

Abstract
Mice lacking the xeroderma pigmentosum group A gene (XPA−/− mice), which have a complete deficiency in nucleotide excision repair (NER), are highly predisposed to tongue squamous cell carcinoma (SCC) when exposed to 4-nitroquinoline 1-oxide (4NQO). To explore the effects of the interaction of the NER machinery with p53 in oral tumorigenesis, we generated an XPA−/− mouse strain carrying mutant alleles for p53. This mouse model of 4NQO carcinogenesis demonstrated that despite the same tumor frequency, XPA−/−p53+/− mice reached 100% SCC incidence at 25 weeks compared with 50 weeks for XPA−/−p53+/+ littermates. XPA−/−p53−/− mice succumbed to spontaneous thymic lymphomas before the development of tongue tumors (before 13 weeks of age). SCC originated in XPA−/−p53+/− mice maintained the p53+/− genotype and the retained wild-type p53 allele appeared to be structurally intact. Only one of 20 XPA−/−p53+/+ SCC showed a missense mutation of p53. Collectively, the accelerated tongue tumor growth may be a consequence of haploinsufficiency but not of mutation of p53 in the context of NER deficiency.
p53 plays a complex and critical role in cellular proofreading in response to many stress signals including potentially oncogenic DNA damage. 1-3 Abrogation of p53 function results in increased genomic instability, decreased growth arrest, deregulated cell proliferation, reduced apoptosis and promoted angiogenesis. Consequently, loss of p53, through clonal expansion of cells in which unrepaired DNA damage would lead to mutation, potentiates tumor development in both humans and mice. The p53 heterozygous (p53+/−) mouse, an animal model for human Li-Fraumeni syndrome, 4 contains a single normal allele of p53 and provides the first opportunity to study the function of p53 in the carcinogenesis initiation, promotion, and progression steps in vivo. 5-7 There are contradictory data concerning the status of the functional p53 allele in tumors derived from p53+/− mice; 6 however, a growing body of evidence to date indicated that haploid loss of p53 is sufficient for driving tumorigenesis 6-14 and may have different potential in different tissues. 7,15,16 Despite the importance of p53 in oral carcinogenesis, 17 the lack of an appropriate genetically based mouse model has left this issue in considerable doubt.
We reported recently that the application of 4-nitroquinoline 1-oxide (4NQO) in the drinking water selectively induced a very high frequency of tongue squamous cell carcinoma (SCC) in the xeroderma pigmentosum group A gene-deficient (XPA−/−) mice. 18 At 50 weeks of exposure, the tumor incidence exceeded 90% for XPA−/− mice, with no tumors occurring in XPA+/− and XPA+/+ littermates. Similar to human oral SCC, a sequence of histopathological lesions develops: mild to severe dysplasia, conversion to carcinoma in situ, and eventually, invasive SCC. Unlike the mouse skin carcinogenesis, papilloma formation, papilloma growth, and progression to SCC are an uncommon event. Thus, this is a unique multistep model of oral cancer. 18 In the present study, we asked whether the introduction of p53 deficiency renders XPA−/− mice more susceptible to 4NQO-induced tongue carcinogenesis.
Materials and Methods
Mouse Model
XPA−/− mice used in this study were developed and maintained on a C3H/HeN background as described previously. 18-21 They were crossed with p53+/− mice on a mixed C57BL/6 (25%) and CBA (75%) genetic background 13,22,23 to generate the following cohorts of mice: XPA−/−p53−/− (n = 20), XPA−/−p53+/− (n = 50) and XPA−/−p53+/+ (n = 40). Littermate controls included p53−/− (n = 35) and p53+/− (n = 50) mice. On entry into the carcinogenicity experiment, about half of the mice were used for the aging study.
