

Abstract
Connective tissue growth factor (CTGF) has been described as a novel fibrotic mediator. CTGF is overexpressed in several kidney diseases and is induced by different factors involved in renal injury. Angiotensin II (AngII) participates in the pathogenesis of kidney damage, contributing to fibrosis; however, whether AngII regulates CTGF in the kidney has not been explored. Systemic infusion of AngII into normal rats for 3 days increased renal CTGF mRNA and protein levels. At day 7, AngII-infused rats presented overexpression of CTGF in glomeruli, tubuli, and renal arteries, as well as tubular injury and elevated fibronectin deposition. Only treatment with an AT1 receptor antagonist, but not an AT2, diminished CTGF and fibronectin overexpression and ameliorated tubular damage. In rats with immune complex nephritis, renal overexpression of CTGF was diminished by the ACE inhibitor quinapril, correlated with a diminution in fibrosis. In cultured renal cells (mesangial and tubular epithelial cells) AngII, via AT1, increased CTGF mRNA and protein production, and a CTGF antisense oligonucleotide decreased AngII-induced fibronectin synthesis. Our data show that AngII regulates CTGF in the kidney and cultured in mesangial and tubular cells. This novel finding suggests that CTGF could be a mediator of the profibrogenic effects of AngII in the kidney.
The connective tissue growth factor (CTGF), a member of the CCN family of early response genes, has been recently described as a new fibrotic mediator. 1 CTGF has been detected in many human tissues and biological fluids, being most abundant in the kidney. 2,3 This growth factor is overexpressed in a variety of fibrotic disorders, such as skin, vascular, and lung diseases. 1-4 Fibrosis is a common final pathway of renal diseases of diverse etiology, including inflammation, hemodynamics, and metabolic injury. In several human kidney diseases, CTGF was strongly up-regulated in the glomeruli and tubulointerstitium in association with scarring and sclerosis. 2,3 In experimental models of renal wound repair and scarring, elevated renal CTGF expression was found correlated with cellular proliferation and extracellular matrix (ECM) accumulation. 2,5-10 CTGF is generated in vitro in multiple cell types, including mesangial, tubular epithelial cells, and renal fibroblasts, by a variety of stimuli involved in renal damage, such as high concentrations of glucose and transforming growth factor-β (TGF-β). 2,6,8,10,11 In renal fibroblasts CTGF modulates cell proliferation, but is not a mitogen for mesangial cells. 8,10,12-15 In many cells, including renal, CTGF increases ECM production 5,6,15 and mediates many of the profibrotic actions of TGF-β. 1,5,7,10,11,16,17 In vivo, the blockade of CTGF synthesis or action reduces TGF-β-induced collagen synthesis. 17 Depending on the cell types, CTGF has diverse bioactivities, including induction of mitogenesis, chemotaxis, cellular adhesion, ECM production, regulation of proliferation/apoptosis, and angiogenesis. 1-4,18,19
Activation of renin angiotensin system (RAS) has been described in human and experimental kidney diseases. 20 Angiotensin II (AngII), the main peptide of RAS, is a renal growth factor that acts through its binding to the specific receptors AT1 and AT2. 20,21 This vasoactive peptide activates mesangial and tubular cells, and interstitial fibroblasts inducing hyperplasia/hypertrophy, depending on the cell type, and increasing the expression and synthesis of ECM. Both AT1 and AT2 receptors are involved in the regulation of cell growth and ECM. 20-24 A close relation between AngII and TGF-β has already been established. Systemic infusion of AngII into normal rats increases glomerular TGF-β. 25 In renal cells AngII increases TGF-β mRNA expression and synthesis as well as TGF-β conversion to its active form. Moreover, neutralizing antibodies to TGF-β remarkably reduce AngII-induced ECM production. 20,22-25 In experimental models of kidney damage, renal RAS activation and renal TGF-β overexpression correlated with increased ECM mRNA expression and deposition have been described. In some of these models, the blockade of AngII actions by angiotensin converting enzyme (ACE) inhibitors and AT1 antagonists reduced TGF-β and fibrosis. 25-27 CTGF appears to play a role in the development and progression of glomerulosclerosis and tubulointerstitial fibrosis. Although emerging evidence suggests that the novel profibrogenic cytokine CTGF may be an important downstream mediator of TGF-β profibrotic activities, 1-4,16,17 the potential link between renal RAS and CTGF has not yet been investigated.
