barbara Parker for manuscript preparation; and Cynthia Biazak, Jin Yao, and Pablito Tejada for laboratory assistance.
Footnotes
Address reprint requests to Cheng Liu, Department of Immunology and Vascular Biology, Mail Code C204, 10550 N. Torrey Pines Rd., La Jolla, CA 92037-1092. E-mail: chengliu@scripps.edu, and Thomas Edgington, E-mail: tse@scripps.edu.
Supported by the National Institutes of Health (grant P01 HL 16411).
References
- 1.Libby P: Changing concepts of atherogenesis. J Intern Med 2000, 247:349-358 [DOI] [PubMed] [Google Scholar]
- 2.Faggiotto A, Ross R: Studies of hypercholesterolemia in the nonhuman primate. II. Fatty streak conversion to fibrous plaque. Arteriosclerosis 1984, 4:341-356 [DOI] [PubMed] [Google Scholar]
- 3.Rosenfeld ME, Tsukada T, Gown AM, Ross R: Fatty streak initiation in Watanabe heritable hyperlipemic and comparably hypercholesterolemic fat-fed rabbits. Arteriosclerosis 1987, 7:9-23 [DOI] [PubMed] [Google Scholar]
- 4.Ross R: The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993, 362:801-809 [DOI] [PubMed] [Google Scholar]
- 5.Ross R: Atherosclerosis: current understanding of mechanisms and future strategies in therapy. Transplant Proc 1993, 25:2041-2043 [PubMed] [Google Scholar]
- 6.Fuster V, Lewis A: Conner Memorial Lecture. Mechanisms leading to myocardial infarction: insights from studies of vascular biology [published erratum appears in Circulation 1995 Jan 1;91(1):256].Circulation 1994, 90:2126-2146 [DOI] [PubMed] [Google Scholar]
- 7.Dong ZM, Chapman SM, Brown AA, Frenette PS, Hynes RO, Wagner DD: The combined role of P- and E-selectins in atherosclerosis. J Clin Invest 1998, 102:145-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cybulsky MI, Gimbrone JR MA: Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science 1991, 251:788-791 [DOI] [PubMed] [Google Scholar]
- 9.Boisvert WA, Black AS, Curtiss LK: ApoA1 reduces free cholesterol accumulation in atherosclerotic lesions of ApoE-deficient mice transplanted with ApoE-expressing macrophages. Arterioscler Thromb Vasc Biol 1999, 19:525-530 [DOI] [PubMed] [Google Scholar]
- 10.Curtiss LK, Boisvert WA: Apolipoprotein E and atherosclerosis. Curr Opin Lipidol 2000, 11:243-251 [DOI] [PubMed] [Google Scholar]
- 11.Pasqualini R, Ruoslahti E: Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380:364-366 [DOI] [PubMed] [Google Scholar]
- 12.Vachon V, Pouliot JF, Laprade R, Beliveau R: Fractionation of renal brush border membrane proteins with Triton X-114 phase partitioning. Biochem Cell Biol 1991, 69:206-211 [DOI] [PubMed] [Google Scholar]
- 13.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS: Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20:3551-3567 [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Catalan C, Bode W, Huber R, Turk D, Calvete JJ, Lichte A, Tschesche H, Maskos K: Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J 1998, 17:5238-5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chesler L, Golde DW, Bersch N, Johnson MD: Metalloproteinase inhibition and erythroid potentiation are independent activities of tissue inhibitor of metalloproteinases-1. Blood 1995, 86:4506-4515 [PubMed] [Google Scholar]
- 16.Corcoran ML, Stetler-Stevenson WG: Tissue inhibitor of metalloproteinase-2 stimulates fibroblast proliferation via a cAMP-dependent mechanism. J Biol Chem 1995, 270:13453-13459 [DOI] [PubMed] [Google Scholar]
- 17.Nishimoto M, Takai S, Kim S, Jin D, Yuda A, Sakaguchi M, Yamada M, Sawada Y, Kondo K, Asada K, Iwao H, Sasaki S, Miyazaki M: Significance of chymase-dependent angiotensin II-forming pathway in the development of vascular proliferation. Circulation 2001, 104:1274-1279 [DOI] [PubMed] [Google Scholar]
- 18.Katugampola SD, Davenport AP: Radioligand binding reveals chymase as the predominant enzyme for mediating tissue conversion of angiotensin I in the normal human heart. Clin Sci (Lond) 2002, 102:15-21 [PubMed] [Google Scholar]
- 19.Miyazaki M, Takai S: Local angiotensin II-generating system in vascular tissues: the roles of chymase. Hypertens Res 2001, 24:189-193 [DOI] [PubMed] [Google Scholar]
- 20.Kolonin M, Pasqualini R, Arap W: Molecular addresses in blood vessels as targets for therapy. Curr Opin Chem Biol 2001, 5:308-313 [DOI] [PubMed] [Google Scholar]
