

Abstract
To address the pathogenesis of diabetic autonomic neuropathy, we have examined the sympathetic nervous system in non-obese diabetic (NOD) and streptozotocin (STZ)-induced diabetic mice, two models of type 1 diabetes, and the db/db mouse, a model of type 2 diabetes. After only 3 to 5 weeks of diabetes, NOD mice developed markedly swollen axons and dendrites (“neuritic dystrophy”) in the prevertebral superior mesenteric and celiac ganglia (SMG-CG), similar to the pathology described in diabetic STZ- and BBW-rat and man. Comparable changes failed to develop in the superior cervical ganglia of the NOD mouse or in the SMG-CG of non-diabetic NOD siblings. STZ-induced diabetic mice develop identical changes, although at a much slower pace and to a lesser degree than NOD mice. NOD-SCID mice, which are genetically identical to NOD mice except for the absence of T and B cells, do not develop diabetes or neuropathology comparable to diabetic NOD mice. However, STZ-treated NOD-SCID mice develop severe neuritic dystrophy, evidence against an exclusively autoimmune pathogenesis for autonomic neuropathy in this model. Chronically diabetic type 2 db/db mice fail to develop neuritic dystrophy, suggesting that hyperglycemia alone may not be the critical and sufficient element. The NOD mouse appears to be a valuable model of diabetic sympathetic autonomic neuropathy with unambiguous, rapidly developing neuropathology which corresponds closely to the characteristic pathology of other rodent models and man.
Autonomic neuropathy is a significant clinical complication of diabetes, whose symptoms range widely from comparatively minor pupillary and sweating problems to significant disturbances in cardiovascular, alimentary, and genitourinary function, which result in increased patient morbidity and mortality. 1-4 Several series of autopsied diabetic patients 5-7 have established the reproducible development of markedly enlarged dystrophic axons and nerve terminals in prevertebral superior mesenteric (SMG) and celiac sympathetic ganglia (CG), thought to represent the morphological residua of aberrant intraganglionic sprouting. 7 Sympathetic neuronal cell bodies, although compressed and distorted by large presynaptic endings, appear otherwise normal, and their number appear relatively well, if not completely, maintained in diabetes. Aged humans also develop intraganglionic dystrophic axons which are comparably distributed and ultrastructurally and immunohistochemically identical to those that appear in diabetic patients, a finding which suggests the possibility of shared pathogenetic mechanisms in aging and diabetes. Dystrophic axons in diabetics appear earlier and in significantly greater numbers than aged non-diabetic humans. 5
Animal models have been sought to address the development of diabetic autonomic neuropathy, particularly its early phases which would be most amenable to therapy, and to provide insight into its pathogenetic mechanisms. We and others have developed and extensively characterized several experimental rodent models of diabetic autonomic neuropathy 7 including the streptozotocin (STZ)-diabetic 8-10 and genetically diabetic BBW rat 11 and Chinese hamster. 12 The regular occurrence of degenerating, regenerating, and pathologically distinctive dystrophic axons and, to a lesser degree, abnormal dendrites has been demonstrated in prevertebral sympathetic ganglia innervating the small bowel of STZ- and genetically diabetic rats and Chinese hamsters. Nonbiased quantitative methods demonstrate that dystrophic axons develop in the absence of neuron loss in STZ-diabetic rat sympathetic ganglia. 13 Thus, there is close correspondence between the neuropathology of diabetic autonomic neuropathy in the sympathetic nervous system of rodents and man.
Although the rat models developed to date have begun to elucidate the mechanisms resulting in diabetic autonomic neuropathy, 7 they are limited by the long duration of diabetes necessary to produce reproducible autonomic neuropathology. Furthermore, the existence of mice with a variety of spontaneous and targeted mutations involving discrete enzymes, pathways, and processes have stimulated interest in the development of a mouse model of diabetic autonomic neuropathy. To better address possible pathogenetic mechanisms, we have extrapolated our previous rodent and human studies to determine whether sympathetic ganglionic neuropathology comparable to rat, hamster, and man develops in several mouse models.
We have selected three mouse models which have important differences in the pathogenesis of diabetes, its severity and application to the wide variety of gene-targeted mice which have been developed on a variety of genetic backgrounds. The non-obese diabetic (NOD) mouse, originally isolated as a spontaneous mutation on an ICR genetic background 14 and maintained for more than 20 years as an inbred commercially available strain, spontaneously develops diabetes as the result of a B- and T-cell-mediated autoimmune attack on its pancreatic islets, a process which begins at approximately 4 to 8 weeks of age, resulting in diabetes with an incidence of ∼80% in female and 20% in male mice by 30 weeks of age. 15 The sibs of NOD mice which do not become diabetic represent the control population. NOD mice become hypoinsulinemic with hyperglycemia (blood glucose values often exceed 600 mg/dl) and typically have a shortened lifespan (5 to 8 weeks after the onset of diabetes). Recent studies show the NOD mouse develops a variety of diabetic complications, dysfunction of the alimentary tract prominent among them. 16-18 A second mouse model of type 1 diabetes, the streptozotocin (STZ)-diabetic mouse, develops diabetes following the administration of a single intravenous injection of the pancreatic β-cell toxin streptozotocin (STZ), resulting in hypoinsulinemia and hyperglycemia typically ranging from 400 to 500 mg/dl, and are capable of extended survival (occasionally as long as 12 months). An important practical difference between STZ-induced diabetes and the NOD mouse is the ability of STZ to produce diabetes in mice with spontaneous or gene-targeted mutations irrespective of genetic background. Finally, we examined the genetically diabetic db/db mouse, in which a leptin mutation results in hyperglycemia (comparable in severity to the STZ-mouse), insulin resistance, and hyperinsulinemia. This animal has been widely used as a model for type 2 diabetes in humans. The studies reported here demonstrate the surprisingly rapid and dramatic development of neuritic dystrophy in prevertebral sympathetic ganglia of NOD mice, far exceeding the severity of involvement of the STZ-diabetic mouse or any other rodent model.
Materials and Methods
Animals
All animals were housed and cared for in accordance with the guidelines of the Washington University Committee for the Humane Care of Laboratory Animals and with National Institutes of Health guidelines on laboratory animal welfare. All mice were allowed standard rat chow and water ad libitum and maintained on a 12-hour light/12-hour dark cycle.
NOD Mice
NOD mice were obtained from a colony kept in pathogen-free conditions at Washington University which were originally purchased from Taconic Laboratories (Germantown, NY). Non-fasting morning plasma glucose levels were determined at weekly intervals (in later experiments, animals were bled twice weekly in order to more precisely determine the onset of diabetes) beginning at approximately 12 weeks of age. Animals were considered diabetic when two consecutive blood glucose measurements each exceeded 250 mg/dl. Using these criteria 80% of female mice become diabetic by 30 weeks of age. Sibs of NOD mice which do not become diabetic represent the control population.
NOD-SCID Mice
Male NOD-SCID mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and were kept in pathogen-free conditions at Washington University. NOD-SCID mice are the result of breeding of the SCID mutation to the NOD background for many generations, such that the NOD-SCID mouse is genetically identical to the NOD mouse save for the absence of DNA-dependent protein kinase, a DNA repair enzyme. 19
STZ-Diabetic Mice
Two-to-three-month-old male and female mice of several strains (C57BL6, B6D2F1, DBA, and C57BL6/NCR) were purchased from Jackson Laboratories. Mice were made diabetic by tail vein injection of freshly made streptozotocin (STZ [Sigma, St. Louis, MO], 200 mg/kg in citrate-saline buffer, pH 4.5) under ketamine/xylazine anesthesia. Within 7 days of STZ injection mice were bled and significantly hyperglycemic animals (plasma glucose >250 mg/dl) were considered diabetic.
db/db mice
Male db/db (C57BL/KsJ) mice and db/m littermates were purchased from the Jackson Laboratory at approximately 2 to 3 months of age, their blood glucose values were confirmed (>250 mg/dl) and animals were allowed to survive without treatment for 5 additional months before sacrifice.