Materials and Methods
Design
The in vivo effect of AngII was evaluated by systemic infusion of AngII (dissolved in saline) into female Wistar rats (subcutaneously by osmotic minipumps, Alza Corp., Palo Alto, CA), at the dose of 50 ng/kg/min. Animals were sacrificed at 3 and 7 days (n = 8 rats in each group). Then, tissue samples were immediately removed and further processed for histological studies and RNA. To determine the role of AngII receptors, a group of rats was treated with the AT1 antagonist Losartan (10 mg/kg/day in the drinking water, n = 8 rats each group) or the AT2 antagonist PD123319 (30 mg/kg/day, subcutaneously by osmotic minipumps, n = 4 rats) from 24 hours before AngII infusion. Losartan was kindly provided by Merck Sharp and Dome (Madrid, Spain), and PD123319 was from Sigma (St. Louis, MO). The doses of Losartan and PD123319 have previously demonstrated to cause an effective blockade of AT1 and AT2, respectively. 28 Control groups of animals of the same age, untreated or treated (AT antagonists and saline-infused), were also studied. Systolic arterial blood pressure was measured in conscious, restrained rats by the tail-cuff sphygmomanometer (NARCO Biosystems, Houston, TX). The blood pressure value for each rat was calculated as the average of three separate measurements at each session.
Immune complex nephritis was induced according to a previously described protocol. 26 Briefly, rats received an initial subcutaneous injection of ovalbumin in complete Freund’s adjuvant, followed by reimmunization 3 weeks later, and 1 week later, daily administration of ovalbumin for 9 weeks was started. When proteinuria appeared, animals were randomly distributed into two groups: untreated and quinapril-treated (ACE inhibitor; 100 mg/L in the drinking water) and studied 3 weeks later.
Renal Histopathological Studies
Kidney samples were studied by staining with hematoxylin and eosin and Masson, and examined by light microscopy. The presence of CTGF in renal tissue was determined by immunohistochemistry with a purified rabbit anti-CTGF antibody, which reacts with mouse and rat CTGF (Torrey Pines Biolabs, San Diego, CA), and a mouse anti-fibronectin monoclonal antibody (Chemicon, Temecula, CA) was used. Paraffin-embedded renal tissue sections (4 μm) were mounted on poly-L-lysine-coated slides. The slides were deparaffinized with xylene and graded concentrations of ethanol and then rehydrated. The endogenous peroxidase was blocked by incubation in 3% H2O2/methanol (1:1) at 25°C for 30 minutes. The slides were subsequently incubated in PBS with 4% bovine serum albumin (BSA) and 6% horse or sheep serum, respectively, for 1 hour at 37°C to reduce nonspecific background staining, and then incubated overnight at 4°C with anti-CTGF or anti-fibronectin antibodies in PBS containing 4% BSA and 1% serum. After being washed with PBS, the sections were incubated with a secondary anti-IgG HRP-conjugated antibody diluted 1:100 in 4% BSA/PBS for 1 hour, and after washing, they were stained with 3,3′-diaminobenzidine (Dako Diagnositcs, Barcelona, Spain) in 0.3% H202 for 10 minutes. The sections were counterstained with Mayer’s hematoxylin and mounted in Pertex (Medite, Burgdorf, Germany). In each experiment, negative controls without the primary antibody, or using an unrelated antibody, were included to check nonspecific staining (not shown).
Morphology was scored by semiquantitative determination as previously reported, 28 and graded as follows: 0, no staining; 1+, mild staining; 2+, moderate staining; 3+, marked staining. Identification of different cell types was based on topographical criteria as published. 28 The mean number of positive cells per glomerular cross-section was determined by evaluating 10 to 15 glomeruli. The whole interstitium was examined from each animal, separately evaluating proximal, distal, and collecting ducts. Tubular damage was defined as flattening of epithelium, lumen increase, vacuolization, desquamations, necrosis, and loss of brush border in proximal tubules, and glomerular damage such as increase in mesangial matrix expansion, as described. 28 Immunohistochemical quantification was evaluated by image analysis using a KZ 300 imaging system 3.0 (Zeiss, Munchen-Hallbergmoos, Germany). Briefly, the percentage of the stained area was calculated as the ratio of suitable binary thresholded image and the total field area. For each sample, the mean staining area was obtained by analysis of 20 different fields (×40). The staining score is expressed as density per mm2. The immunohistochemistry experiments were performed from two to four kidney sections from each experimental animal, which were stained and analyzed to obtain a mean score for each of them. In all cases, evaluations were performed by two independent observers in a blinded fashion, and the mean score value was then calculated for each rat.
Cell Cultures
Mesangial cells were cultured from isolated rat glomeruli by sequential sieving and differential centrifugation. 29 Cells were grown in RPMI 1640 medium, pH 7.4, supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mmol/L glutamine, and 10% fetal calf serum (FCS), at 37°C in 5% CO2. These cells were characterized by phase-contrast microscopy, positive staining for desmin and vimentin, and negative staining for keratin and factor VIII antigen, excluding epithelial and endothelial contamination, respectively. 29 The murine proximal tubuloepithelial cells (MCT line), kindly supplied by Dr. E. G. Neilson (University of Pennsylvania, Philadelphia, PA), were grown in RPMI. Mesangial and tubular epithelial cells have AT1 and AT2 receptors. 28,29 After confluence, cells were growth-arrested by incubation with 0.5% FCS for 48 hours before the experiments. The experiments were done with different preparations of cultured mesangial cells with 0 or 1 passage.