4NQO-Induced Carcinogenesis and Tumor Analyses
Six-week-old mice had free access to drinking water containing 10 ppm 4NQO (Iwai Chemical, Tokyo, Japan) as detailed in our previous report. 18 At necropsy, each tumor was divided into halves; one was fixed in 10% phosphate-buffered formalin for histological and immunohistochemical studies, and the other flash-frozen in liquid nitrogen and stored at −80°C for later extraction of DNA. Tumors and autopsied tissues were prepared for routine examination of hematoxylin and eosin (H&E)-stained sections. p53 expression in tongue tumors was investigated with a polyclonal antibody (NCL-p53-CM5p, Novocastra, Newcastle-on-Tyne, UK). Specific immune complexes were detected by the streptavidin-biotin peroxidase complex method. 13,18 Data for male and female mice were combined, as there was no significant difference in both mortality and tumorigenicity data between the sexes (preliminary observations).
PCR-Based and Southern Blot-Based Genotyping Analyses
Loss of the wild-type p53 allele in XPA−/−p53+/− tumors (n = 20) was detected by polymerase chain reaction (PCR)-based genotyping analysis as detailed previously. 13,22,23 These results were also confirmed by Southern blot-based genetic analysis. 22 Briefly, DNA was digested with SstI, separated by electrophoresis in 1% agarose gels and transferred to nitrocellulose. Hybridization was performed in 50% formamide/10X Denhardt’s reagent/4X SSC containing 20 μg per ml of denatured salmon sperm DNA at 42°C followed by washing in 0.2X SSC/0.1% SDS at 50°C. The probes used were the random-primed SstI-XmnI fragment. 22
Mutational Analysis of p53
Total genomic DNA extracted from tongue tumors of XPA−/−p53+/− (n = 20) and XPA−/−p53+/+ (n = 20) mice was subjected to PCR-based cold single-strand conformation polymorphism (SSCP) analysis of exons 5–8 of the mouse p53 as described elsewhere. 14,16 Briefly, exon-specific PCR products were denatured at 95°C and then subjected to rapid chilling on ice. This mixture was loaded onto a 20% TBE gel and electrophoresed under exon-specific optimized conditions at 300V. After electrophoresis, gels were stained with 0.5 μg/ml ethidium bromide (1X TBE) and visualized using a 340 nm UVR. Samples with a mobility shift in PCR-SSCP analysis were confirmed by sequencing after subcloning as described previously. 18 Briefly, DNA fragments from the SSCP gel were subcloned into the EcoRV site of the T-tailed pBluescript II SK(-) (Stratagene, La Jolla, CA). Sequences of inserted DNA from more than 50 recombinant mixed colonies were determined for both of the strands by the dideoxyribonucleotide chain termination method, using a T7 sequence kit (Pharmacia, Uppsala, Sweden). Each analysis was repeated at least twice for confirmation.
Results
Life Expectancy
Viability of XPA−/−p53−/− (n = 10), XPA−/−p53+/− (n = 28), XPA−/−p53+/+ (n = 20), p53−/− (n = 20), and p53+/− (n = 30) mice was closely monitored.As shown in Kaplan-Meier survival plots (Figure 1A) ▶ ,XPA−/−p53−/− and p53−/− mice died from spontaneous lymphomas of the thymus and spleen or unknown causes within 13 or 20 weeks of age and also about 10% of mice with p53+/− genotype succumbed to lymphoma by 50 weeks. Consistent with our previous reports, 13,23 nullizygosity for p53 significantly reduced mouse survival. This confounded the present chronic carcinogenicity regimen.
Figure 1.

A: Survival curves in 10 XPA−/−p53−/− (□), 28 XPA−/−p53+/− (▪), 20 XPA−/−p53+/+ (⊚), 20 p53−/− (○), and 30 p53+/− (▵) mice. B: Tongue tumor incidence in 22 XPA−/−p53+/− (▪), 20 XPA−/−p53+/+ (⊚), and 20 p53+/− (▵) mice. Points represent mice that had tongue tumors visible to the naked eye.