Tissue Preparation
Animals were anesthetized with ketamine/xylazine and perfused with 50 ml of heparinized saline followed by 100 to 200 ml of 3% glutaraldehyde in 0.1 mol/L phosphate buffer, pH 7.3, containing 0.45 mmol/L Ca+2. The superior mesenteric-celiac ganglia (SMG-CG) were dissected as a single block, cleaned of extraneous tissue while maintaining the superior mesenteric artery with the ganglionic block, and fixation continued overnight at 4°C in the same buffer. Tissue samples were post-fixed in phosphate-buffered 2% OsO4 containing 1.5% potassium ferricyanide, dehydrated in graded concentrations of alcohol, and embedded in Epon with propylene oxide as an intermediary solvent. One-μm thick plastic sections were examined by light microscopy after staining with toluidine blue. Ultra-thin sections of individual SMG-CG were cut onto formvar-coated slot grids (EMS, Fort Washington, PA), which permits visualization of entire ganglionic cross-sections. Tissues were subsequently stained with uranyl acetate and lead citrate and examined with a JEOL 1200 electron microscope (JEOL, Peabody, MA).
Quantitative Histological Methods
Dystrophic elements are typically intimately related to neuronal perikarya and, therefore, we routinely express their frequency as the ratio of numbers of lesions to nucleated neuronal cell bodies. This method, used in many of our previous studies, substantively reduced the variance in assessments of intraganglionic lesion frequency. In addition, its simplicity permits the quantitative ultrastructural examination of relatively large numbers of ganglia. In our current animal studies an entire cross-section of the SMG-CG was scanned at ×12,000 magnification and the number of dystrophic neurites and synapses was determined. Dystrophic neurites were divided into several morphological classes based on their content of: tubulovesicular aggregates; admixed normal and degenerating subcellular organelles, multivesicular and dense bodies; neurofilaments; and pure aggregates of otherwise normal appearing mitochondria, which were further subdivided into neurites <5 μm and ≥5 μm in diameter since in some studies small axonal collections of mitochondria have been considered to be normal ganglionic constituents. The number of nucleated neurons (range, 50 to 200 neurons examined in each ganglionic cross-section) was then determined by recounting at ×6000 magnification. The frequency of ganglionic neuritic dystrophy was expressed as the ratio of number of dystrophic neurites to the number of nucleated neurons in the same cross-section.
Statistical Analysis
All results are expressed as means ± SEM. Analysis of variance was performed with the SAS general linear models procedure. 20
Results
NOD Mice
NOD mice were monitored weekly by glucometer beginning at approximately 12 weeks of age for development of diabetes, as defined by blood glucose readings of >250 mg/dl on two separate occasions. Diabetic animals were sacrificed and examined 3 to 5 weeks after the onset of diabetes (ie, at 23 to 25 weeks of age) at which time they were markedly hyperglycemic (blood glucose values >600 mg/dl, Table 1 ▶ ). Age-matched non-diabetic sibs were used as controls.
Table 1.
Neuritic Dystrophy in the Prevertebral Sympathetic Ganglia of NOD and NOD-SCID Mice
| Mouse | n | Blood glucose (mg%) | SMG-CG dystrophy (no. dystrophic neurites/no. neuron) |
|---|---|---|---|
| Diabetic NOD | 9 | ≥600 | 1.25 ± 0.35* |
| Controls (non-DM sibs) | 5 | 104 ± 14 | 0.06 ± 0.01 |
| NOD-SCID | 6 | 106 ± 8 | 0.22 ± 0.03† |
Dystrophic neurites (expressed as number per nucleated neuron) in the SMG-CG of female NOD mice diabetic for 3–5 weeks show a dramatic increase in frequency in comparison to non-diabetic siblings. Dystrophic neurites in SMG-CG of 25-week-old female NOD-SCID mice (ie, age-matched to diabetic NOD mice) show a marked decrease in frequency compared to diabetic NOD mice. Values represent the means ± SEM. Statistical comparison:
*p ≤ 0.01 vs. control;
†p<0.01 vs. diabetic NOD.
SMG-CG Neuropathology
Examination of 1-μm thick plastic sections of SMG-CG in 3 to 5 week diabetic NOD (Figure 1A) ▶ and age-matched non-diabetic sibs (Figure 1B) ▶ showed an apparently well preserved complement of principal sympathetic neurons surrounded by neuropil composed of an admixture of axons and dendritic elements. There was no evidence of active neuronal degeneration (specifically, no apoptosis), nodules of Nageotte (tombstones of prior neuron loss) or chromatolysis. None of the diabetic ganglia contained an inflammatory infiltrate or an association of individual lymphocytes or macrophages with neuronal perikarya.
Figure 1.
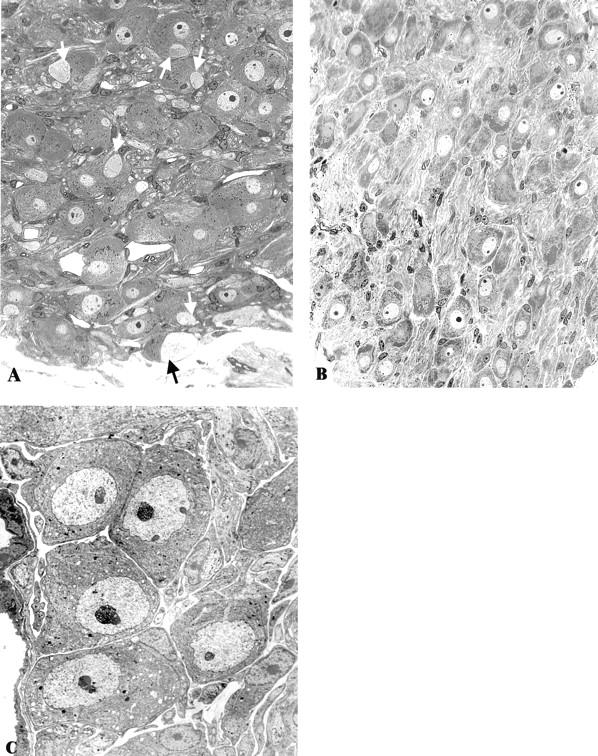
Light microscopic appearance of the SMG-CG of 4 week diabetic NOD mouse (A) and an age-matched non-diabetic sibling (B). Numerous dystrophic neurites (arrows, A) are accompanied by normal appearing neuronal perikarya in the NOD diabetic mouse. The superior cervical ganglion (SCG) of a diabetic NOD mouse (C) shows a normal complement of neurons without dystrophic neurites even though the SMG-CG of the same animal (Figure 2A) ▶ shows marked neuritic dystrophy. Original magnifications: ×300, (A and B); ×1950, (C).
Large swollen neurites were prominent in light microscopic examination of one-micron thick toluidine-blue-stained plastic sections (arrows, Figure 1A ▶ ) in the SMG-CG of NOD mice diabetic for 3 to 5 weeks, often with a patchy distribution as though they may involve selected subpopulations of neurons. Dystrophic neurites were frequently located immediately adjacent to neuronal cell bodies, often within their satellite cell sheaths, resulting in displacement and distortion of perikaryal contours of targeted neurons. Dystrophic neurites were not observed by light microscopic examination in non-diabetic age-matched siblings (Figure 1B) ▶ .
