

Abstract
Calmodulin (CaM) antagonists have been shown to inhibit tumor cell invasion and metastasis and to induce apoptosis in various tumor models, but the molecular mechanism of CaM antagonist-mediated apoptosis is poorly understood. Here, we demonstrate that interferon (IFN)-γ induces susceptibility to CaM antagonist-mediated apoptosis in human cholangiocarcinoma cells weakly expressing Fas (Fas-low cells). During CaM antagonist-mediated apoptosis in IFN-γ-pretreated Fas-low cells, cleavage of caspases-8, -9, and -3 and Bid, release of cytochrome c from the mitochondria and an increase in the free cytosolic calcium concentration were observed. CaM antagonists also caused depolarization of the mitochondrial membrane independent of caspase activation. Although a broad-range caspase inhibitor partially blocked CaM antagonist-mediated apoptosis, the neutralizing Fas antibody had no effect, suggesting that CaM antagonist-mediated apoptosis does not require interaction between CaM antagonists and surface Fas. CaM antagonists induce apoptosis via mechanisms other than inhibition of CaM-dependent protein kinase II and calcineurin, as their inhibitors, KN93 and cyclosporine A, had no effect on apoptosis. Taken together, these results indicate that CaM antagonists induce apoptosis in both caspase-dependent and -independent manners, and that susceptibility to CaM antagonists is modulated by IFN-γ. The combination of IFN-γ and CaM antagonists, including tamoxifen, may be a potential therapeutic modality for cholangiocarcinoma and possibly other malignancies.
Apoptosis, a form of programmed cell death, plays an essential role in embryonic development and maintenance of cellular and tissue homeostasis. 1 Enhanced or diminished apoptosis is associated with many human diseases including neurodegenerative and autoimmune disorders, AIDS, and cancers. Cells from a wide variety of human malignancies show a decreased ability to undergo apoptosis in response to various stimuli, which may contribute to the clonal expansion of cancer cells. 2 Decreased apoptosis of tumor cells results from either a deficiency of proapoptotic molecules or expression of inhibitors of apoptotic pathways. Therefore, understanding and modulating apoptotic pathways in tumor cells may provide a potential for therapeutic intervention.
Apoptosis is most commonly regulated by the caspases, the cysteine proteases with specificity for aspartic acid residues. 3 The initiator caspases, such as caspase-8 and caspase-10, are activated through induced proximity on ligand binding to death receptors, such as Fas (APO-1/CD95), which can then cleave and activate the executioner caspases such as caspase-3 and caspase-7 4 that cleave a variety of proteins, thus, killing the cells. In response to extracellular cues and/or internal insults such as DNA damage, the mitochondrial death pathway is also involved. At the mitochondria, pro- and anti-apoptotic Bcl-2 family proteins regulate the release of cytochrome c that associates with Apaf-1 and activates caspase-9. Crosstalk between death receptors and the mitochondrial pathway is mediated by Bid, a proapoptotic Bcl-2 family protein. 4 Recently, there is increasing evidence suggesting the existence of caspase-independent programmed cell death. 5,6 Despite lack of caspase activation, dying cells present several characteristics of apoptosis, ie, rounding, shrinkage, and detachment of cells as well as DNA fragmentation. 7 Overexpression of c-myc, and treatment with some pharmacological agents, such as dexamethasone, induce cell death in the presence of caspase inhibitors. 8,9 The release of endonuclease G from the mitochondria and its translocation to the nucleus is also known to induce DNA fragmentation and cell death, independent of caspases. 10
Calmodulin (CaM) is an 18-kd multifunctional protein and is the major intracellular Ca2+-binding protein. The molecule consists of two globular lobes connected by a long exposed α-helix. Two calcium ions bind to each lobe through helix-loop-helix domains similar to those of other calcium-binding proteins. The long helix between the lobes is involved in the interactions of CaM with proteins and drugs. 11 Various aromatic molecules have been found to act as CaM antagonists, including the phenothiazine drugs such as the anti-psychotic trifluoperazine (TFP), 12 the anti-estrogen tamoxifen (TMX), 13,14 some naphthalenesulfonamide derivatives such as N-(6-aminohexyl)-5-chloro-1-naphthalene sulfonamide (W7), 15 and cationic derivatives of phenyl-substituted thiazole. 16 CaM not only mediates the effects of changes in cytoplasmic Ca2+ concentration ([Ca2+]c), but is also involved in the control of [Ca2+]c by regulating the activity of Ca2+ pumps and channels, such as the plasma membrane Ca2+ pumps, ryanodine receptors, 17 inositol 1,4,5-triphosphate receptors, 18 cyclic nucleotide-gated Ca2+ channels 19 and Ca2+ channels encoded by the trp gene. 20
Because of the importance of Ca2+ in progression through the cell cycle, CaM also plays a critical role in the regulation of cell proliferation. 21,22 It has been reported that diseases characterized by pathological, unregulated cell growth, such as cancer, are associated with elevated levels of Ca2+-bound CaM. 23,24 In addition, CaM antagonists have been shown to inhibit tumor cell invasion in vitro 25 and metastasis in vivo, 26 suggesting that the CaM antagonists are promising chemotherapeutic agents for malignancies.
Several studies have implicated CaM in mediating apoptosis. Induction of CaM gene expression was observed during glucocorticoid- and Fas-mediated apoptosis. 27,28 In CD4+ T cells from patients with AIDS, the CaM antagonist, TFP, protects cells from apoptosis. 29 In contrast, it induces apoptosis in cancer cell lines. 30 Growth inhibition and induction of apoptosis by TMX, an anti-estrogen that is also a potent CaM inhibitor, have been observed in human cancers and cancer cell lines. 30-32 However, the molecular mechanisms of CaM antagonist-mediated apoptosis remain poorly understood.
Cholangiocarcinoma is a highly malignant, generally fatal neoplasm originating from the bile duct epithelial cells or cholangiocytes of the intra- and extrahepatic biliary system. 33 This tumor has an increasingly frequent diagnosis worldwide, but information about the molecular pathogenesis of cholangiocarcinoma is lacking. In this regard, curative therapeutic intervention is limited by the advanced disease stage of most patients at initial presentation and the lack of effective chemotherapy. 34 The overall survival after diagnosis of cholangiocarcinoma is less than 12% at 5 years. 35,36 Previously, we reported that a human cholangiocarcinoma cell line, Sk-ChA-1, expresses Fas heterogeneously resulting in two subpopulations, Fas-high cells that strongly express Fas, and Fas-low cells that weakly express Fas. 30 Fas-high cells are sensitive to Fas-mediated apoptosis and are nontumorigenic in nude mice, whereas Fas-low cells are completely resistant to Fas-mediated apoptosis and are tumorigenic. Furthermore, the CaM antagonists, TMX and TFP, induce apoptosis in Fas-high cells, but not in Fas-low cells. 30 We also reported that the cytokine, interferon (IFN)-γ, up-regulates a broad range of apoptosis-related molecules and renders Fas-low cholangiocarcinoma cells sensitive to Fas-mediated apoptosis. 37
Here, we show that IFN-γ sensitizes Fas-low cholangiocarcinoma cells to CaM antagonist-mediated apoptosis. Furthermore, we describe the molecular events that occur during CaM antagonist-mediated apoptosis in IFN-γ-pretreated cells, which are in many aspects similar to those of Fas-mediated apoptosis.
Materials and Methods
Cells and Cell Culture
The human cholangiocarcinoma cell line, Sk-ChA-1 was provided by Dr. A. Knuth (Ludwig Institute for Cancer Research, London, UK). This cell line was separated into the two subpopulations, Fas-high and Fas-low cells as described previously. 30 Cells were grown in RPMI 1640 (Life Technologies, Inc., Gaithersburg, MD) supplemented with 2 mmol/L of l-glutamine, penicillin (5 U/ml), streptomycin (5 μg/ml), and 10% heat-inactivated fetal bovine serum.