Tongue Tumor Incidence, Latency, and Pathology
The incidence and latency times of tongue tumor development are summarized in Figure 1B ▶ . There was no difference in total intake of 4NQO per mouse among mice examined (data not shown). All tongue tumors, regardless of the genotype, were diagnosed as invasive, well-differentiated SCC (Figure 2A) ▶ . Tumors were first detected in a XPA−/−p53+/− mouse line at 16 weeks. Of 22 XPA−/−p53+/− mice, two died before the development of tongue tumor and by 25 weeks, 100% (20 of 20) of mice bore tumors. Interestingly, half of XPA−/−p53+/− mice had multiple SCC in the upper aerodigestive tract. In XPA−/−p53+/+ mice, tumors began to appear at 31 weeks and by the end of 50 weeks, 100% (20 of 20) of mice developed tumors. All of 10 XPA−/−p53−/− mice died until 7 weeks of 4NQO administration, with no tumor detected by gross examination (data not shown). Although adequate necropsy could not performed because of either a long delay in identification or cagemate cannibalism, at least three moribund XPA−/−p53−/− mice showed histological evidence of dysplastic changes in the tongue (Figure 2B) ▶ . p53+/− and p53−/− littermates were all tumor-free when followed for >1.5 years or 20 weeks, the time of death for p53−/− mice (Figure 2C) ▶ . Nuclear accumulation of p53 protein was detected in all of XPA−/−p53+/− SCC (Figure 2D) ▶ and the pattern was largely heterogeneous, and ranged from slight (a few focal areas) to diffuse (a wide portion of the tumor). In XPA−/−p53+/+ mice, 10 of 20 SCC contained clusters of p53-positive cells in variable proportions.
Figure 2.
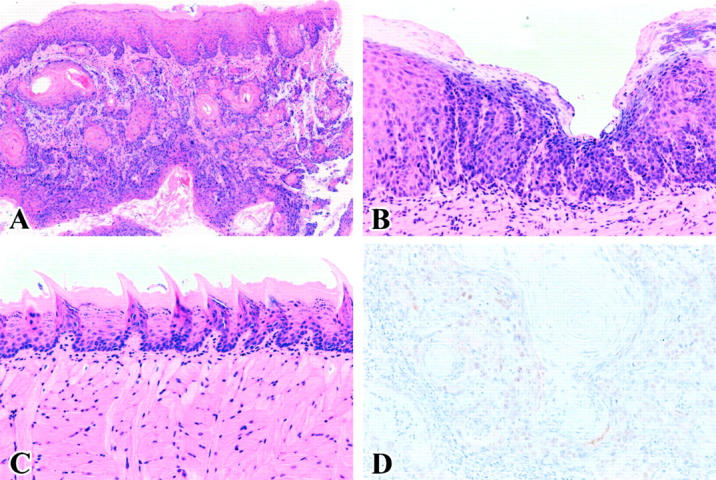
A: Tongue SCC of XPA−/−p53+/− mice. B: Moderate epithelial dysplasia of tongue in XPA−/−p53−/− mice. C: No evidence of neoplastic changes in p53+/− tongue. H&E staining. D: p53 immunohistochemistry in tongue SCC of XPA−/−p53+/− mice. Hematoxylin counterstain. Original magnifications: ×20 (A), ×100 (B), ×40 (C), and ×200 (D).
p53 Status in XPA−/−p53+/− Tumors
A combination of PCR and Southern blotting procedures revealed that tongue SCC of XPA−/−p53+/− mice maintained the p53+/− genotype, as both the wild-type and mutant p53 alleles were identified (Figure 3, A and B) ▶ . There were no polymorphisms (mutated-shifted bands) in tumors that retained the p53 wild-type allele by PCR-SSCP analysis (data not shown).
Figure 3.

A and B: Genotyping analysis of 20 XPA−/−p53+/− tongue SCC by PCR (A) and Southern blot (B) showing no loss of the wild-type p53 allele. M, molecular weight standards; N, normal tissue; T, tumor tissue. C: Sequence analysis of p53 in a XPA−/−p53+/+ tongue SCC showing a CCT-to-CAT mutation at codon 139 of exon 5.
p53 Mutation in XPA−/−p53+/+ Tumors
Only 1 of 20 XPA−/−p53+/+ tongue SCC showed a mobility-shifted band in PCR-SSCP analysis (data not shown). By sequencing after subcloning, p53 missense mutation at codon139, CCT to CAT, resulting in an amino acid substitution of proline for histidine was detected (Figure 3C) ▶ .