Ultrastructural examination confirmed the light microscopic appearance, demonstrating that swollen dystrophic elements were numerous in SMG-CG of NOD mice (arrows, ▶ Figure 2A ▶ ▶ ) and rare in non-diabetic age-matched NOD siblings (Figure 2B) ▶ . Dystrophic elements exhibited a variety of ultrastructural patterns based on differences in their content of subcellular organelles (Figure 2) ▶ . The most typical appearance (45 ± 7% of the total number of dystrophic neurites, Table 2 ▶ ) consisted of neuritic swellings containing large numbers of tubulovesicular elements (arrow, Figure 2C ▶ ), which ranged from delicate (Figure 2D) ▶ to coarse with varied degrees of compaction. Dystrophic neurites were typically completely enclosed within the cytoplasm of Schwann or satellite cells (Figure 2D) ▶ and also separated from adjacent perikarya by interposed satellite cell processes (arrows, Figure 2D ▶ ). A second subgroup of dystrophic swellings consisted of collections of large numbers of otherwise normal mitochondria that were tightly aggregated without a significant amount of intervening axoplasm (Figure 2, E and F) ▶ . These mitochondrial collections were separated into two groups: neurites <5 μm in diameter (32 + 3%, Table 2 ▶ ) and those ≥5 μm (17 + 4%, Table 2 ▶ ) since small axons containing mitochondria have previously been considered normal “sensory endings” in sympathetic ganglia. Less frequently, neurites contained mixed collections of organelles (mitochondria, autophagosomes, neurofilaments, and multivesicular bodies, Figure 2, G and H ▶ ). Some dystrophic swellings appeared to represent dendrites on which synapses were identified (arrow, Figure 2I ▶ ) or contained ribosome-like structures or lipopigment. However, in many cases it was difficult to confidently identify dystrophic elements as either axons or dendrites and, thus, we have referred to dystrophic processes simply as involving neuritic elements and the process as neuritic dystrophy. Occasionally collections of minute regenerative axonal sprouts (arrows, Figure 2J ▶ ), not separated by Schwann cell processes, were encountered.
Figure 2.
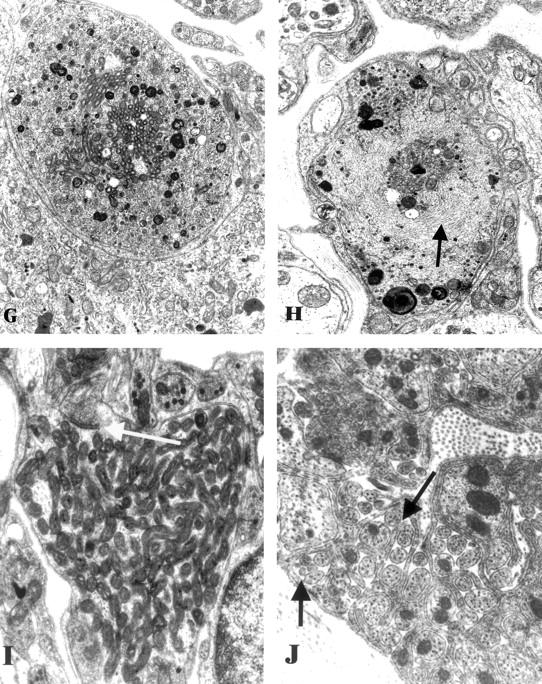
Ultrastructural appearance of the SMG-CG of 3 to 5 week diabetic NOD and non-diabetic control mice. A and B: Numerous dystrophic neurites (arrows, A) are scattered through the ganglionic neuropil and perineuronal space in the diabetic NOD mouse SMG-CG in comparison to the appearance of the normal ganglion of non-diabetic NOD mouse sibling (B). Original magnifications: ×980 (A); ×1950 (B). C and D: The most common ultrastructural appearance of dystrophic neurites in the diabetic NOD mouse consists of dilatations containing tubulovesicular elements. The contours of the sympathetic perikaryon adjacent to a large dystrophic element (arrow, C) are distorted but the neuron appears otherwise normal. A dystrophic neurite containing large numbers of tubulovesicular elements (D) is separated from the adjacent neuronal cell body (N) by satellite cell processes (arrows) along part of its circumference. Original magnifications: ×2600 (C); ×13,000 (D). E and F: Markedly enlarged neurites containing nearly pure collections of mitochondria represent a second major category of neuritic dystrophy Original magnifications: ×7800 (E); ×19,500 (F). G and H: Dystrophic neurites may also contain a variety of admixed organelles including mitochondria, tubulovesicular elements, and dense bodies (G) or neurofilaments (arrow, H). Original magnifications: ×15,600 (G and H). I: Dystrophic neurites occasionally exhibit synaptic specializations (arrow). Original magnification: ×24,200. J: The ganglionic neuropil may contain large numbers of axonal sprouts, suggesting a regenerative component. Original magnification: ×29,000.
Figure 2A.
Continues
Table 2.
Subpopulations of Dystrophic Neurites in SMG-CG of NOD, Non-Diabetic Controls and NOD-SCID Mice
| Mouse | Mixed organelles (%) | Neurofilaments (%) | Tubulovesicles (%) | Mitochondria A (%) | Mitochondria B (%) |
|---|---|---|---|---|---|
| Diabetic NOD | 4 ± 1 | 1 ± 1 | 45 ± 7* | 32 ± 3 | 17 ± 4 |
| Controls (non-DM sibs) | 21 ± 9 | 0 | 12 ± 7 | 44 ± 14 | 23 ± 10 |
| NOD-SCID | 8 ± 3 | 0 | 3 ± 3† | 38 ± 8 | 51 ± 4† |
Dystrophic neurites of NOD mice diabetic for 3 to 5 weeks, their sibs and age-matched NOD-SCID mice were separated into subpopulations for each animal based on ultrastructural content of dystrophic neurites and expressed as a percentage of total numbers of dystrophic neurites calculated for each animal. Mitochondria-engorged neurites are presented as two groups on the basis of size of the dystrophic neurite: A < 5μ and B ≥ 5μ. Values represent the means ± SEM. Statistical comparison:
* p ≤ 0.01 vs. control;
† p ≤ 0.01 vs. diabetic NOD.
Since dystrophic neurites were present in both diabetic and control mice, it was necessary to apply an ultrastructural quantitative method to accurately compare their relative numbers. The numbers of dystrophic neurites were counted and expressed as a ratio (numbers of dystrophic elements/numbers of nucleated neurons). This analysis established that dystrophic neurites were increased 20-fold in 3 to 5 week diabetic NOD mouse SMG-CG compared to age-matched non-diabetic siblings (Table 1) ▶ .
Comparison of the frequency of the subpopulation of small (< 5 μm) mitochondria-laden neurites (“Mitochondria A”, Table 2 ▶ ) in NOD diabetic (32% × 1.25 ± 0.35 = 0.40 ± 0.11 lesions/neuron, n = 9 mice) and non-diabetic sibling controls (44% × 0.06 ± 0.01 = 0.030 ± 0.004, n = 5, P ≤ 0.001) provides evidence that, in this setting, small mitochondria filled neurites are clearly pathological and associated with the diabetic state.