Antibodies and Reagents
Tamoxifen (TMX), TFP, and W7 were obtained from Sigma (St. Louis, MO). Recombinant human IFN-γ was purchased from R&D Systems (Minneapolis, MN). Human activating Fas Ab (CH11) and neutralizing Fas Ab (ZB4) were obtained from Upstate Biotechnology (Lake Placid, NY). Antibodies to caspase-8, caspase-9, caspase-3, and Bid were purchased from Cell Signaling Technology (Beverly, MA), and the cytochrome c antibody was from PharMingen (San Diego, CA). The caspase inhibitor, z-VAD-fmk [benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone], cyclosporine A, KN-93, and valinomycin were obtained from Calbiochem (La Jolla, CA). 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and Indo-1 acetoxymethyl ester were from Molecular Probes (Eugene, OR).
Preparation of Whole Cell Lysates and Cytosolic Extracts
For whole cell lysates, cells were washed with phosphate-buffered saline (PBS) and lysed in sodium dodecyl sulfate lysis buffer (100 mmol/L Tris-HCl, pH 8.0, 150 mmol/L NaCl, 1% sodium dodecyl sulfate, 10% glycerol, 5 mmol/L EDTA, 5 mmol/L EGTA, 2 mmol/L phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin and leupeptin). To extract cytosolic proteins for detection of cytochrome c release, cells (4 × 106) were harvested and washed twice with ice-cold PBS and resuspended in 300 μl of ice-cold buffer (20 mmol/L HEPES-KOH, pH 7.0, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 250 mmol/L sucrose, 1 μg/ml of leupeptin and pepstatin, 2 μg/ml of aprotinin). After incubation on ice for 15 minutes, cells were homogenized with a Dounce homogenizer (B pestle/25 strokes) and centrifuged at 1000 × g for 10 minutes to separate nuclei and unbroken cells. The supernatants were centrifuged at 14,000 × g for 15 minutes in a microcentrifuge to pellet membranes including mitochondria. The resulting supernatants were used as cytosolic extracts.
Western Blotting
Whole cell lysates or cytosolic extracts (20 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to Immobilon P membranes (Millipore, Bedford, MA). Membranes were blocked in 2% nonfat milk and incubated with primary antibodies, followed by incubation with anti-mouse or anti-rabbit horseradish peroxidase-conjugated antibodies (Amersham Pharmacia Biotech, Piscataway, NJ). Blots were developed using enhanced chemiluminescence Western blotting detection reagents (Amersham Pharmacia Biotech).
Detection of Apoptotic Cell Death
Apoptosis was determined using annexin V and propidium iodide (PI) staining using an annexin V-fluorescein isothiocyanate apoptosis detection kit (Medical & Biological Laboratories, Nagoya, Japan). After incubations as indicated in legends, 1 × 105 cells were harvested and resuspended in 200 μl of binding buffer (Medical & Biological Laboratories). Annexin V-fluorescein isothiocyanate and PI were added, followed by incubation at room temperature for 5 minutes. Annexin V binding and PI staining were analyzed by flow cytometry (FACSCalibur; Becton Dickinson, Mountain View, CA).
Measurement of the Mitochondrial Membrane Potential
Cells (1 to 2 × 105) were trypsinized and resuspended in Hanks’ balanced salt solution (Life Technologies, Inc., Grand Island, NY). Then, cells were loaded with 10 μg/ml of JC-1 at 37°C for 15 minutes and washed twice with Hanks’ balanced salt solution. Samples were analyzed by flow cytometry (FACScalibur). JC-1 monomers and aggregates were assessed by excitation at 488 nm and measurement of emission at 525 nm and 590 nm, respectively.
Measurement of the Free Cytosolic Calcium Concentration
Cells were pelleted and resuspended in Hanks’ balanced salt solution containing 0.1% bovine serum albumin, then loaded with 2 μmol/L of Indo-1 acetoxymethyl ester (Molecular Probes, Eugene, OR) for 30 minutes at room temperature. After washing twice, cells were analyzed in a spectrofluorometer (Photon Technology International, Lawrenceville, NJ) at 338 nm excitation and alternating emission wavelengths of 405 and 485 nm. Free cytosolic Ca2+ concentrations were calculated according to the equation of Grynkiewicz and colleagues; 38 [Ca2+]c = Kd × (R − Rmin)/(Rmax −R) × Sf2/Sb2, where Kd is the Indo-1 dissociation constant for Ca2+ (250 nmol/L), R is the ratio of the intensities at 405 nm and 485 nm, and Rmin and Rmax are the R values at 0 and saturating levels of Ca2+, respectively. Sf2/Sb2 is the ratio of the intensities at 485-nm emission under Rmin and Rmax conditions.
Statistical Analysis
Results are expressed as mean ± SE. All statistical analysis was performed using Microsoft Excel software (Microsoft). Statistical significance was determined using the two-tailed unpaired t-test. P < 0.05 was considered significant.
Results
IFN-γ Enhances CaM Antagonist-Mediated Apoptosis in Cholangiocarcinoma Cells
Our previous study demonstrated that subpopulations of the Sk-ChA-1 human cholangiocarcinoma cell line show different sensitivities to Fas- and CaM antagonist-mediated apoptosis; Fas-high cells (cells expressing high levels of Fas) are sensitive to both Fas- and CaM antagonist-mediated apoptosis, but Fas-low cells (cells expressing low levels of Fas) are resistant. 30 We also showed that IFN-γ up-regulates Fas and other apoptosis-related molecules and enhances Fas-mediated apoptosis in both Fas-high and Fas-low cells. 37 We were thus interested in investigating the effect of IFN-γ on CaM antagonist-mediated apoptosis. Three CaM antagonists, TMX, TFP, and W7, were used. One of these, TMX, has been shown to be anti-tumorigenic for cholangiocarcinoma. 31 Although TMX is known as an anti-estrogen, it is also a CaM antagonist. 13 Additionally, our cholangiocarcinoma cell line is estrogen receptor-negative, 31 and the pure anti-estrogen, ICI 182780, does not induce apoptosis in this cell line, 30 suggesting that TMX induces apoptosis by its anti-CaM action. We first pretreated both Fas-high and Fas-low cholangiocarcinoma cells with 250 U/ml of IFN-γ or medium alone for 18 hours, IFN-γ was removed, and then TMX (10 μmol/L), TFP (15 μmol/L), or W7 (50 μmol/L) were added. In 20 hours, apoptotic cells were detected by flow cytometry after annexin V/PI staining. Activating Fas antibody (Ab) (CH11)-treated cells were also analyzed for comparison. Figure 1A ▶ is representative data from flow cytometric analysis after annexin V/PI staining, and the results of six separate experiments are summarized in Figure 1B ▶ . The results show that Fas-low cells are resistant or only slightly sensitive to apoptosis, whereas Fas-high cells undergo apoptosis when challenged with either CaM antagonists or activating Fas Ab. However, when pretreated with IFN-γ, Fas-low cells also showed susceptibility to CaM antagonists and activating Fas Ab. IFN-γ also enhanced the sensitivities to CaM antagonists and Fas Ab in Fas-high cells. Induction of apoptosis by CaM antagonists in IFN-γ-pretreated cells is more effective in Fas-high cells than in Fas-low cells (Figure 1, A and B) ▶ , suggesting that there are defects in apoptotic signaling pathways in Fas-low cells, as we discussed in our previous report. 37 Because Fas-high cells respond to CaM antagonists and Fas Ab, even in the absence of IFN-γ, 30 we were more interested in apoptosis induced by combined treatment with IFN-γ and CaM antagonists in Fas-low cells, which are completely resistant to apoptosis if not pretreated with IFN-γ. Therefore, the remainder of these studies was focused on the effects of combined treatment with IFN-γ and CaM antagonists in Fas-low cells.
Figure 1.