Discussion
SCC of the oral cavity is common worldwide, being among the third most common cancer in developing nations and the sixth most common in the general population and responsible for significant mortality and morbidity. 17,24,25 Substantial efforts have been made to explore the pivotal role of p53 in human oral cancer and there are several classic hamster and rat models to induce oral SCC; however, these studies have limitations. 25 To date, no conclusive evidence exists to link p53 deficiency to oral cancer development. In this study, we used a well-characterized cancer-prone XPA mouse model to examine the effects of loss of p53 on oral carcinogenesis. 18 We found that the onset of tongue SCC was drastically accelerated in XPA−/−p53+/− mice and that the structurally intact wild-type p53 allele continued to be expressed in tumors. Taken together, haploinsufficiency of p53 by itself has a profound effect on nucleotide excision repair (NER)-deficient XPA mouse carcinogenesis with gene dosage effect in the tongue.
Tumorigenesis is a multistep process that involves a series of genetic and epigenetic alterations. 26 During tumor development, the disruption of p53 signaling is deemed critical. 1-3 Loss of p53 occurs at various stages and its effect is organ/tissue-specific. 3,7,15,16 Since p53 mutations can be detected in premalignant dysplastic lesions, p53 might be expected to play a role in the early event in oral carcinogenesis. 17,18 Contrary to this expectation, it was surprising that none of p53-deficient mice harbored tongue tumors in the present experimental setup. As described above, both p53+/− (followed for >1.5 years) and 53−/− (all less than 20 weeks of age) mice orally exposed to 4NQO showed no histological evidence of a tumorigenic response in the tongue. A simple explanation may be that the oral mucosa of mice is far less susceptible to chemical carcinogenesis than that of hamsters and rats. 18 In support of this notion, these mice were also resistant to tongue carcinogenesis by 7,12-dimethylbenz[a]anthracene, one of the most powerful and widely used carcinogen to induce oral SCC in rodents (unpublished results).
It is becoming increasingly clear that p53 mutations are infrequent in chemically induced epithelial tumors of p53+/− mice. 7,8,10,14 Since CM5 used in this experiment recognizes both wild-type and mutant p53, p53-positive immunohistochemical results in XPA−/−p53+/− tumors are only suggestive of an increased stability or half-life of p53, which might be due to either up-regulation of MDM2 or p19ARF dysregulation and not mutation. Indeed, the PCR, Southern blot, and SSCP data suggested that p53 allele is structurally intact. Similarly, our previous analysis indicated that the mutational frequency of p53 was low in 4NQO tongue carcinogenesis. 18 In conjunction with the absence of p53 mutation in 4NQO-induced rat tongue SCC, 27 p53 is not a primary target for gene alteration in oral tumorigenesis. Conceivably, additional genetic alterations such as activation of oncogenes and/or inactivation of other tumor-suppressor genes are essential for neoplastic transformation of p53-deficient cells. The lack of obvious tongue tumor formation in XPA−/−p53−/− mice might be best explained by the very limited time window (<7 weeks of 4NQO exposure); however, we could not exclude the possibility that a reduction of p53 levels has a more dramatic affect than the complete absence of p53. We are now trying to establish cell lines derived from tongue epithelium of XPA−/−p53−/− mice for further investigation.
XPA−/− mice used in this study are characterized by a complete deficiency in both global genome repair (GGR) and transcription-coupled repair (TCR) of NER. 19 Since DNA repair is not successively accomplished in these mice, functional p53 may promote death of cells with unrepaired DNA damage by triggering apoptosis. To clarify the synergism of p53 and NER in tumorigenesis, we and others have generated a XPA/p53-deficient mouse strain. 28-30 It has been suggested that p53 play a role in regulating NER, especially the GGR subpathway. 31-36 As shown in our experiment, a heterozygous loss of p53 appears to act synergistically to a NER defect in tongue tumor induction. The bulky DNA lesions elicited by 4NQO are known to substrate for GGR and 4NQO-DNA damage per se provokes up-regulation of p53. 37,38 The hypersensitivity of human and mouse XPA cells to killing by 4NQO may be associated with increased activity of p53, leading to augmented p53-mediated apoptosis. 37 Although 4NQO-induced NER is p53-dependent, 37,38 it remains ambiguous that the modulating effect of p53 on NER is gene-dosage-dependent.