SMG-CG versus Superior Cervical Ganglion (SCG) in NOD mice
In man 5 and rat 8 there is a dramatic and as yet unexplained difference in the frequency of diabetes- and age-induced neuritic dystrophy between prevertebral SMG and CG, which are prominently involved by neuritic dystrophy, in comparison to the SCG, the largest of the paravertebral chain ganglia, which are minimally involved. Therefore, we compared the frequency of neuritic dystrophy in the SMG-CG of 4 to 5 week diabetic NOD mice to the SCG of the same animals. Dystrophic neurites were not identified in the diabetic NOD SCG (Figure 1C) ▶ . The frequency of dystrophic change in diabetic SMG-CG was three orders of magnitude greater than that in the SCG of the same animals [SMG-CG: 1.38 ± 0.51 dystrophic neurites/nucleated neuron, mean ± SEM, n = 6 mice versus SCG: 0.01 ± 0.01, P ≤ 0.0001].
STZ-Diabetic Mice
Adult male and female mice were injected with STZ, developing diabetes within a few days, and were allowed to survive for various intervals (Table 3) ▶ . The severity of hyperglycemia at the time of sacrifice was significantly less in STZ-diabetic mice than in NOD mice (compare Tables 1, 2, and 3 ▶ ).
Table 3.
Effect of Streptozotocin-Induced Diabetes on Neuritic Dystrophy as a Function of Mouse Strain, Sex, and Duration of Diabetes
| Strain | Mice (n) | Sex | Duration of diabetes | Blood glucose (mg%) | SMG-CG dystrophy (no. dystrophic neurites/no. neuron) |
|---|---|---|---|---|---|
| B6D2F1 | |||||
| Control | 8 | M | 5 months | 122 ± 18 | 0.01 ± 0.003 |
| Diabetic | 7 | 480 ± 48 | 0.30 ± 0.07* | ||
| Control | 3 | M | 12 months | 147 ± 8 | 0.08 ± 0.02 |
| Diabetic | 1 | 558 | 0.83 | ||
| C57BL6 | |||||
| Control | 5 | M | 2–3 months | 129 ± 13 | 0.039 ± 0.017 |
| Diabetic | 5 | 426 ± 23 | 0.112 ± 0.017† | ||
| Control | 4 | M | 5 months | 224 ± 10 | 0.02 ± 0.01 |
| Diabetic | 3 | 397 ± 85 | 0.18 ± 0.03† | ||
| DBA/2J | |||||
| Control | 5 | M | 2–3 months | 136 ± 6 | 0.006 ± 0.004 |
| Diabetic | 8 | 525 ± 34 | 0.128 ± 0.03* | ||
| BL6/NCR | |||||
| Control | 3 | F | 3 weeks | 142 ± 12 | 0.003 ± 0.003 |
| Diabetic | 7 | 508 ± 29 | 0.036 ± 0.005‡ |
Values represent the means ± SEM of n mice. Statistical comparison.
*p, ≤ 0.01; †p, ≤ 0.05, ‡ p, ≤ 0.001 vs. matched control group.
SMG-CG Neuropathology
Streptozotocin-diabetic mice exhibited identical lesions to those in the NOD mouse (Figure 3A-C) ▶ , although they were significantly less frequent and developed over a much longer time course than diabetic NOD mice (Table 3) ▶ .
Figure 3.
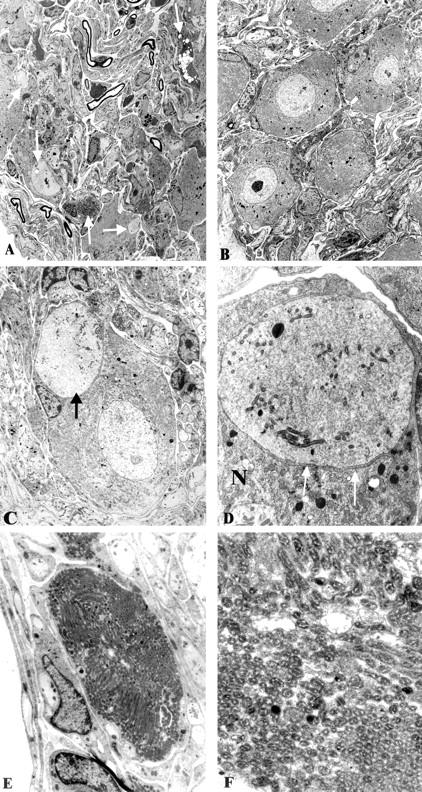
Neuritic dystrophy in the STZ-diabetic mouse is characterized by the same neuropathology as the diabetic NOD mouse, although at a significantly decreased frequency. A dystrophic neurite (arrow, A) at higher magnification (arrow, B) shows scattered tubulovesicular elements as well as an intervening satellite cell process (arrowheads, B). A more substantial accumulation of tubulovesicular elements is seen in other dystrophic neurites (arrows, C). B and C: N = neuronal cell body. Original magnifications: ×3250 (A); ×15,600 (B); ×9750 (C).
In previous studies of aged mouse SMG-CG we have found substantial differences in the development of age-related ganglionic neuritic dystrophy in various mouse strains. Determination of neuritic dystrophy in the SMG-CG of 24-month-old DBA/2 mice (0.32 ± 0.06 dystrophic neurites/nucleated neuron, mean ± SEM, n = 4) significantly exceeded that of mice of the C57BL6 strain (0.02 ± 0.007, n = 4, P ≤ 0.01). To determine whether there is a similar mouse strain effect on the frequency of neuritic dystrophy in STZ-diabetic mice, we induced diabetes in adult male C57BL6 and DBA/2 mice and sacrificed them after 2 to 3 months of diabetes. We found that both strains showed a significant and comparable effect of diabetes, but that neither strain developed neuritic dystrophy equivalent in severity to 3 to 5 week diabetic NOD mice (compare Tables 1, 2, and 3 ▶ ). Specifically, the frequency of dystrophic neurites in these STZ-diabetic mice was only ∼10% of that observed in NOD mice.
Most of our previous experiments with STZ-diabetes have used male mice and rats; 8-10,21 however, in NOD-diabetes, as in many autoimmune diseases, female animals are most consistently and severely diabetic. To try to reproduce the conditions (ie, female sex and short duration of diabetes) in the diabetic NOD mouse as closely as possible in the STZ-mouse paradigm, we examined female mice made diabetic with STZ and killed after 4 weeks of diabetes. Diabetic female mice in this experiment showed a 10-fold increase in neuritic dystrophy in SMG-CG (diabetic, 0.036 ± 0.004 dystrophic neurites/neuron, n = 7) compared to age- and gender-matched controls (0.003 ± 0.003, n = 3, P < 0.01). However, in NOD mice with a comparable duration of diabetes dystrophic changes were 35-fold more frequent (compare Table 1 ▶ and Table 3 ▶ ). Therefore, the NOD versus STZ difference in severity of dystrophic changes in SMG-CG does not simply reflect the effect of gender or short duration of diabetes.
NOD-SCID Mice
SMG-CG Neuropathology
It has been proposed that human diabetic sympathetic autonomic neuropathy may be the result of an autoimmune process which targets the nervous system. 22 To determine the role played by the immune system in the development of ganglionic neuropathology observed in the NOD mouse, we examined NOD-SCID mice before and following STZ-induced diabetes. The NOD-SCID mouse, which lacks T and B cells, fails to develop autoimmune diabetes (Table 1) ▶ .
We examined female NOD-SCID mice at the age of 25 weeks, ie, closely approximating the age of 3 to 5 week diabetic NOD mice (23 to 25 weeks). NOD-SCID mice exhibited 82% fewer dystrophic neurites than 3 to 5 week diabetic NOD mice (Table 1) ▶ of the same age. Although they developed dramatically fewer lesions in comparison to diabetic NOD mice, the frequency of neuritic dystrophy in NOD-SCID mice was significantly increased compared with non-diabetic NOD sibs (Table 1) ▶ and age-matched control mice in STZ-treatment series (Table 3) ▶ . In NOD-SCID mice dystrophic mitochondria-filled neurites accounted for 89% of dystrophic neurites (including both mitochondria A and B groups, Table 2 ▶ ) in comparison to diabetic NOD mice (Table 2) ▶ in which they represented only 49% of the total (P ≤ 0.001).