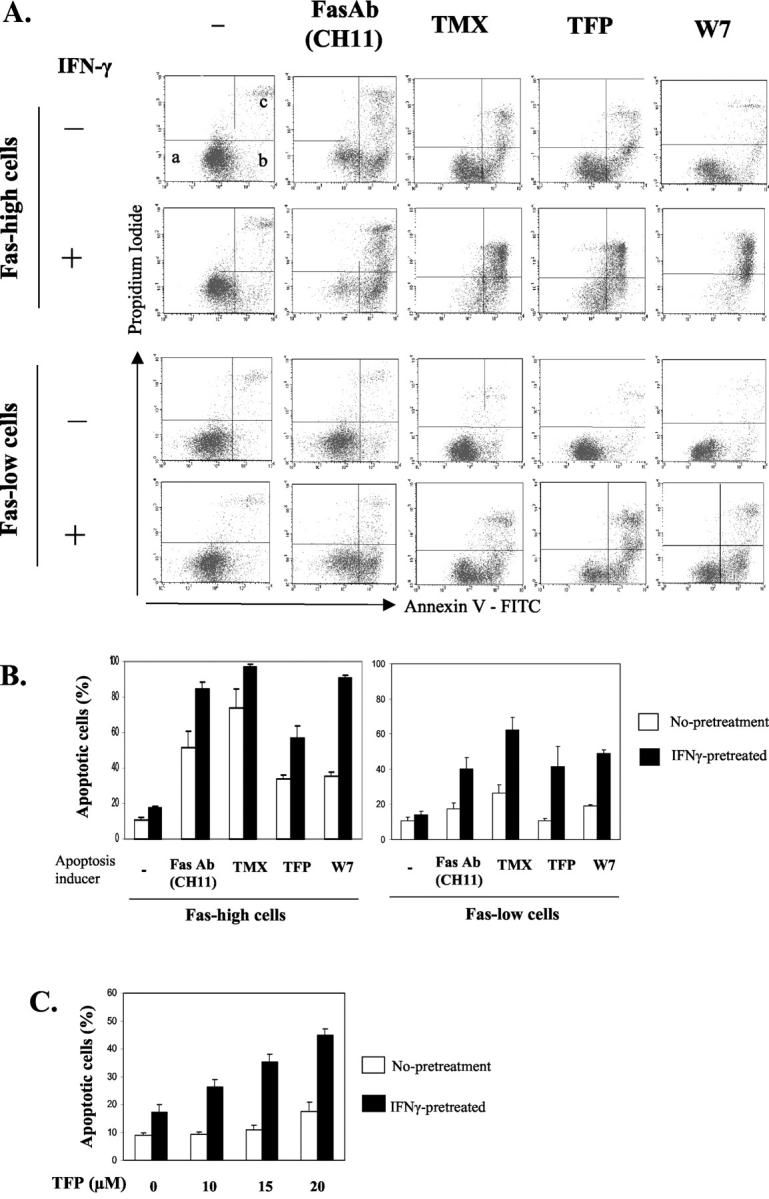
IFN-γ enhances CaM antagonist-mediated apoptosis in cholangiocarcinoma cells. A: Fas-high and Fas-low Sk-ChA-1 cholangiocarcinoma cells were incubated with IFN-γ (250 U/ml) or medium alone for 18 hours followed by an additional 20 hours of incubation with activating Fas antibody (Fas Ab, CH11, 250 ng/ml) or the three CaM antagonists, TMX (10 μmol/L), TFP (15 μmol/L), and W7 (50 μmol/L). Apoptotic cell death was analyzed by flow cytometry after staining with fluorescein isothiocyanate-conjugated annexin V and PI. Cells in the different quadrants represent: a, viable cells; b, early apoptotic cells; c, late apoptotic/necrotic cells. B: Mean percentage of apoptotic cells ± SE (n = 6). Annexin V-positive cells (cells in area b and c) were counted as apoptotic cells. C: Different concentrations of TFP were added to Fas-low Sk-ChA-1 cholangiocarcinoma cells (Fas-low cells), with or without IFN-γ-pretreatment. Apoptotic cells were counted and graphed as described in B (n = 4).
The effect of IFN-γ pretreatment was then tested at different concentrations of TFP. Nonpretreated Fas-low cholangiocarcinoma cells undergo apoptosis only slightly at 20 μmol/L TFP, but none at 10 or 15 μmol/L TFP. In contrast, IFN-γ-pretreated Fas-low cells have a slight increase in apoptosis in the absence of TFP, and show a dose-dependent enhancement of apoptosis at all concentrations of TFP tested (10, 15, and 20 μmol/L) (Figure 1C) ▶ .
The synergism of IFN-γ and CaM antagonists in the induction of apoptosis was also observed in the pancreatic carcinoma cell lines, HPAC and HPAF-II. Although IFN-γ, TMX, or TFP alone did not induce apoptosis, combined treatment with IFN-γ and TMX increased apoptosis by 80 ± 10% (data not shown). The combination of IFN-γ and TFP also effectively induced apoptosis in these pancreatic carcinoma cell lines (data not shown).
CaM Antagonists Induce Cleavage of Caspases and Bid, and Release of Cytochrome c in IFN-γ-Pretreated Cells
We previously showed that enhanced Fas-mediated apoptosis in IFN-γ-pretreated cells results from an increase in apoptosis-related molecules such as Fas, caspases, and Bak, which facilitate caspase cleavage and cytochrome c release from the mitochondria to the cytosol. 37 Because IFN-γ renders Fas-low cholangiocarcinoma cells sensitive to both activating Fas Ab and CaM antagonists, the downstream mechanisms of CaM antagonist-mediated apoptosis were investigated. Caspase-8, one of the key initiator caspases, is cleaved by a two-step mechanism. Initial cleavage generates a p43 and a p12 fragment. Subsequent cleavage of the p43 generates a prodomain, p26, and an active fragment, p18. 39 To determine caspase-8 activation, Fas-low cells were pretreated with IFN-γ or medium alone for 18 hours, then stimulated with activating Fas Ab, TMX, or TFP for 12 hours. Western blot analysis of cell lysates reveals cleavage of caspase-8 producing the intermediate products p41/43 and the active fragment p18 only when cells were pretreated with IFN-γ and challenged with Fas Ab, TMX, or TFP (Figure 2) ▶ . In nonpretreated cells, only weak p41/43 bands were detected and p18 fragments were undetectable.
Figure 2.

CaM antagonists induce cleavage of caspases-8, -9, and -3; cleavage of Bid; and release of cytochrome c in IFN-γ-pretreated cells. Fas-low cells were incubated with IFN-γ (250 U/ml) or medium alone for 18 hours followed by an additional 12 hours of incubation with activating Fas Ab (CH11, 250 ng/ml), TMX (10 μmol/L), or TFP (15 μmol/L). To detect caspase and Bid cleavage, whole cell lysates were prepared and immunoblotted for caspases or Bid. For the detection of cytochrome c release, cytosolic extracts were prepared and immunoblotted for cytochrome c.
We were also interested in the involvement of the mitochondria in CaM antagonist-mediated apoptosis. Among the proapoptotic Bcl-2 family, Bid, a BH3-only member, connects the death receptor pathway and the mitochondrial pathway. Bid is a direct substrate of caspase-8 and is activated by the Fas/TNFR-1 pathway. Cleaved C-terminal Bid translocates to the mitochondria and potently induces cytochrome c release. 40,41 We found that the CaM antagonists, TMX and TFP, induce cleavage of Bid, producing truncated fragments (p15), and cause release of cytochrome c from the mitochondria to the cytosol (Figure 2) ▶ . Without IFN-γ pretreatment, CaM antagonists, as well as activating Fas Ab alone, did not induce Bid cleavage or cytochrome c release, consistent with the resistance to apoptosis of these Fas-low cells.
Because released cytochrome c leads to the formation of an Apaf-1/caspase-9 complex, in the presence of ATP, and subsequent activation of caspase-9, 42 we next assessed caspase-9 activation. Western blot analysis shows the generation of cleaved caspase-9 fragments by treatment with the CaM antagonists, TMX and TFP, in IFN-γ-pretreated cells (Figure 2) ▶ . In addition, parallel effects on caspase-3 activation were shown by an increase in the 19/17-kd cleaved form of caspase-3 (Figure 2) ▶ . These observations demonstrate that combined treatment with IFN-γ and CaM antagonists induces apoptosis by mechanisms similar to those involved in Fas-mediated apoptosis, involving caspase activation and cytochrome c release.