The precise mechanism of p53 loss in tumors is uncertain but is likely to occur by a two-step process. Our XPA−/−p53+/− mice have already undergone a first step of the process, the inactivation of one p53 allele. The recent investigations indicated that loss of the remaining p53 allele is not a prerequisite for tumor formation. 6,7,11,16 The present data also provide a strong argument that p53 haploinsufficiency creates a condition that drive tongue carcinogenesis. Further functional assays including the activation of p21WAF1/CIP1 and MDM2 expression and inducibility of apoptosis are underway to identify whether structurally intact wild-type p53 protein in XPA−/−p53+/− tumors is functioning normally.
In summary, we believe that XPA/p53-deficient mice provide an attractive model for studying the histopathological and genetic changes and molecular mechanisms underlying oral SCC development and also allow for the investigation of the relationship between DNA damage, cell cycle regulation, apoptosis, and cancer in vivo.
Footnotes
Address reprint requests to Dr. Fumio Ide, Department of Oral Pathology, Meikai University School of Dentistry, 1-1 Keyakidai, Sakado, Saitama 350-0283, Japan. E-mail: idef@dent.meikai.ac.jp.
References
- 1.Vogelstein B, Lane DP, Levine AJ: Surfing the p53 network. Nature 2000, 408:307-310 [DOI] [PubMed] [Google Scholar]
- 2.Evan GI, Vousden KH: Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411:342-348 [DOI] [PubMed] [Google Scholar]
- 3.Guimaraes DP, Hainaut P: TP53: a key gene in human cancer. Biochimie 2002, 84:83-93 [DOI] [PubMed] [Google Scholar]
- 4.Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, Naber SP, Jerry DJ: Development of spontaneous mammary tumors in BALB/cp53 heterozygous mice. A model for Li-Fraumeni syndrome. Am J Pathol 2000, 157:2151-2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lozano G, Liu G: Mouse models dissect the role of p53 in cancer and development. Semin Cancer Biol 1998, 8:337-344 [DOI] [PubMed] [Google Scholar]
- 6.Boley SE, Anderson EE, French JE, Donehower LA, Walker DB, Recio L: Loss of p53 in benzene-induced thymic lymphomas in p53+/- mice: evidence of chromosomal recombination. Cancer Res 2000, 60:2831-2835 [PubMed] [Google Scholar]
- 7.French J, Storer RD, Donehower LA: The nature of the heterozygous Trp53 knockout model for identification of mutagenic carcinogens. Toxicol Pathol 2001, 29:24-29 [DOI] [PubMed] [Google Scholar]
- 8.Jerry DJ, Butel JS, Donehower LA, Paulson EJ, Cochran C, Wiseman RW, Medina D: Infrequent p53 mutations in 7, 12-dimethylbenz[a]anthracene-induced mammary tumors in BALB/c and p53 hemizygous mice. Mol Carcinogen 1994, 9:175-183 [DOI] [PubMed] [Google Scholar]
- 9.Maroulakou IG, Shibata M-A, Jorcyk CL, Chen X-X, Green JE: Reduced p53 dosage associated with mammary tumor metastases in C3(1)/TAG transgenic mice. Mol Carcinogen 1997, 20:168-174 [DOI] [PubMed] [Google Scholar]
- 10.Ozaki K, Sukata T, Yamamoto S, Uwagawa S, Seki T, Kawasaki H, Yoshitake A, Wanibuchi H, Koide A, Mori Y, Fukushima S: High susceptibility of p53(+/-) knockout mice in N-butyl-N-(4-hydroxybutyl)nitrosamine urinary bladder carcinogenesis and lack of frequent mutation in residual allele. Cancer Res 1998, 58:3806-3811 [PubMed] [Google Scholar]