NOD-SCID Mice Made Diabetic with STZ
To selectively address the role of diabetes in NOD-SCID animals distinct from the possible pathogenetic effect of immune deficiency, NOD-SCID animals were treated with STZ at the age of 8 weeks, which resulted in a severely diabetic state (Table 4) ▶ comparable to diabetic NOD mouse and more hyperglycemic than STZ-treated mice of other strains examined (Table 3) ▶ . STZ-treated NOD-SCID mice demonstrated severe dystrophic changes (otherwise identical in ultrastructure to those developing in NOD- or STZ-treated mice) in the SMG-CG after only 2 and 4 weeks (Figures 4 and 5 ▶ , Table 4 ▶ ), which represents a dramatic amplification of the severity of neuritic dystrophy and acceleration in the time course expected in mouse STZ-diabetes. These results may reflect an exaggerated response of the NOD-SCID mouse strain, resulting in an increased baseline of neuritic dystrophy in untreated controls (compared to non-diabetic NOD, Table 1 ▶ ) and producing a dramatic increase with STZ-induction of diabetes. The diabetes-induced amplification of neuritic dystrophy in non-diabetic NOD-SCID mice may be a reflection of the genetic defect, which impairs the ability to repair double-stranded DNA breaks.
Table 4.
Effect of STZ-Diabetes on NOD-SCID Mice
| Mouse | n | Blood glucose (mg%) | SMG-CG dystrophy (no. dystrophic neurites/no. neuron) |
|---|---|---|---|
| NOD-SCID+ STZ | |||
| 2 weeks | 5 | >600 | 2.2 ± 0.3* |
| 4 weeks | 3 | >600 | 1.5 ± 0.2† |
| NOD-SCID | 4 | 97 ± 6 | 0.3 ± 0.1 |
Male NOD-SCID mice were treated with streptozotocin at the age of 8 weeks and allowed to survive for an additional 2 or 4 weeks before sacrifice. NOD-SCID controls, sibs which were not treated with streptozotocin, were sacrificed at the same time as the 2 week STZ-diabetic group. Values represent the means ± SEM of n mice. Statistical comparison:
*p, ≤ 0.01;
†,p ≤ 0.05 vs. NOD-SCID controls.
Figure 4.
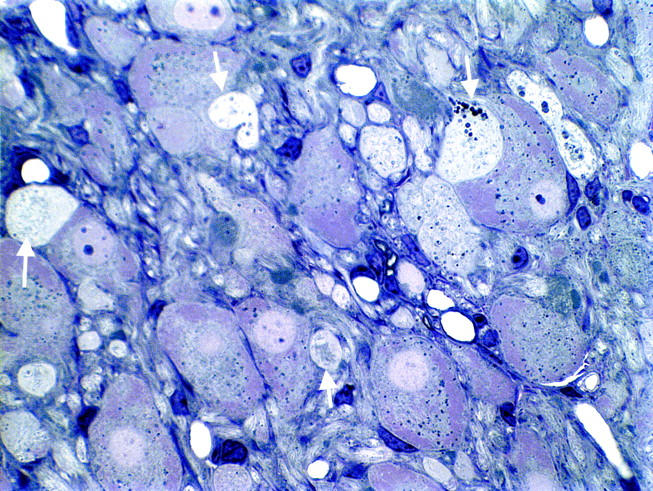
Light microscopic appearance of marked neuritic dystrophy (arrows) in the SMG-CG of a NOD-SCID mouse made diabetic with STZ 2 weeks earlier. Original magnification: ×660.
db/db Mouse
Recent studies in STZ- and Zucker diabetic fatty (ZDF)-diabetic rat models 21 have suggested that loss of the neurotrophic effects of insulin and/or IGF-I on sympathetic neurons and not hyperglycemia per se, may underlie the development of sympathetic neuritic dystrophy. To further test this idea with a mouse model, we have examined adult db/db mice diabetic for 5 months. db/db mice are hyperglycemic, obese and hyperinsulinemic. Although the degree of hyperglycemia in db/db mice was comparable to that of STZ-diabetic mice (Table 5) ▶ , db/db mice failed to develop dystrophic neurites in excess of those in non-diabetic, age-matched db/m controls (Table 5) ▶ .
Table 5.
Neuritic Sympathetic Dystrophy in the SMG-CG of db/db Type 2 Diabetic Mice
| Mouse | n | Weight (gm) | Blood glucose (mg%) | SMG-CG dystrophy (no. dystrophic neurites/ no. neuron) |
|---|---|---|---|---|
| Control | 3 | 30.5 ± 1.0 | 100 ± 10 | 0.11 ± 0.1 |
| Diabetic | 4 | 42.5 ± 1.6 | 453 ± 44 | 0.14 ± 0.03 |
The frequency of neuritic dystrophy is shown in male db/db mice diabetic for 5 months before sacrifice and age-matched littermate controls (db/m). Values represent means ± SEM for “n” mice.
Discussion
Comparison of Mouse Models of Diabetic Autonomic Neuropathy: Advantages and Disadvantages
NOD Mouse
Our studies demonstrate that the NOD mouse develops striking neuropathologic findings in the sympathetic SMG-CG comparable to those described in diabetic Chinese hamster, rat, and human subjects 7 and with a time course of a few weeks rather than the 6 to 9 month interval usually required for robust and reproducible numbers of lesions to develop in rats and Chinese hamsters. The extent of involvement of sympathetic ganglia by dystrophic pathology in NOD mice is far in excess of that seen in either STZ-treated or BB/W rats diabetic for the same period; nonetheless, the anatomical distribution and range of ultrastructural pathology are identical. The rapid development of unambiguous neuropathologic findings observed in the SMG-GC of diabetic NOD mice provides a potentially useful model for studies of diabetic autonomic neuropathy, particularly intensive therapeutic prevention or intervention studies which can be conducted over the course of a few weeks rather than months required in rat models. The degree of involvement should also facilitate electrophysiologic studies of the effect of neuritic dystrophy since in the most severely involved animals, numerous lesions are associated with individual neurons.
There are disadvantages, however, with the use of the NOD model. The NOD mouse develops diabetes spontaneously over a time course which may range from 12 to 30 weeks of age; therefore, NOD mice with the same duration of diabetes may actually represent animals which differ significantly in age which may complicate the interpretation of some age-sensitive phenomena. Unfortunately, the necessity of using the NOD background inbred strain complicates the practical application of the NOD model to the wealth of available mice with transgenic or spontaneous mutations and knockouts of selected genes which have been generated on a variety of other genetic backgrounds.
NOD-SCID Mice
NOD-SCID mice do not develop diabetes spontaneously at any time yet determined. The ability to synchronize the development of diabetes by STZ-treatment of NOD-SCID mice and their development of severe neuritic dystrophy within a few weeks represent significant advantages over the NOD model, obviating the need for weekly measurements of blood sugar values to establish the time of onset of diabetes and avoiding possible mouse-to-mouse differences in the time course from initial onset of diabetes to maximum serum glucose values. Thus, the STZ-treated NOD-SCID mouse model provides a more uniform population of animals for analysis.