- 21.Tressler RJ, Belloni PN, Nicolson GL: Correlation of inhibition of adhesion of large cell lymphoma and hepatic sinusoidal endothelial cells by RGD-containing peptide polymers with metastatic potential: role of integrin-dependent and -independent adhesion mechanisms. Cancer Commun 1989, 1:55-63 [DOI] [PubMed] [Google Scholar]
- 22.Gething MJ: Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol 1999, 10:465-472 [DOI] [PubMed] [Google Scholar]
- 23.Triantafilou M, Fradelizi D, Triantafilou K: Major histocompatibility class one molecule associates with glucose regulated protein (GRP) 78 on the cell surface. Hum Immunol 2001, 62:764-770 [DOI] [PubMed] [Google Scholar]
- 24.Misra UK, Gonzalez-Gronow M, Gawdi G, Hart JP, Johnson CE, Pizzo SV: The role of Grp 78 in alpha 2-macroglobulin-induced signal transduction. Evidence from RNA interference that the low density lipoprotein receptor-related protein is associated with, but not necessary for, GRP 78-mediated signal transduction. J Biol Chem 2002, 277:42082-42087 [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharjee G, Misra UK, Gawdi G, Cianciolo G, Pizzo SV: Inducible expression of the alpha2-macroglobulin signaling receptor in response to antigenic stimulation: a study of second messenger generation. J Cell Biochem 2001, 82:260-270 [DOI] [PubMed] [Google Scholar]
- 26.Misra UK, Gonzalez-Gronow M, Gawdi G, Pizzo SV: Up-regulation of the alpha2-macroglobulin signaling receptor on rheumatoid synovial fibroblasts. J Biol Chem 1997, 272:497-502 [DOI] [PubMed] [Google Scholar]
- 27.Asplin IR, Misra UK, Gawdi G, Gonzalez-Gronow M, Pizzo SV: Selective upregulated expression of the alpha2-macroglobulin signaling receptor in highly metastatic 1-LN prostate carcinoma cells. Arch Biochem Biophys 2000, 383:135-141 [DOI] [PubMed] [Google Scholar]
- 28.Jorgensen MM, Jensen ON, Holst HU, Hansen JJ, Corydon TJ, Bross P, Bolund L, Gregersen N: Grp78 is involved in retention of mutant low density lipoprotein receptor protein in the endoplasmic reticulum. J Biol Chem 2000, 275:33861-33868 [DOI] [PubMed] [Google Scholar]
- 29.Triantafilou K, Fradelizi D, Wilson K, Triantafilou M: GRP78, a coreceptor for coxsackievirus A9, interacts with major histocompatibility complex class I molecules which mediate virus internalization. J Virol 2002, 76:633-643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS: A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest 2001, 107:1255-1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kinlay S, Libby P, Ganz P: Endothelial function and coronary artery disease. Curr Opin Lipidol 2001, 12:383-389 [DOI] [PubMed] [Google Scholar]
- 32.Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, Yoffe B: Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 2002, 36:592-601 [DOI] [PubMed] [Google Scholar]
- 33.van der Vlies D, Pap EH, Post JA, Celis JE, Wirtz KW: Endoplasmic reticulum resident proteins of normal human dermal fibroblasts are the major targets for oxidative stress induced by hydrogen peroxide. Biochem J 2002, 366:825-830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyata T, Kokame K, Agarwala KL, Kato H: Analysis of gene expression in homocysteine-injured vascular endothelial cells: demonstration of GRP78/BiP expression, cloning and characterization of a novel reducing agent-tunicamycin regulated gene. Semin Thromb Hemost 1998, 24:285-291 [DOI] [PubMed] [Google Scholar]
- 35.Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK, Malinow MR, Austin RC: Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest 2001, 107:1263-1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linnik KM, Herscovitz H: Multiple molecular chaperones interact with apolipoprotein B during its maturation. The network of endoplasmic reticulum-resident chaperones (ERp72, GRP94, calreticulin, and BiP) interacts with apolipoprotein b regardless of its lipidation state. J Biol Chem 1998, 273:21368-21373 [DOI] [PubMed] [Google Scholar]