CaM Antagonists Cause the Loss of Mitochondrial Membrane Potential in IFN-γ-Pretreated Cells
Although the mechanism by which cytochrome c is released is still controversial, an associated event is the collapse of the mitochondrial inner membrane potential, indicating the opening of a large conductance channel, known as the mitochondrial permeability transition pore. 43 To investigate whether a mitochondrial membrane potential change is involved in apoptosis induced by CaM antagonists after IFN-γ treatment, Fas-low cells were pretreated with IFN-γ or medium alone, stimulated with Fas Ab, TMX, TFP, or W7, and then loaded with JC-1. JC-1 is a cationic dye that is commonly used to detect the mitochondrial membrane potential. JC-1 displays two major emission peaks (590 nm and 525 nm, with excitation at 488 nm) that correspond to red-fluorescent J-aggregates and green fluorescent monomers, respectively. Because, this dye forms aggregates in polarized mitochondria and exists as monomers in depolarized mitochondria, a decreased ratio of red/green fluorescence intensity indicates mitochondrial depolarization. Therefore, the detection of the fluorescence ratio allows us to determine the percentage of depolarized mitochondria within the population. 44 In the flow cytometric data shown in Figure 3 ▶ , the upper left quadrant represents polarized mitochondria, the lower right represents depolarized mitochondria, and the upper right represents an intermediate stage. Treatment with the potassium ionophore, valinomycin, serves as a positive control causing depolarization of the mitochondrial membrane. Figure 3A ▶ is representative of three different experiments and the numbers represent the mean percentage of cells in each quadrant. The three CaM antagonists, TMX, TFP, and W7, as well as activating Fas Ab, induced mitochondrial depolarization when cells were pretreated with IFN-γ, while valinomycin produces the same effect in both nonpretreated and IFN-γ-pretreated cells (Figure 3A) ▶ . IFN-γ treatment alone does not induce depolarization of the mitochondrial membrane; the majority of cells (75.6%) maintains a high mitochondrial membrane potential similar to a nontreated control group (77.5%). These results indicate that induction of susceptibility to CaM antagonists in IFN-γ-pretreated cells is accompanied by the loss of the mitochondrial membrane potential, possibly resulting in cytochrome c release.
Figure 3.
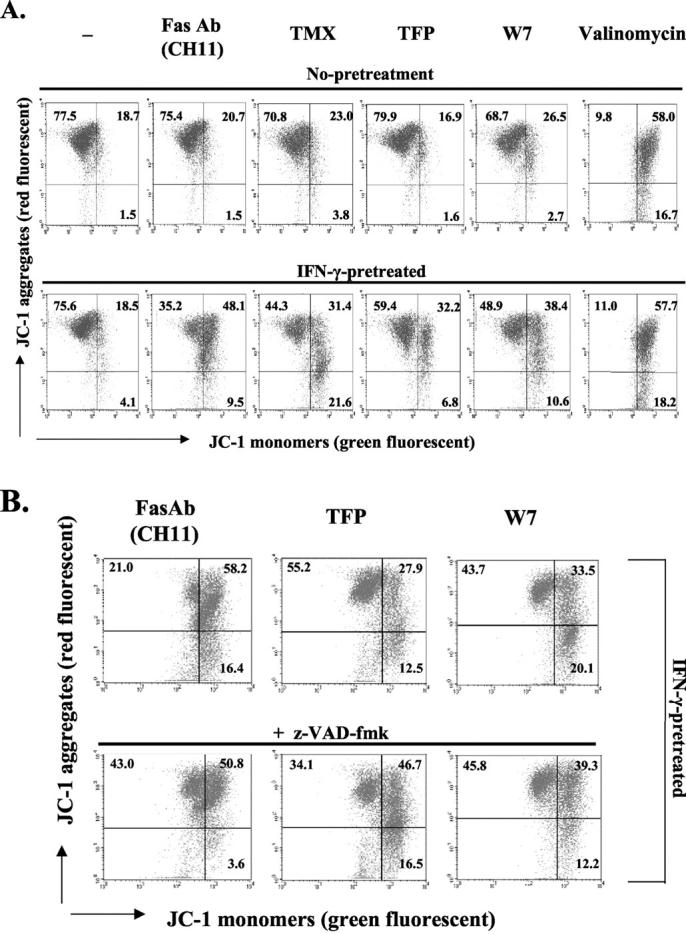
CaM antagonists induce depolarization of the mitochondrial membrane in IFN-γ-pretreated cells. A: Fas-low cells were incubated with IFN-γ (250 U/ml) or medium alone (no pretreatment) for 18 hours, followed by an additional 12 hours of incubation with activating Fas Ab (CH11, 250 ng/ml), TMX (10 μmol/L), TFP (15 μmol/L), or W7 (50 μmol/L). Exposure of cells to valinomycin (500 nmol/L) served as a positive control. B: IFN-γ-pretreated (for 18 hours) Fas-low cells were treated with z-VAD-fmk (100 μmol/L) or medium alone for 1 hour, and then stimulated with Fas Ab (CH11), TFP, or W7 for 15 hours. Mitochondrial membrane potential was measured with JC-1 dye as described in Materials and Methods. Cells in the top left quadrant exhibit high mitochondrial membrane potential, while cells in the bottom right quadrant exhibit low mitochondrial membrane potential and top right quadrant represent an intermediate stage. Shown is a representative result of three independent experiments and the numbers in each quadrant represent the average percentage of cells in that quadrant.
To investigate whether depolarization of the mitochondrial membrane is a caspase-dependent event, we pretreated IFN-γ-primed cells with or without 100 μmol/L z-VAD-fmk, a broad-range caspase inhibitor, before additions of activating Fas Ab, TFP, or W7. JC-1 staining of the cells showed that z-VAD-fmk did not block CaM antagonist-mediated depolarization of the mitochondria, while Fas-mediated depolarization was partially inhibited (Figure 3B) ▶ .
CaM Antagonists Cause an Increase in the Free Cytosolic Calcium Concentration in IFN-γ-Pretreated Cells
Alteration of intracellular Ca2+ homeostasis has been implicated in apoptosis. Specific Ca2+-channel blockers abrogate apoptosis in prostate cells 45 and Ca2+-mobilizing agents (eg, Ca2+ ionophores or the endoplasmic/sarcoplasmic reticulum Ca2+-ATPase pump inhibitor, thapsigargin) can induce apoptosis in many cell types. 46-48 Moreover, the CaM antagonists, TMX, TFP, and W7, have been shown to increase free cytosolic Ca2+ concentration ([Ca2+]c), 32,49-51 suggesting a possible involvement of increased [Ca2+]c during CaM antagonist-mediated apoptosis. To investigate whether IFN-γ pretreatment affects CaM antagonist-mediated [Ca2+]c change, we measured [Ca2+]c after the addition of CaM antagonists to IFN-γ-pretreated or nonpretreated Fas-low cells. In the unstimulated state, the basal [Ca2+]c of IFN-γ-pretreated cells (177 ± 4 nmol/L) is slightly, but significantly, higher than that of nonpretreated cells (150 ± 7 nmol/L, Figure 4 ▶ , at time point 0 in all panels). When cells were not pretreated with IFN-γ, [Ca2+]c was not changed by CaM antagonists or activating Fas Ab treatment (Figure 4 ▶ , open circles), which is consistent with resistance to apoptosis. In contrast, in IFN-γ-pretreated cells, the three CaM antagonists, TMX, TFP, W7 and activating Fas Ab increased [Ca2+]c by 70%, 54%, 78%, and 100%, respectively, at 8 hours. At 4 hours, only activating Fas Ab increased [Ca2+]c significantly (Figure 4) ▶ and neither CaM antagonists nor activating Fas Ab triggered immediate [Ca2+]c change (data not shown). These data indicate that IFN-γ treatment results in cells that respond to CaM antagonists, as well as activating Fas Ab, with a slow, prolonged increase in [Ca2+]c.
Figure 4.