The increase in the baseline frequency of neuritic dystrophy in NOD-SCID mice compared to non-diabetic NOD sibs (Table 1) ▶ and non-diabetic control mice (Table 3) ▶ suggests that NOD-SCID mice may be particularly sensitive to the development of neuritic dystrophy, a result which may reflect the direct contribution of the SCID mutation to the pathogenesis of dystrophic neurites. It is known that SCID mice exhibit a mutation in a component of the enzyme DNA-dependent protein kinase which is required for the repair of double-stranded DNA breaks. 19 As a result V(D)(J) recombination is defective resulting in defects in the generation of T and B-cell receptors and, hence, T and B cells. 23 It is possible that the inability to repair damaged DNA-related processes in sympathetic neurons may participate in the genesis of neuritic dystrophy.
Streptozotocin-Diabetic Mouse
Although STZ-diabetic mice develop fewer dystrophic neurites over a longer time course, the distribution and ultrastructural appearance of the ganglionic neuropathology are identical to lesions developing in NOD and STZ-treated NOD-SCID models, further demonstrating that the ganglionic neuritic dystrophy we have described is the result of the diabetic state. The explanation for the differences in the severity of neuritic dystrophy in NOD and STZ-models is unclear. The use of STZ to induce diabetes in mice lacking a genetic predilection for the spontaneous development of diabetes represents a significant advantage of this model over the NOD and STZ-treated NOD-SCID mouse models. Specifically, STZ-induction of diabetes can be used in mice with a variety of spontaneous mutations as well as in knockout and transgenic mice that have been developed on a variety of genetic backgrounds.
db/db Mouse
db/db mice exhibit a mutant form of the leptin receptor in the hypothalamus which results in early obesity, insulin resistance, and hyperglycemia. 24 Thus, db/db mice are known to be hyperglycemic and hyperinsulinemic. The failure of db/db mice to develop neuritic dystrophy in the presence of significant hyperglycemia suggests that hyperglycemia alone is not sufficient to produce neuritic dystrophy.
Insights into the Pathogenesis of Diabetic Autonomic Neuropathy
Mitochondrial Neuritic Dystrophy and Oxidative Stress
The demonstration of swollen neurites containing nearly pure collections of mitochondria is a prominent finding in diabetic NOD- and STZ-diabetic mice whose significance is unknown, although the dramatic amplification of the number and size of mitochondria-engorged neurites in diabetic mice suggest a possible pathogenetic role. Such collections may reflect or induce alterations in axonal transport. Collections of mitochondria may themselves produce an abnormal subcellular environment, resulting in a local exaggeration of oxidative stress and the development of self-propagating neuritic dystrophy. A prominent role for overproduction of superoxide by the mitochondrial electron transport chain has been recently proposed by Brownlee as a pathogenetic mechanism in the development of many diabetic complications. 25 It is postulated that the four main molecular mechanisms proposed in the development of diabetic neuropathy, ie, increased polyol pathway flux, formation of advanced-glycosylation endproducts (AGE), activation of protein kinase C, and the hexosamine pathway, all reflect a single hyperglycemia-induced process of mitochondrial overproduction of superoxide and the generation of a variety of downstream oxidant stressors. 25 The ability to localize sites of production of reactive oxygen species in vivo may identify dystrophic swellings as hotspots of oxidative stress.
An Autoimmune Pathogenesis for Diabetic Autonomic Neuropathy?
The presence of lymphocytic infiltrates in the celiac sympathetic ganglia of five patients with symptomatic diabetic autonomic neuropathy 6 has been repeatedly interpreted as evidence for an autoimmune pathogenesis of diabetic autonomic neuropathy. However, our autopsy series of 347 adult patients 26 showed that lymphocytic infiltration was a common alteration in human sympathetic ganglia obtained at autopsy and was neither more frequent nor of greater intensity in diabetic subjects compared to non-diabetics, although none of the subjects in this series were as symptomatic for autonomic neuropathy as those in the previous study. 6 In addition, autoantibodies directed against adrenal gland, sympathetic ganglia, and vagus nerve have been proposed in some studies to be associated with an increased incidence of autonomic neuropathy in human subjects, 22,27 although other studies have failed to find an association or have identified an inverse correlation of autoantibodies and autonomic neuropathy. 28-30 Although abnormal regional myocardial 123I-MIBG uptake (a measure of sympathetic innervation of the heart) correlated with the presence of anti-SCG autoantibodies in long-term IDDM patients, 31 examination of newly diagnosed diabetics provided little evidence that autoantibodies produce diabetic sympathetic autonomic neuropathy. 31
The NOD mouse develops anti-islet antibodies early in its life and then develops diabetes as a result of cellular autoimmune attack on pancreatic β-cells. 32,33 Recently, investigators have determined that autoimmune targeting of peri-islet Schwann cells, resulting in their degeneration before pancreatic islet β-cell death, is an early and integral part of this process. 34 We reasoned that a possible autoimmune process directed against Schwann and/or neuroendocrine cells composing pancreatic islets might also cross-react with sympathetic neurons, intraganglionic Schwann cells or perineuronal satellite cells to produce neuritic dystrophy or neuronal degeneration. However, in NOD mice 3 to 5 weeks after the onset of diabetes there was no evidence of an inflammatory infiltrate or active neuronal degeneration in SMG-CG, nor were immune effector cells localized to perineuronal sites or adjacent to dystrophic segments. To determine whether an autoimmune attack directed against sympathetic ganglia had developed and resolved during the earliest phases of diabetogenesis, we also examined the sympathetic ganglia of NOD mice at 4 and 8 weeks of age (data not shown), at which time an autoimmune attack on islets has begun but has not progressed to systemic diabetes, and failed to find evidence of an inflammatory infiltrate, active neuronal degeneration, or dystrophic changes comparable to those developing later in the SMG-CG NOD mice. In addition, the sympathetic SCG consistently failed to develop an inflammatory infiltrate, neuronal degeneration, or development of dystrophic changes in the same animals in which the SMG-CG was strikingly affected, evidence against a systemic autoimmune pathogenesis. The failure of immunodeficient NOD-SCID mice (which are incapable of mounting a B- and T-cell mediated autoimmune attack) to develop neuritic dystrophy comparable to immunologically intact NOD diabetic mice does not discriminate between the loss of a direct immune pathogenetic process affecting prevertebral sympathetic ganglia or the simple failure of the NOD-SCID animals to become diabetic. However, the subsequent STZ-induction of diabetes in NOD-SCID mice results in the rapid development of large numbers of dystrophic neurites identical to those developing in diabetic NOD mice, evidence supporting a role for diabetes-induced pathogenesis rather than an autoimmune causation. The NOD-SCID mice which did not receive STZ are normoglycemic but do show a greater degree of neuritic dystrophy than typical NOD non-diabetic controls which may reflect a difference between NOD-SCID and NOD mice in a general susceptibility to the development of neuritic dystrophy which is secondary to the genetic defect in DNA repair present in this mouse strain.
Role of Hyperglycemia in the Development of Neuritic Dystrophy
Although hyperglycemia directly results in a variety of abnormal metabolic reactions in nerve (eg, disordered polyol and phosphoinositide metabolism, increase in glycated proteins, and exaggerated oxidative stress), which may contribute to the development of neuropathy, other processes may also play a role. One such mechanism may involve the neurotrophic action of insulin or insulin-like growth factor-I (IGF-I), independent of their glycemic effects, an idea initially proposed by Ishii and colleagues in their studies of cultured neurons and somatic neuropathy in experimental diabetes. 35,36 We have previously demonstrated 37 that 6-month STZ-diabetic rats (ie, a duration of diabetes resulting in established neuroaxonal dystrophy) treated for 2 additional months with systemic recombinant human IGF-I resulted in nearly complete normalization of neuroaxonal dystrophy in the SMG and ileal mesenteric nerves in the absence of an effect on the metabolic severity of diabetes. Our recent studies of type 1 STZ-diabetic and type 2 ZDF-diabetic rat models 21 have shown marked differences in neuroaxonal dystrophy in the SMG and ileal mesenteric nerves between the hypoinsulinemic, IGF-I-deficient STZ-diabetic rat and the chronically diabetic ZDF rat which is hyperinsulinemic and has normal levels of IGF-I. 21 Future studies will determine whether the failure of chronically diabetic db/db mice to develop sympathetic neuritic dystrophy could involve a neurotrophic role for insulin and/or IGF-I or reflect an as yet unidentified diabetes-induced substance or biochemical pathway as the critical determinant.