- 11.Venkatachalam S, Shi Y-P, Jones SN, Vogel H, Bradley A, Pinkel D, Donehower LA: Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J 1998, 16:4657-4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tyner SD, Choi J, Lauciria R, Ford RJ, Donehower LA: Increased tumor cell proliferation in murine tumors with decreasing dosage of wild-type p53. Mol Carcinogen 1999, 24:197-208 [DOI] [PubMed] [Google Scholar]
- 13.Ide F, Kitada M, Sakashita H, Kusama K: Reduction of p53 dosage renders mice hypersensitive to 7, 12-dimethylbenz(a)anthracene-induced salivary gland tumorigenesis. Anticancer Res 2003, 22:201-204 [PubMed] [Google Scholar]
- 14.Salim EI, Wanibuchi H, Morimura K, Wei M, Mitsuhashi M, Yoshida K, Endo G, Fukushima S: Carcinogenicity of dimethylarsinic acid in p53 heterozygous knockout and wild-type C57BL/6J mice. Carcinogenesis 2003, 24:335-342 [DOI] [PubMed] [Google Scholar]
- 15.Sukata T, Ozaki K, Uwagawa S, Seki T, Wanibuchi H, Yamamoto S, Okuno Y, Fukushima S: Organ-specific, carcinogen-induced increases in cell proliferation in p53-deficient mice. Cancer Res 2000, 60:74-79 [PubMed] [Google Scholar]
- 16.French JE, Lacks GD, Trempus C, Dunnick JK, Foley J, Mahler J, Tice RR, Tennant RW: Loss of heterozygosity frequency at the Trp53 locus in p53-deficient (+/-) mouse tumors is carcinogen- and tissue-dependent. Carcinogenesis 2001, 21:99-106 [DOI] [PubMed] [Google Scholar]
- 17.Blons H, Laurent-Puig P: TP53 and head and neck neoplasms. Hum Mutat 2003, 21:252-257 [DOI] [PubMed] [Google Scholar]
- 18.Ide F, Oda H, Nakatsuru Y, Kusama K, Sakashita H, Tanaka K, Ishikawa T: Xeroderma pigmentosum group A gene action as a protection factor against 4-nitroquinoline 1-oxide-induced tongue carcinogenesis. Carcinogenesis 2001, 22:567-572 [DOI] [PubMed] [Google Scholar]
- 19.Nakane H, Takeuchi S, Yuba S, Saijo M, Nakatsu Y, Murai H, Nakatsuru Y, Ishikawa T, Hirota S, Kitamura Y, Kato Y, Tsunoda Y, Miyauchi H, Horio T, Tokunaga T, Matsunaga T, Nikaido O, Nishimune Y, Okada Y, Tanaka K: High incidence of ultraviolet-B-or chemical-carcinogen-induced skin tumours in mice lacking the xeroderma pigmentosum group A gene. Nature 1995, 377:165-168 [DOI] [PubMed] [Google Scholar]
- 20.Ide F, Iida N, Nakatsuru Y, Oda H, Tanaka K, Ishikawa T: Mice deficient in the nucleotide excision repair gene XPA have elevated sensitivity to benzo[a]pyrene induction of lung tumors. Carcinogenesis 2000, 21:1263-1265 [PubMed] [Google Scholar]
- 21.Takahashi Y, Nakatsuru Y, Zhang S, Shimizu Y, Kume H, Tanaka K, Ide F, Ishikawa T: Enhanced spontaneous and aflatoxin-induced liver tumorigenesis in xeroderma pigmentosum group A gene-deficient mice. Carcinogenesis 2002, 23:627-633 [DOI] [PubMed] [Google Scholar]
- 22.Tsukada T, Tomooka Y, Takai S, Ueda Y, Nishikawa S, Yagi T, Tokunaga T, Takeda N, Suda Y, Abe S, Matsuo I, Ikawa Y, Aizawa S: Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene 1993, 8:3313-3322 [PubMed] [Google Scholar]
- 23.Oda H, Zhang S, Tsurutani N, Shimizu S, Nakatsuru Y, Aizawa S, Ishikawa T: Loss of p53 is an early event in induction of brain tumors in mice by transplacental carcinogen exposure. Cancer Res 1997, 57:646-650 [PubMed] [Google Scholar]