CaM antagonists increase the free cytosolic calcium concentration in IFN-γ-pretreated cells. Fas-low cells were pretreated with IFN-γ (250 U/ml, filled circles) or medium alone (open circles) for 18 hours and activating Fas Ab (CH11, 250 ng/ml), TMX (10 μmol/L), TFP (15 μmol/L), or W7 (50 μmol/L) were added. After 4 and 8 hours, cells were loaded with Indo-1-Indo-1 acetoxymethyl ester and the free cytosolic calcium concentration was measured and calculated as described in Materials and Methods. Error bars represent SE. *, P = 0.01 (n = 8). **, P < 0.05 compared with time point 0 (n = 4).
Neutralizing Fas Antibody Can Not Block CaM Antagonist-Mediated Apoptosis
Previous data suggest that CaM antagonist-mediated apoptosis correlates with the presence or amount of Fas. Fas-high cells are more sensitive to CaM antagonists than Fas-low cells, 30 and IFN-γ treatment induces Fas expression 37 and the concomitant susceptibility to CaM antagonists (Figure 1) ▶ . We were thus interested in determining whether the interaction of CaM antagonists with surface Fas is required for CaM antagonists to stimulate apoptosis. In addition, it has been reported that cytotoxic drugs commonly used in cancer therapy can induce apoptosis through enhanced expression of Fas 52 and FasL, 53 resulting in Fas-FasL interaction. Because up-regulated Fas in IFN-γ-treated cells might interact with FasL that is expressed in Fas-low cells (unpublished observation), it is important to determine whether the Fas-FasL interaction is necessary to initiate apoptosis. To investigate this possibility, Fas-low cells were grown in the presence of IFN-γ for 18 hours, and pretreated with neutralizing Fas Ab (ZB4) for 2 hours before the addition of activating Fas Ab, TMX, or TFP. Neutralizing Fas Ab almost completely blocked apoptosis induced by activating Fas Ab (CH11), but had no effect on TMX- or TFP-mediated apoptosis (Figure 5) ▶ . These results indicate that apoptosis induced by CaM antagonists, after IFN-γ treatment, does not involve either the interaction between surface Fas and the CaM antagonist or between surface Fas and FasL.
Figure 5.

Neutralizing Fas antibody does not block CaM antagonist-mediated apoptosis. IFN-γ-pretreated (250 U/ml, 18 hours) Fas-low cells were incubated without (control) or with neutralizing Fas antibody (ZB4, 2 μg/ml) for 2 hours before the addition of activating Fas Ab (CH11, 250 ng/ml), TMX (10 μmol/L), or TFP (15 μmol/L). After 20 hours, apoptotic cell death was determined as described in Figure 1 ▶ . Shown are means ± SE (n = 4).
Caspase Inhibitor Partially Blocks CaM Antagonist-Mediated Apoptosis
Because caspases-8, -9, and -3 are cleaved during IFN-γ/CaM antagonist-mediated apoptosis (Figure 2) ▶ , we investigated the effect of a caspase inhibitor on apoptosis. Fas-low cells were first treated with IFN-γ for 18 hours, and incubated with 100 μmol/L of z-VAD-fmk, a broad-range caspase inhibitor, for 2 hours before the addition of activating Fas Ab, TMX, or TFP. Analysis of apoptotic cells revealed that z-VAD-fmk partially inhibits activating Fas Ab-, TMX-, and TFP-mediated apoptosis (Figure 6) ▶ . Because IFN-γ up-regulates several caspases, 37 it is possible that 100 μmol/L of z-VAD-fmk is not sufficient for complete inhibition, resulting in partial inhibition. To investigate this possibility, we used 200 μmol/L of zVAD-fmk and found that it still inhibits apoptosis only partially (data not shown). We also used Fas-high Sk-ChA-1 cholangiocarcinoma cells that are sensitive to both activating Fas Ab and CaM antagonists, even without IFN-γ pretreatment. 30 In nonpretreated Fas-high cells, 100 μmol/L of zVAD-fmk inhibits Fas-mediated apoptosis completely, while it only partially inhibits TMX- and TFP-mediated apoptosis (data not shown), suggesting that CaM antagonists induce cell death through both caspase-dependent and -independent pathways.
Figure 6.

Caspase inhibitor partially blocks CaM antagonist-mediated apoptosis. IFN-γ-pretreated (250 U/ml, 18 hours) Fas-low cells were incubated without (control) or with z-VAD-fmk (100 μmol/L) for 2 hours before the addition of activating Fas Ab (CH11, 250 ng/ml), TMX (10 μmol/L), or TFP (15 μmol/L). In 20 hours, apoptotic cell death was determined as described in Figure 1 ▶ . Shown are means ± SE (n = 5). *, P < 0.008.
CaM Antagonists Induce Apoptosis in IFN-γ-Pretreated Cells Independent of CaMKII and Calcineurin
CaM-dependent protein kinase II (CaMKII) is a major mediator of cellular Ca2+ effects, and has been implicated in both apoptosis 54,55 and survival. 56 Calcineurin, a serine/threonine protein phosphatase, has also been demonstrated to induce apoptosis or inhibit apoptosis through NF-AT activation 57 and dephosphorylation of Bad. 58 Because both CaMKII and calcineurin are CaM-dependent enzymes, it is possible that CaM antagonists induce apoptosis through inhibition of CaMKII or calcineurin activation. To determine whether inhibition of CaMKII or calcineurin induces apoptosis, we treated IFN-γ-primed cells with KN93, a CaMKII inhibitor, or cyclosporine A (CysA), a calcineurin inhibitor. Results show that neither KN93 (10 μmol/L) nor CysA (1 μmol/L) induced apoptosis, while TFP (15 μmol/L) did (Figure 7) ▶ , suggesting that the CaM antagonists induce apoptosis, in IFN-γ-pretreated cells, by mechanisms other than inhibition of CaMKII or calcineurin.
Figure 7.

KN93 and cyclosporine A do not either induce apoptosis or affect TFP-mediated apoptosis. IFN-γ-pretreated (250 U/ml, 18 hours) Fas-low cells were incubated with KN93 (10 μmol/L), cyclosporine A (CysA, 1 μmol/L), or TFP (15 μmol/L). For the combined treatment, cells were pretreated with KN93 or CysA for 1 hour, and then TFP was added. After 20 hours, apoptotic cell death was determined as described in Figure 1 ▶ . Shown are means ± SE (n = 3).
Because both CaMKII and calcineurin are fully activated on binding to the Ca2+-bound form of CaM, it is also possible that CaMKII and calcineurin are activated by the increased [Ca2+]c during TFP-mediated apoptosis (Figure 4) ▶ . To determine whether CaMKII or calcineurin activity are required during TFP-mediated apoptosis, we treated IFN-γ-primed cells with TFP in the presence of KN93 or CysA and observed no effect of KN93 or CysA on TFP-mediated apoptosis (Figure 7) ▶ . We also observed no effect of KN93 or CysA on apoptosis mediated by other CaM antagonists, TMX and W7 (data not shown). These results suggest that neither CaMKII nor calcineurin activity are required during CaM antagonist-mediated apoptosis.
Discussion
CaM has been recognized as a primary transducer of Ca2+-dependent signals and a regulator of many essential cellular functions. Because Ca2+ is important in cell-cycle regulation, CaM antagonists have been recognized as anti-proliferative agents. 21 CaM antagonists have also been reported to inhibit tumor cell invasion in vitro 25 and inhibit metastasis in vivo. 26 Although CaM has been reported to regulate apoptosis, 27,28,30 the underlying molecular mechanisms are unknown. In addition, some cancer cells are resistant to apoptosis when treated with anti-CaM drugs. 30 In this study, CaM antagonists were used, in combination with IFN-γ pretreatment, to induce cell death in Fas-low cholangiocarcinoma cells, which are otherwise resistant to apoptosis. IFN-γ is a pleiotropic cytokine that plays a central role in promoting innate and adaptive mechanisms of host defense. 59 The ability of IFN-γ to enhance Fas-mediated apoptosis has been extensively studied. The up-regulation of Fas by IFN-γ in HT29 human colon adenocarcinoma cells, 60 melanoma cells, 61 squamous cell carcinoma, 62 and cholangiocarcinoma cells 37 has been reported, and enhanced Fas-mediated apoptosis was observed in many cases.