Does Neuritic Dystrophy Produce Autonomic Dysfunction?
The prevertebral sympathetic SMG-CG represent complex peripheral integrative centers of gut reflexes in which nerve terminals ending on principal sympathetic neurons originate from neurons located in the intermediolateral column of the spinal cord, dorsal root sensory ganglia, parasympathetic ganglia, other sympathetic ganglia and include a prominent retrograde projection from intrinsic myenteric ganglia. 38 Recent studies of the alimentary tract of NOD mice have identified delayed gastric emptying (a model of diabetic “gastroparesis”), impaired electrical pacemaking, and reduced neurotransmission. 16-18 Dystrophic changes in the presynaptic axonal and postsynaptic dendritic elements may effectively isolate and disconnect sympathetic neurons in the absence of neuron loss. Although the structural changes described in NOD prevertebral ganglia could contribute to abnormal integration of alimentary reflexes and result in abnormality of gastrointestinal tract function, the issue is complicated by reported damage to the interstitial cells of Cajal within the NOD mouse gastric wall 16 and the loss of nitric oxide mediated non-adrenergic non-cholinergic (NANC) relaxation in the pylorus of the NOD mouse stomach, 17 a defect shared with the ileum. Immunohistochemical studies of NOD mouse gut also show changes in intrinsic enteric neuropeptides that vary from site to site within the gastric antrum, duodenum, and colon. 18
Summary
The studies we have reported have established several models of autonomic neuropathy developing in mice with experimental diabetes. Spontaneously diabetic NOD and STZ-induced NOD-SCID mice develop severe sympathetic ganglionic neuropathology over a short time course which may permit the rapid determination of the effect of therapeutic agents given in preventative and interventional paradigms as well as testing a variety of proposed pathogenetic mechanisms. 39 STZ-induction of diabetes in a variety of mouse strains results in the development of neuritic dystrophy similar to that in NOD and STZ-treated NOD-SCID mice, although to a lesser degree and developing over a extended time course. Nonetheless, STZ-induction of diabetes can be accomplished in mice harboring a variety of spontaneous and gene-targeted mutations superimposed on many genetic backgrounds, and may permit investigators to selectively isolate and directly address each of these pathogenetic mechanisms (and others as they evolve) to separate critical pathogenetic processes from epiphenomena and permit the development of rational forms of therapy.
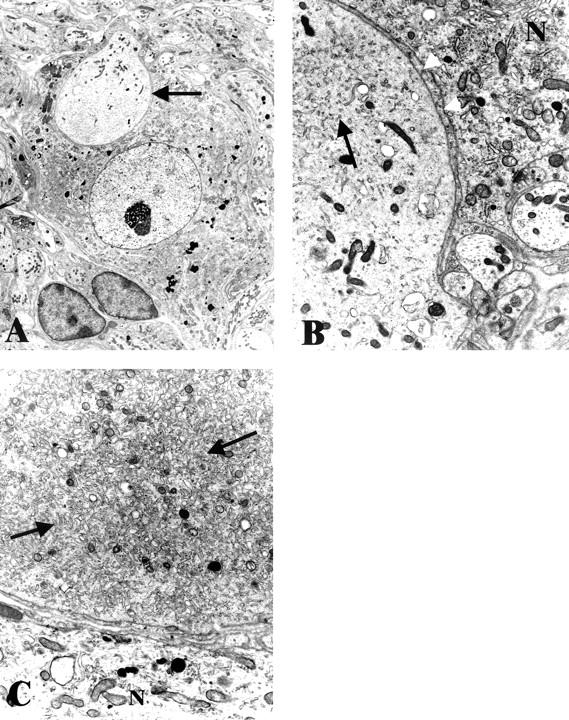
Figure 5.
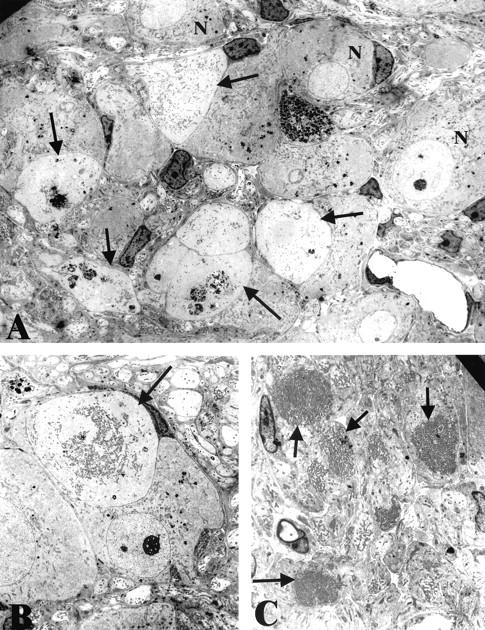
Ultrastructural appearance of NOD-SCID mice with 2 to 4 weeks of STZ-induced diabetes. A: Numerous dystrophic neurites (arrows) are intimately admixed with normal appearing neuronal cell bodies (N). Original magnification: ×2600. B: Distortion of an otherwise normal neuronal cell body by a dystrophic neurite (arrow) containing large numbers of mitochondria and tubulovesicular elements. Original magnification: ×2600. C: Numerous mitochondria-laden dystrophic neurites (arrows) in the intraganglionic neuropil. Original magnification: ×3900.
Acknowledgments
We thank Drs. Emil Unanue and Eugene M. Johnson for their critical interest and support.
Footnotes
Address reprint requests to Robert E. Schmidt, M.D., Ph.D., Department of Pathology and Immunology (Box 8118), Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail: reschmidt@pathology.wustl.edu.
Supported by National Institutes of Health awards R01 AG10299 and R37 DK19645 and a Clinical Investigator Faculty Award from the Howard Hughes Medical Institute to Washington University (to M.G.L.).
We are saddened to report the passing of Dr. Santiago Plurad, our colleague for more than 20 years, who contributed to this work.