- 24.Patel V, Leethanakul C, Gutkind JS: New approaches to the understanding of the molecular basis of oral cancer. Crit Rev Oral Biol Med 2001, 12:55-63 [DOI] [PubMed] [Google Scholar]
- 25.Opitz OG, Harada H, Suliman Y, Rhoades B, Sharpless NE, Kent R, Kopelovich L, Nakagawa H, Rustgi AK: A mouse model of human oral-esophageal cancer. J Clin Invest 2002, 110:761-769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA: The hallmarks of cancer. Cell 2000, 100:57-70 [DOI] [PubMed] [Google Scholar]
- 27.Suzui M, Yoshimi N, Tanaka T, Mori H: Infrequent Ha-ras mutations and absence of Ki-ras, N-ras, and p53 mutations in 4-nitroquinoline 1-oxide-induced rat oral lesions. Mol Carcinogen 1995, 14:294-298 [DOI] [PubMed] [Google Scholar]
- 28.van Steeg H, de Vries A, van Oostrom CTH, van Benthem J, Beems R, van Kreijl CF: DNA repair-deficient Xpa and Xpa/p53+/- knockout mice: nature of the models. Toxicol Pathol 2001, 29:109-116 [DOI] [PubMed] [Google Scholar]
- 29.van Steeg H: The role of nucleotide excision repair and loss of p53 in mutagenesis and carcinogenesis. Toxicol Lett 2001, 120:209-219 [DOI] [PubMed] [Google Scholar]
- 30.Hoogervorst EM, de Vries A, Beems RB, van Oostrom CTH, Wester PW, Vos JG, Bruins W, Roodbergen M, Cassee FR, Vijg J, van Schooten F-J, van Steeg H: Combined oral benzo[a]pyrene and inhalatory ozone exposure have no effect on lung tumor development in DNA repair-deficient Xpa mice. Carcinogenesis 2003, 24:613-619 [DOI] [PubMed] [Google Scholar]
- 31.Li G, Ho VC, Mitchell DL, Trotter MJ, Tron VA: Differentiation-dependent p53 regulation of nucleotide excision repair in keratinocytes. Am J Pathol 1997, 150:1457-1464 [PMC free article] [PubMed] [Google Scholar]
- 32.Wani MA, Zhu QZ, El-Mahdy M, Wani AA: Influence of p53 tumor suppressor protein on bias of DNA repair and apoptotic response in human cells. Carcinogenesis 1999, 20:765-772 [DOI] [PubMed] [Google Scholar]
- 33.Geske FJ, Nelson AC, Lieberman R, Strange R, Sun T, Gerschenson LE: DNA repair is activated in early stages of p53-induced apoptosis. Cell Death Differ 2000, 7:393-401 [DOI] [PubMed] [Google Scholar]
- 34.Smith ML, Ford JM, Hollander MC, Bortnick RA, Amundson SA, Seo YR, Deng C-X, Hanawalt PC, Fornance AJ: p53-mediated DNA repair responses to UV radiation: studies of mouse cells lacking p53, p21, and/or gadd45 genes. Mol Cell Biol 2000, 20:3705-3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amundson SA, Patterson A, Do KT, Fornace AJ: A nucleotide excision repair master-switch: p53 regulated coordinate induction of global genome repair genes. Cancer Biol Ther 2002, 1:145-149 [DOI] [PubMed] [Google Scholar]
- 36.Rubbi CP, Milner J: p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J 2003, 22:975-986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo Y-R, Lee S-H, Han S-S, Ryu J-C: Effect of p53 tumor suppressor on nucleotide excision repair in human colon carcinoma cells treated with 4-nitroqunoline 1-oxide. Res Commun Mol Pathol Pharmacol 1999, 104:157-164 [PubMed] [Google Scholar]
- 38.Mirzayans R, Bashir S, Murray D, Paterson MC: Inverse correlation between p53 protein levels and DNA repair efficiency in human fibroblast strains treated with 4-nitroquinoline 1-oxide: evidence that lesions other than DNA strand breaks trigger the p53 response. Carcinogenesis 1999, 20:941-946 [DOI] [PubMed] [Google Scholar]