In this report, we first demonstrate that the combination of IFN-γ and CaM antagonists induces apoptosis in apoptosis-resistant cholangiocarcinoma cells. Previously, we reported that the human cholangiocarcinoma cell line, Sk-ChA-1, expresses Fas heterogeneously, and the Fas expression level correlates with sensitivity to Fas- and CaM antagonist-mediated apoptosis. 30 Because the effect of IFN-γ on up-regulation of Fas has been shown in many cell types, we treated Fas-low cholangiocarcinoma cells, which are resistant to Fas-mediated apoptosis, with IFN-γ and found increases in Fas and other apoptosis-related molecules and induction of susceptibility to Fas-mediated apoptosis. 37 Interestingly, IFN-γ also renders Fas-low cholangiocarcinoma cells sensitive to CaM antagonist-mediated cell death through apoptosis as demonstrated in this report. After combined treatment with IFN-γ and CaM antagonists, we observed morphological changes typical of apoptosis, eg, rounding, formation of apoptotic bodies, and cell shrinkage (data not shown). Moreover, we show that, in IFN-γ-pretreated cells, CaM antagonists induce caspase cleavage, a hallmark of apoptosis; cleavage of caspases-8, -9, and -3 was detected (Figure 2) ▶ . In addition, cleavage of Bid, one of the direct substrates of caspase-8, was clearly detected (Figure 2) ▶ . We also demonstrate that CaM antagonists induce cytochrome c release from the mitochondria (Figure 2) ▶ and the collapse of the mitochondrial membrane potential in IFN-γ-pretreated cells (Figure 3A) ▶ .
One of the molecular events we identified during CaM antagonist-mediated apoptosis is the elevation of the free cytosolic Ca2+ concentration ([Ca2+]c) (Figure 4) ▶ that has been implicated in both caspase-dependent and -independent cell death. The role of Ca2+ in cell death involving caspase activation has been extensively examined. 63,64 After a variety of cellular insults, Ca2+ has also been shown to be necessary for apoptotic endonuclease activation, causing DNA cleavage. 65,66 Growing evidence suggests that the Ca2+-dependent protease, calpain, is frequently activated in apoptosis associated with elevated [Ca2+]c. 67,68 Sustained increases in [Ca2+]c leads to Ca2+ overload in the mitochondria, causing the collapse of the mitochondrial membrane potential. 69 In this regard, the increase in [Ca2+]c associated with CaM antagonist treatment is likely to be involved in diverse processes that induce apoptosis in both caspase-dependent and -independent manners. The source of the increased [Ca2+]c and its exact role in CaM antagonist-mediated apoptosis merits further investigation.
Although many events during Fas- and CaM antagonist-mediated apoptosis are similar, there are important differences. First, the generation of p18 fragments of caspase-8 and release of cytochrome c by the CaM antagonists are less extensive than seen during Fas-mediated apoptosis (Figure 2) ▶ , whereas apoptotic cell death mediated by CaM antagonists and activating Fas Ab are comparable (Figure 1B) ▶ . Therefore, it is possible that CaM antagonists initiate multiple pathways to induce apoptosis. Cleavage of initiator caspases, such as caspase-8, and induction of cytochrome c release may be a part of those multiple pathways. Secondly, we show that a broad-range caspase inhibitor does not block CaM antagonist-induced mitochondrial membrane potential change, but partially blocks Fas Ab-mediated mitochondrial membrane potential change, in IFN-γ-pretreated cells (Figure 3B) ▶ . This result suggests that CaM antagonists are able to affect mitochondrial function independent of caspase activation. Consistent with this, the broad-range caspase inhibitor only partially blocks CaM antagonist-mediated apoptosis (Figure 6) ▶ , indicating that CaM antagonists trigger both caspase-dependent and -independent pathways, and the depolarization of the mitochondrial membrane potential by CaM antagonists represents one possible caspase-independent mechanism. Third, neutralizing Fas Ab has no effect on CaM antagonist-mediated apoptosis whereas it blocks Fas-mediated apoptosis completely (Figure 5) ▶ . Both Fas- and CaM antagonist-mediated apoptosis are partially inhibited by the broad-range caspase inhibitor, z-VAD-fmk (Figure 6) ▶ . These results suggest that even though Fas- and CaM antagonist-mediated apoptosis share some common molecular events, the interaction of CaM antagonists with surface Fas is not required for CaM antagonists to stimulate apoptosis.
In this report, we have focused on the induction of apoptosis in Fas-low cholangiocarcinoma cells because they are resistant to Fas- and CaM antagonist-mediated apoptosis, if not treated with IFN-γ, and are tumorigenic in nude mice. Although IFN-γ renders Fas-low cells susceptible to CaM antagonist-mediated apoptosis as shown in this report, IFN-γ-pretreated Fas-low cells are still less sensitive than IFN-γ-pretreated Fas-high cells (Figure 1, A and B) ▶ . Previously, we compared the expression levels of many apoptosis-related molecules in Fas-high and Fas-low cells, showing there is no difference in levels of caspases and Bcl-2 family members in these two subpopulations. 37 The in vitro proliferation rates of Fas-high and Fas-low cells were also the same (data not shown). It is possible that other inhibitors of apoptotic pathways, such as c-FLIP, may be involved in resistance to apoptosis in Fas-low cells, as we speculated in our previous report. 37
To investigate the effect of CaM antagonists, we used three compounds, TMX, TFP, and W7. TMX is commonly used as a chemotherapeutic and chemopreventive agent for breast cancer. Although, it has been primarily used for the treatment of hormone-dependent cancers, several studies have shown that TMX also inhibits the growth of hormone-independent tumors. 31,70,71 Animal studies and clinical trials have shown that TMX may be useful in the treatment of a much wider range of diseases, including atherosclerosis, autoimmune diseases, and osteoporosis. 72 The effect of TMX on this wide range of diseases results from its multiple functions. Its interaction with the estrogen receptor was initially believed to be its sole action. It is now clear that TMX has mixed properties, being both an agonist and an antagonist of the estrogen receptor. In addition, it has many effects independent of estrogen receptor-related pathways. These include modulation of signaling proteins such as protein kinase C, CaM, transforming growth factor-β, and the proto-oncogene, c-myc. 14,72 Recent studies also demonstrate that TMX increases the spatial expansion of calcium waves 71 and mitochondrial depolarization 73 in an estrogen receptor-independent manner. We speculate that TMX induces apoptosis via its CaM antagonism, because the molecular events after TMX treatment were identical with those induced by other classical CaM antagonists (TFP and W7). The morphological changes in cells treated with TMX and other CaM antagonists were also identical, ie, rounding, shrinkage, and detachment (data not shown).
The effective induction of apoptosis by combined treatment with IFN-γ and CaM antagonists was also confirmed in some pancreatic carcinoma cell lines, HPAC and HPAF-II, suggesting the dual treatment may be useful for inducing apoptosis in several types of cancer cells. However, we found that the combined treatment did not enhance apoptosis in other cell lines (pancreatic carcinoma cell lines MIaPaCa-2 and PANC-1 and the breast cancer cell line MCF-7). The underlying mechanism of differing sensitivity to combined treatment with IFN-γ and CaM antagonists in different cell lines requires further investigation. The in vivo effects of combined treatment with IFN-γ and TMX are also under investigation in our laboratory. Because both IFN-γ and TMX have been approved for clinical use, combined treatment with this dual therapeutic modality may be a useful strategy against cholangiocarcinoma and potentially other malignancies.
Acknowledgments
We thank Tracey McGuire and Enid Keyser (Clinical Immunology/Rheumatology Flow Cytometry Core Facility, the University of Alabama at Birmingham) for their technical help and Dr. Richard B. Marchase and Zachary Kneass (Department of Cell Biology, the University of Alabama at Birmingham) for advice on the measurement of calcium concentration.
Footnotes
Address reprint requests to Jay M. McDonald, M.D., Professor and Chair, Department of Pathology, Director, UAB Center for Metabolic Bone Disease, The University of Alabama at Birmingham, 509 LHRB, 1530 3rd Ave. South, Birmingham, AL 35294-0007. E-mail: mcdonald@path.uab.edu.