References
- 1.Rundles RW: Diabetic neuropathy: general review with report of 125 cases. Medicine (Baltimore) 1945, 24:111-160 [Google Scholar]
- 2.Hosking DJ, Bennett T, Hampton JR: Diabetic autonomic neuropathy. Diabetes 1978, 27:1043-1055 [DOI] [PubMed] [Google Scholar]
- 3.Ewing DJ, Campbell IW, Clarke BF: The natural history of diabetic autonomic neuropathy. Q J Med 1980, 49:95-108 [PubMed] [Google Scholar]
- 4.Sampson MJ, Wilson S, Karagiannis P, Edmonds M, Watkins PJ: Progression of diabetic autonomic neuropathy over a decade in insulin-dependent diabetics. Q J Med 1990, 278:635-646 [PubMed] [Google Scholar]
- 5.Schmidt RE, Plurad SB, Parvin CA, Roth KA: The effect of diabetes and aging on human sympathetic autonomic ganglia. Am J Pathol 1993, 143:143-153 [PMC free article] [PubMed] [Google Scholar]
- 6.Duchen LW, Anjorin A, Watkins PJ, MacKay JD: Pathology of autonomic neuropathy in diabetes. Ann Intern Med 1980, 92:301-303 [DOI] [PubMed] [Google Scholar]
- 7.Schmidt RE: Neuropathology and pathogenesis of diabetic autonomic neuropathy. Tomlinson DR eds. Neurobiology of Diabetic Neuropathy. 2002:pp 267-292 Academic Press, Amsterdam [DOI] [PubMed]
- 8.Schmidt RE, Plurad SB: Ultrastructural and biochemical characterization of autonomic neuropathy in rats with chronic streptozotocin diabetes. J Neuropathol Exp Neurol 1986, 45:525-544 [DOI] [PubMed] [Google Scholar]
- 9.Schmidt RE, Johnson EM, Jr, Nelson JS: Experimental diabetic autonomic neuropathy. Am J Pathol 1981, 103:210-225 [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt RE, Plurad SB, Modert CW: Experimental diabetic autonomic neuropathy: characterization in streptozotocin-diabetic Sprague-Dawley rats. Lab Invest 1983, 49:538-552 [PubMed] [Google Scholar]
- 11.Yagihashi S, Sima AAF: The distribution of structural changes in sympathetic nerves of the BB rat. Am J Pathol 1985, 121:138-147 [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt RE, Plurad DA, Plurad SB, Cogswell BE, Diani AR, Roth KA: Ultrastructural and immunohistochemical characterization of autonomic neuropathy in genetically diabetic Chinese hamsters. Lab Invest 1989, 61:77-92 [PubMed] [Google Scholar]
- 13.Schmidt RE: Neuronal preservation in the sympathetic ganglia of rats with chronic streptozotocin-induced diabetes. Brain Res 2001, 921:256-259 [DOI] [PubMed] [Google Scholar]
- 14.Kikutani H, Makino S: The murine autoimmune diabetes model: NOD and related strains. Adv Immunol 1992, 51:285-322 [DOI] [PubMed] [Google Scholar]
- 15.Sreenan S, Pick AJ, Levisetti M, Baldwin AC, Pugh W, Polonsky KS: Increased β-cell proliferation and reduced mass before diabetes onset in the non-obese diabetic mouse. Diabetes 1999, 48:989-996 [DOI] [PubMed] [Google Scholar]
- 16.Ordog T, Takayama I, Cheung WKT, Ward SM, Sanders KM: Remodeling of networks of interstitial cells of Cajal in a murine model of diabetic gastroparesis. Diabetes 2000, 49:1731-1739 [DOI] [PubMed] [Google Scholar]
- 17.Watkins CC, Sawa A, Jaffrey S, Blackshaw S, Barrow RK, Snyder SH, Ferris CD: Insulin restores neuronal nitric oxide synthase expression and function that is lost in diabetic gastropathy. J Clin Invest 2000, 106:373-384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spangeus A, Suhr O, El-Salhy : Diabetic state affects the innervation of gut in an animal model of human type 1 diabetes. Histol Histopathol 2000, 15:739-744 [DOI] [PubMed] [Google Scholar]
- 19.Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, Jeggo PA: Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci USA 1996, 93:10285-10290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.: SAS Institute: SAS User’s Guide: The NPAR1WAY Procedure: Statistics, Version 5, Chapter 26. 1985:pp 607-614 SAS Institute Cary, NC
- 21.Schmidt RE, Dorsey DA, Beaudet LN, Peterson RG: Analysis of the Zucker diabetic fatty (ZDF) rat model suggests a neurotrophic role for insulin/IGF-I in diabetic autonomic neuropathy. Am J Pathol 2003, 163:21-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabinowe SL, Brown FM, Watts M, Smith AM: Complement-fixing antibodies to sympathetic and parasympathetic tissues in IDDM. Diabetes Care 1990, 13:1084-1088 [DOI] [PubMed] [Google Scholar]
- 23.Bosma GC, Custer RP, Bosma MJ: A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301:527-530 [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP: Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996, 84:491-495 [DOI] [PubMed] [Google Scholar]
- 25.Brownlee M: Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414:813-820 [DOI] [PubMed] [Google Scholar]
- 26.Schmidt RE: The neuropathology of human sympathetic autonomic ganglia. Microsc Res Tech 1996, 35:107-121 [DOI] [PubMed] [Google Scholar]
- 27.Muhr D, Mollenhauer U, Ziegler A-G, Maslbeck M, Standl E, Schnell O: Autoantibodies to sympathetic ganglia GAD, or tyrosine phosphatase in long term IDDM with and without ECG-based cardiac autonomic neuropathy. Diabetes Care 1997, 20:1009-1012 [DOI] [PubMed] [Google Scholar]
- 28.Husebye ES, Winqvist O, Sundkvist G, Kampe O, Karlsson FA: Autoantibodies against adrenal medulla in type 1 and type 2 diabetes mellitus: no evidence for an association with autonomic neuropathy. J Intern Med 1996, 239:139-146 [DOI] [PubMed] [Google Scholar]
- 29.Stroud CR, Heller SR, Ward JD, Hardisty CA, Weetman AP: Analysis of antibodies against components of the autonomic nervous system in diabetes mellitus. Q J Med 1997, 90:577-585 [DOI] [PubMed] [Google Scholar]
- 30.Sundkvist G, Velloso LA, Kampe O, Rabinowe SL, Ivarsson SA, Lilja B, Karlsson FA: Glutamic acid decarboxylase antibodies, autonomic nerve antibodies, and autonomic neuropathy in diabetic patients. Diabetologia 1994, 37:293-299 [DOI] [PubMed] [Google Scholar]
- 31.Schnell O, Muhr D, Dresel S, Tatsch K, Ziegler AG, Haslbeck M, Standl E: Autoantibodies against sympathetic ganglia and evidence of cardiac sympathetic dysinnervation in newly diagnosed and long-term IDDM patients. Diabetologia 1996, 39:970-975 [DOI] [PubMed] [Google Scholar]
- 32.Castano L, Eisenbarth GS: Type-1 diabetes: a chronic autoimmune disease of human, mouse, and rat. Annu Rev Immunol 1990, 8:647-679 [DOI] [PubMed] [Google Scholar]
- 33.Notkins AL, Lernmark A: Autoimmune type 1 diabetes: resolved and unresolved issues. J Clin Invest 2001, 108:1247-1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, Sampson A, Afifiyan F, Elford A, Jackowski G, Becker DJ, Santamaria P, Ohashi P, Dosch H-M: Autoimmune islet destruction in spontaneous type 1 diabetes is not β-cell exclusive. Nat Med 2003, 9:198-205 [DOI] [PubMed] [Google Scholar]
- 35.Recio-Pinto E, Rechler MM, Ishii DN: Effects of insulin, insulin-like growth factor-II, and nerve growth factor in neurite formation and survival in cultured sympathetic and sensory neurons. J Neurosci 1986, 6:1211-1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishii DN: Implication of insulin-like growth factors in the pathogenesis of diabetic neuropathy. Brain Res Rev 1995, 20:47-67 [DOI] [PubMed] [Google Scholar]
- 37.Schmidt RE, Dorsey DA, Beaudet LN, Plurad SB, Parvin CA, Miller MS: Insulin-like growth factor I reverses experimental diabetic autonomic neuropathy. Am J Pathol 1999, 155:1651-1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sejnowski TJ: Peptidergic synaptic transmission in sympathetic ganglia. Fed Proc 1982, 41:2923-2928 [PubMed] [Google Scholar]
- 39.Vinik AI, Holland MT, LeBeau JM, Liuzzi FJ, Stansberry KB, Colen LB: Diabetic neuropathies. Diabetes Care 1992, 15:1926-1975 [DOI] [PubMed] [Google Scholar]