- 37.Watson LM, Chan AK, Berry LR, Li J, Sood SK, Dickhout JG, Xu L, Werstuck GH, Bajzar L, Klamut HJ, Austin RC: Overexpression of the 78-kDa glucose-regulated protein/immunoglobulin-binding protein (GRP78/BiP) inhibits tissue factor procoagulant activity. J Biol Chem 2003, 278:17438-17447 [DOI] [PubMed] [Google Scholar]
- 38.Bello L, Lucini V, Carrabba G, Giussani C, Machluf M, Pluderi M, Nikas D, Zhang J, Tomei G, Villani RM, Carroll RS, Bikfalvi A, Black PM: Simultaneous inhibition of glioma angiogenesis, cell proliferation, and invasion by a naturally occurring fragment of human metalloproteinase-2. Cancer Res 2001, 61:8730-8736 [PubMed] [Google Scholar]
- 39.Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP: Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol 1997, 74:111-122 [PubMed] [Google Scholar]
- 40.George SJ: Therapeutic potential of matrix metalloproteinase inhibitors in atherosclerosis. Expert Opin Investig Drugs 2000, 9:993-1007 [DOI] [PubMed] [Google Scholar]
- 41.van der Wal AC, Becker AE, van der Loos CM, Das PK: Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994, 89:36-44 [DOI] [PubMed] [Google Scholar]
- 42.Lendon CL, Davies MJ, Born GV, Richardson PD: Atherosclerotic plaque caps are locally weakened when macrophages density is increased. Atherosclerosis 1991, 87:87-90 [DOI] [PubMed] [Google Scholar]
- 43.Krieger M, Acton S, Ashkenas J, Pearson A, Penman M, Resnick D: Molecular flypaper, host defense, and atherosclerosis. Structure, binding properties, and functions of macrophage scavenger receptors. J Biol Chem 1993, 268:4569-4572 [PubMed] [Google Scholar]
- 44.Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA: CD36 is a receptor for oxidized low density lipoprotein. J Biol Chem 1993, 268:11811-11816 [PubMed] [Google Scholar]
- 45.Ottnad E, Parthasarathy S, Sambrano GR, Ramprasad MP, Quehenberger O, Kondratenko N, Green S, Steinberg D: A macrophage receptor for oxidized low density lipoprotein distinct from the receptor for acetyl low density lipoprotein: partial purification and role in recognition of oxidatively damaged cells. Proc Natl Acad Sci USA 1995, 92:1391-1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramprasad MP, Fischer W, Witztum JL, Sambrano GR, Quehenberger O, Steinberg D: The 94- to 97-kDa mouse macrophage membrane protein that recognizes oxidized low density lipoprotein and phosphatidylserine-rich liposomes is identical to macrosialin, the mouse homologue of human CD68. Proc Natl Acad Sci USA 1995, 92:9580-9584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M: Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271:518-520 [DOI] [PubMed] [Google Scholar]
- 48.Gu X, Trigatti B, Xu S, Acton S, Babitt J, Krieger M: The efficient cellular uptake of high density lipoprotein lipids via scavenger receptor class B type I requires not only receptor-mediated surface binding but also receptor-specific lipid transfer mediated by its extracellular domain. J Biol Chem 1998, 273:26338-26348 [DOI] [PubMed] [Google Scholar]
- 49.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA: Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem 1997, 272:13597-13607 [DOI] [PubMed] [Google Scholar]
- 50.Witztum JL, Berliner JA: Oxidized phospholipids and isoprostanes in atherosclerosis. Curr Opin Lipidol 1998, 9:441-448 [DOI] [PubMed] [Google Scholar]
- 51.Berliner J, Leitinger N, Watson A, Huber J, Fogelman A, Navab M: Oxidized lipids in atherogenesis: formation, destruction and action. Thromb Haemost 1997, 78:195-199 [PubMed] [Google Scholar]
- 52.Gimbrone MA, Jr, Topper JN, Nagel T, Anderson KR, Garcia-Cardena G: Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann NY Acad Sci 2000, 902:230-240 [DOI] [PubMed] [Google Scholar]
- 53.Gimbrone MA, Jr, Nagel T, Topper JN: Biomechanical activation: an emerging paradigm in endothelial adhesion biology. J Clin Invest 1997, 99:1809-1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carlos TM, Harlan JM: Leukocyte-endothelial adhesion molecules. Blood 1994, 84:2068-2101 [PubMed] [Google Scholar]
- 55.Frenette PS, Wagner DD: Adhesion molecules—Part 1. N Engl J Med 1996, 334:1526-1529 [DOI] [PubMed] [Google Scholar]
- 56.Frenette PS, Wagner DD: Adhesion molecules—Part II: blood vessels and blood cells. N Engl J Med 1996, 335:43-45 [DOI] [PubMed] [Google Scholar]