Supported by the Office of Research and Development, Department of Veterans Affairs (to J. M. M.).
References
- 1.Thompson CB: Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267:1456-1462 [DOI] [PubMed] [Google Scholar]
- 2.Soini Y, Paakko P, Lehto VP: Histopathological evaluation of apoptosis in cancer. Am J Pathol 1998, 153:1041-1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thornberry NA, Lazebnik Y: Caspases: enemies within. Science 1998, 281:1312-1316 [DOI] [PubMed] [Google Scholar]
- 4.Hengartner MO: The biochemistry of apoptosis. Nature 2000, 407:770-776 [DOI] [PubMed] [Google Scholar]
- 5.Kitanaka C, Kuchino Y: Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ 1999, 6:508-515 [DOI] [PubMed] [Google Scholar]
- 6.Borner C, Monney L: Apoptosis without caspases: an inefficient molecular guillotine? Cell Death Differ 1999, 6:497-507 [DOI] [PubMed] [Google Scholar]
- 7.Mathiasen IS, Lademann U, Jaattela M: Apoptosis induced by vitamin D compounds in breast cancer cells is inhibited by Bcl-2 but does not involve known caspases or p53. Cancer Res 1999, 59:4848-4856 [PubMed] [Google Scholar]
- 8.McCarthy NJ, Whyte MK, Gilbert CS, Evan GI: Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J Cell Biol 1997, 136:215-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brunet CL, Gunby RH, Benson RS, Hickman JA, Watson AJ, Brady G: Commitment to cell death measured by loss of clonogenicity is separable from the appearance of apoptotic markers. Cell Death Differ 1998, 5:107-115 [DOI] [PubMed] [Google Scholar]
- 10.Li LY, Luo X, Wang X: Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412:95-99 [DOI] [PubMed] [Google Scholar]
- 11.Babu YS, Sack JS, Greenhough TJ, Bugg CE, Means AR, Cook WJ: Three-dimensional structure of calmodulin. Nature 1985, 315:37-40 [DOI] [PubMed] [Google Scholar]
- 12.Motohashi N: Phenothiazines and calmodulin (review). Anticancer Res 1991, 11:1125-1164 [PubMed] [Google Scholar]
- 13.Lopes MC, Vale MG, Carvalho AP: Ca2+-dependent binding of tamoxifen to calmodulin isolated from bovine brain. Cancer Res 1990, 50:2753-2758 [PubMed] [Google Scholar]
- 14.Mandlekar S, Kong AN: Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001, 6:469-477 [DOI] [PubMed] [Google Scholar]
- 15.Hidaka H, Sasaki Y, Tanaka T, Endo T, Ohno S, Fujii Y, Nagata T: N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide, a calmodulin antagonist, inhibits cell proliferation. Proc Natl Acad Sci USA 1981, 78:4354-4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakai TT, Krishna NR: Synthesis and properties of some novel anti-calmodulin drugs. Bioorg Med Chem 1999, 7:1559-1565 [DOI] [PubMed] [Google Scholar]
- 17.Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G: Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J Biol Chem 2001, 276:20144-20153 [DOI] [PubMed] [Google Scholar]
- 18.Patel S, Morris SA, Adkins CE, O’Beirne G, Taylor CW: Ca2+-independent inhibition of inositol trisphosphate receptors by calmodulin: redistribution of calmodulin as a possible means of regulating Ca2+ mobilization. Proc Natl Acad Sci USA 1997, 94:11627-11632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu M, Chen TY, Ahamed B, Li J, Yau KW: Calcium-calmodulin modulation of the olfactory cyclic nucleotide-gated cation channel. Science 1994, 266:1348-1354 [DOI] [PubMed] [Google Scholar]
- 20.Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX: Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem 2001, 276:21303-21310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasmussen CD, Lu KP, Means RL, Means AR: Calmodulin and cell cycle control. J Physiol Paris 1992, 86:83-88 [DOI] [PubMed] [Google Scholar]
- 22.Bachs O, Agell N, Carafoli E: Calcium and calmodulin function in the cell nucleus. Biochim Biophys Acta 1992, 1113:259-270 [DOI] [PubMed] [Google Scholar]
- 23.Wei JW, Morris HP, Hickie RA: Positive correlation between calmodulin content and hepatoma growth rates. Cancer Res 1982, 42:2571-2574 [PubMed] [Google Scholar]
- 24.Hait WN, Lazo JS: Calmodulin: a potential target for cancer chemotherapeutic agents. J Clin Oncol 1986, 4:994-1012 [DOI] [PubMed] [Google Scholar]
- 25.Dewhurst LO, Gee JW, Rennie IG, MacNeil S: Tamoxifen, 17beta-oestradiol and the calmodulin antagonist J8 inhibit human melanoma cell invasion through fibronectin. Br J Cancer 1997, 75:860-868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito H, Wang JZ, Shimura K: Inhibition of lung metastasis by a calmodulin antagonist, N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide (W-7), in mice bearing Lewis lung carcinoma. Anticancer Res 1991, 11:249-252 [PubMed] [Google Scholar]
- 27.Dowd DR, MacDonald PN, Komm BS, Haussler MR, Miesfeld R: Evidence for early induction of calmodulin gene expression in lymphocytes undergoing glucocorticoid-mediated apoptosis. J Biol Chem 1991, 266:18423-18426 [PubMed] [Google Scholar]
- 28.Gerner C, Frohwein U, Gotzmann J, Bayer E, Gelbmann D, Bursch W, Schulte-Hermann R: The Fas-induced apoptosis analyzed by high throughput proteome analysis. J Biol Chem 2000, 275:39018-39026 [DOI] [PubMed] [Google Scholar]
- 29.Pan G, Zhou T, Radding W, Saag MS, Mountz JD, McDonald JM: Calmodulin antagonists inhibit apoptosis of CD4+ T-cells from patients with AIDS. Immunopharmacology 1998, 40:91-103 [DOI] [PubMed] [Google Scholar]
- 30.Pan G, Vickers SM, Pickens A, Phillips JO, Ying W, Thompson JA, Siegal GP, McDonald JM: Apoptosis and tumorigenesis in human cholangiocarcinoma cells. Involvement of Fas/APO-1 (CD95) and calmodulin. Am J Pathol 1999, 155:193-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sampson LK, Vickers SM, Ying W, Phillips JO: Tamoxifen-mediated growth inhibition of human cholangiocarcinoma. Cancer Res 1997, 57:1743-1749 [PubMed] [Google Scholar]
- 32.Kim JA, Kang YS, Jung MW, Lee SH, Lee YS: Involvement of Ca2+ influx in the mechanism of tamoxifen-induced apoptosis in HepG2 human hepatoblastoma cells. Cancer Lett 1999, 147:115-123 [DOI] [PubMed] [Google Scholar]
- 33.Pitt HA, Dooley WC, Yeo CJ, Cameron JL: Malignancies of the biliary tree. Curr Probl Surg 1995, 32:1-90 [DOI] [PubMed] [Google Scholar]
- 34.Pitt HA, Nakeeb A, Abrams RA, Coleman J, Piantadosi S, Yeo CJ, Lillemore KD, Cameron JL: Perihilar cholangiocarcinoma. Postoperative radiotherapy does not improve survival. Ann Surg 1995, 221:788-797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saunders K, Longmire W, JR, Tompkins R, Chavez M, Cates J, Roslyn J: Diffuse bile duct tumors: guidelines for management. Am Surg 1991, 57:816-820 [PubMed] [Google Scholar]
- 36.Yeo CJ, Pitt HA, Cameron JL: Cholangiocarcinoma. Surg Clin North Am 1990, 70:1429-1447 [DOI] [PubMed] [Google Scholar]
- 37.Ahn EY, Pan G, Vickers SM, McDonald JM: IFN-gamma upregulates apoptosis-related molecules and enhances Fas-mediated apoptosis in human cholangiocarcinoma. Int J Cancer 2002, 100:445-451 [DOI] [PubMed] [Google Scholar]
- 38.Grynkiewicz G, Poenie M, Tsien RY: A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 1985, 260:3440-3450 [PubMed] [Google Scholar]
- 39.Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME: FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J 1997, 16:2794-2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X: Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94:481-490 [DOI] [PubMed] [Google Scholar]
- 41.Li H, Zhu H, Xu CJ, Yuan J: Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94:491-501 [DOI] [PubMed] [Google Scholar]
- 42.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X: Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91:479-489 [DOI] [PubMed] [Google Scholar]
- 43.Green DR, Reed JC: Mitochondria and apoptosis. Science 1998, 281:1309-1312 [DOI] [PubMed] [Google Scholar]
- 44.Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A: JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett 1997, 411:77-82 [DOI] [PubMed] [Google Scholar]
- 45.Martikainen P, Isaacs J: Role of calcium in the programmed death of rat prostatic glandular cells. Prostate 1990, 17:175-187 [DOI] [PubMed] [Google Scholar]
- 46.Jiang S, Chow SC, Nicotera P, Orrenius S: Intracellular Ca2+ signals activate apoptosis in thymocytes: studies using the Ca2+-ATPase inhibitor thapsigargin. Exp Cell Res 1994, 212:84-92 [DOI] [PubMed] [Google Scholar]
- 47.Choi MS, Boise LH, Gottschalk AR, Quintans J, Thompson CB, Klaus GG: The role of bcl-XL in CD40-mediated rescue from anti-mu-induced apoptosis in WEHI-231 B lymphoma cells. Eur J Immunol 1995, 25:1352-1357 [DOI] [PubMed] [Google Scholar]
- 48.Jackisch C, Hahm HA, Tombal B, McCloskey D, Butash K, Davidson NE, Denmeade SR: Delayed micromolar elevation in intracellular calcium precedes induction of apoptosis in thapsigargin-treated breast cancer cells. Clin Cancer Res 2000, 6:2844-2850 [PubMed] [Google Scholar]
- 49.Jan CR, Lu CH, Chen YC, Cheng JS, Tseng LL, Jun-Wen W: Ca2+ mobilization induced by W-7 in MG63 human osteosarcoma cells. Pharmacol Res 2000, 42:323-327 [DOI] [PubMed] [Google Scholar]
- 50.Jan CR, Yu CC, Huang JK: N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide hydrochloride) (W-7) causes increases in intracellular free Ca2+ levels in bladder female transitional carcinoma (BFTC) cells. Anticancer Res 2000, 20:4355-4359 [PubMed] [Google Scholar]
- 51.Radding W, Jordan SE, Hester RB, Blair HC: Intracellular calcium puffs in osteoclasts. Exp Cell Res 1999, 253:689-696 [DOI] [PubMed] [Google Scholar]
- 52.Micheau O, Solary E, Hammann A, Martin F, Dimanche-Boitrel MT: Sensitization of cancer cells treated with cytotoxic drugs to fas-mediated cytotoxicity. J Natl Cancer Inst 1997, 89:783-789 [DOI] [PubMed] [Google Scholar]
- 53.Friesen C, Herr I, Krammer PH, Debatin KM: Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells. Nat Med 1996, 2:574-577 [DOI] [PubMed] [Google Scholar]
- 54.Wright SC, Schellenberger U, Ji L, Wang H, Larrick JW: Calmodulin-dependent protein kinase II mediates signal transduction in apoptosis. EMBO J 1997, 11:843-849 [DOI] [PubMed] [Google Scholar]
- 55.Fladmark KE, Brustugun OT, Mellgren G, Krakstad C, Boe R, Vintermyr OK, Schulman H, Doskeland SO: Ca2+/calmodulin-dependent protein kinase II is required for microcystin-induced apoptosis. J Biol Chem 2002, 277:2804-2811 [DOI] [PubMed] [Google Scholar]
- 56.Xiao C, Yang BF, Asadi N, Beguinot F, Hao C: Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-FLIP and PED/PEA-15 in glioma cells. J Biol Chem 2002, 277:25020-25025 [DOI] [PubMed] [Google Scholar]
- 57.Crabtree GR: Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell 1999, 96:611-614 [DOI] [PubMed] [Google Scholar]
- 58.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC: Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999, 284:339-343 [DOI] [PubMed] [Google Scholar]
- 59.Farrar MA, Schreiber RD: The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol 1993, 11:571-611 [DOI] [PubMed] [Google Scholar]
- 60.Ossina NK, Cannas A, Powers VC, Fitzpatrick PA, Knight JD, Gilbert JR, Shekhtman EM, Tomei LD, Umansky SR, Kiefer MC: Interferon-gamma modulates a p53-independent apoptotic pathway and apoptosis-related gene expression. J Biol Chem 1997, 272:16351-16357 [DOI] [PubMed] [Google Scholar]
- 61.Ugurel S, Seiter S, Rappl G, Stark A, Tilgen W, Reinhold U: Heterogenous susceptibility to CD95-induced apoptosis in melanoma cells correlates with bcl-2 and bcl-x expression and is sensitive to modulation by interferon-gamma. Int J Cancer 1999, 82:727-736 [DOI] [PubMed] [Google Scholar]
- 62.Moers C, Warskulat U, Muschen M, Even J, Niederacher D, Josien R, Koldovsky U, Beckmann MW, Haussinger D: Regulation of CD95 (Apo-1/Fas) ligand and receptor expression in squamous-cell carcinoma by interferon-gamma and cisplatin. Int J Cancer 1999, 80:564-572 [DOI] [PubMed] [Google Scholar]
- 63.Wertz IE, Dixit VM: Characterization of calcium release-activated apoptosis of LNCaP prostate cancer cells. J Biol Chem 2000, 275:11470-11477 [DOI] [PubMed] [Google Scholar]
- 64.Petersen A, Castilho RF, Hansson O, Wieloch T, Brundin P: Oxidative stress, mitochondrial permeability transition and activation of caspases in calcium ionophore A23187-induced death of cultured striatal neurons. Brain Res 2000, 857:20-29 [DOI] [PubMed] [Google Scholar]
- 65.Gaido ML, Cidlowski JA: Identification, purification, and characterization of a calcium-dependent endonuclease (NUC18) from apoptotic rat thymocytes. NUC18 is not histone H2B J Biol Chem 1991, 266:18580-18585 [PubMed] [Google Scholar]
- 66.Urbano A, McCaffrey R, Foss F: Isolation and characterization of NUC70, a cytoplasmic, hematopoietic apoptotic endonuclease. J Biol Chem 1998, 273:34820-34827 [DOI] [PubMed] [Google Scholar]
- 67.Nakagawa T, Yuan J: Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol 2000, 150:887-894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mathiasen IS, Sergeev IN, Bastholm L, Elling F, Norman AW, Jaattela M: Calcium and calpain as key mediators of apoptosis-like death induced by vitamin D compounds in breast cancer cells. J Biol Chem 2002, 277:30738-30745 [DOI] [PubMed] [Google Scholar]
- 69.Duchen MR: Mitochondria and calcium: from cell signalling to cell death. J Physiol 2000, 529:57-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Menard S, Aiello P, Tagliabue E, Rumio C, Lollini PL, Colnaghi MI, Balsari A: Tamoxifen chemoprevention of a hormone-independent tumor in the proto-neu transgenic mice model. Cancer Res 2000, 60:273-275 [PubMed] [Google Scholar]
- 71.Zhang W, Couldwell WT, Song H, Takano T, Lin JH, Nedergaard M: Tamoxifen-induced enhancement of calcium signaling in glioma and MCF-7 breast cancer cells. Cancer Res 2000, 60:5395-5400 [PubMed] [Google Scholar]
- 72.Grainger DJ, Metcalfe JC: Tamoxifen: teaching an old drug new tricks? Nat Med 1996, 2:381-385 [DOI] [PubMed] [Google Scholar]
- 73.Dietze EC, Caldwell LE, Grupin SL, Mancini M, Seewaldt VL: Tamoxifen but not 4-hydroxytamoxifen initiates apoptosis in p53(-) normal human mammary epithelial cells by inducing mitochondrial depolarization. J Biol Chem 2001, 276:5384-5394 [DOI] [PubMed] [Google Scholar]