RNA Studies
Total RNA was isolated using a standard method with Trizol reagent (Gibco-BRL, Grand Island, NY) and CTGF mRNA expression was analyzed by reverse transcription-polymerase chain reaction (RT-PCR) and/or Northern blot. 6 Northern blot analysis was carried out as described. 23 Briefly, 40 μg of total RNA were denatured and electrophoresed on 1% agarose-formaldehyde gels, and then transferred to nylon membranes (Genescreen; New England Nuclear). Membranes were hybridized with the α[32P]-labeled DNA probes, and were exposed to autoradiographic films. G3PDH was used as control of RNA loading. For RT-PCR specific primers used were: CTGF (sense: 5′-GAGTGGGTGTGTGACGAGCCCAAGG-3′, antisense: 5′-ATGTCTCCGTACATCTTCCTGTAGT-3′) and G3PDH (sense: 5′-AATGCATCCTGCACCACCAA-3′, antisense: 5′-GTAGCCATATTCATTGTCATA-3′),which yield a 376 bp and 515 bp product, and PCR (1 minute at 94°C, 1 minute at 63°/54°C and 2 minutes at 68°C; 25 cycles), respectively. In control experiments we have determined the CTGF mRNA levels of control and AngII-treated samples, using different amount of mRNA and analyzing several PCR cycles (from 20 to 40, in 5-cycle interval), and then 25 cycles were chosen to compare groups because they provide submaximal cDNA amplifications. Other additional controls were done: RT-PCR with RNA samples but without AMV reverse transcriptase. In parallel experiments, CTGF PCR products were purified, sequences were confirmed and then used as DNA probes for Northern blot. The PCR products were analyzed on 6% polyacrylamide gels, which were dried and then exposed to X-OMAT AS films (Eastman Kodak, Rochester, NY). The autoradiographs were scanned using the GS-800 Calibrated Densitometer (Quantity One, Bio-Rad, Spain), obtaining densitometric arbitrary units. Data were normalized against those of the corresponding G3PDH data. To compare different groups, ratio CTGF/G3PDH in control rats is arbitrary equal to 1. In addition, all samples were analyzed in duplicate to obtain each sample mRNA value. Then, results were calculated as n-fold increase over control and expressed as mean ± SEM of n animals of each group. In another set of control experiments using RNA samples from cells, Northern blot, and RT-PCR analysis were compared. We have obtained similar results with both methods (data not shown), confirming the validation of RT-PCR for analysis of CTGF gene levels in renal samples, where the total amount of RNA obtained is not enough to perform Northern blot.
Western Blot
Cells were homogenized in lysis buffer (170 mmol/L TrisHCl, 22% glycerol, 2,2% sodium dodecyl sulfate (SDS), 0,1 mmol/L phenylmethylsulfonyl fluoride, and a protease inhibitor cocktail) and then separated by 12% SDS-polyacrylamide gel electrophoresis under reducing conditions. After electrophoresis, samples were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA). The membranes were blocked in PBS containing 0.1% Tween-20, and 5% dry skimmed milk for 1 hour at 37°C, and then incubated in the same buffer with specific CTGF or fibronectin antibodies for 18 hours at 4°C. After washing, detection was made by incubation with peroxidase-conjugated secondary antibody, and developed using an ECL chemiluminescence kit (Amersham). In all experiments protein content was determined by BCA method. In addition, Red Ponceau staining was used to show quality of proteins and the efficacy of protein transfer to the membrane (not shown). In experiments of cell-associated proteins α-tubuline was used as a loading control.
To neutralize TGF-β bioactivity we have used a neutralizing antibody against active TGF-β (R&D Systems, UK). The neutralization dose (10 μg/ml) was selected following the manufacturer instructions. To block CTGF actions, we used a CTGF antisense oligonucleotide, constructed with a 16 mer derived from the starting translation site, which contained the initial ATG whose sequence is 5′-TACTGGCGGCGGTCAT-3′. 18 The CTGF antisense oligonucleotide (20 μg/ml) was added directly to the serum-free medium without any transfection compounds. CTGF sense oligonucleotide was used as a control.
Statistical Analysis
Results are expressed as n-fold increase over control in densitometric arbitrary units, normalized by internal controls, and expressed as mean ± SEM of the experiments done. Significance was established by the GraphPAD Instat using Student’s t-test (GraphPAD Software), Mann-Whitney test (non-parametric t-test) and analysis of variance nonparametric (Kruskal-Wallis test), and differences were considered significant if the P value was less than 0.05.
Results
Angiotensin II Infusion Increases CTGF in the Kidney
CTGF transcripts have been detected in the kidney by RT-PCR and in situ hybridization, showing that CTGF expression transcripts were low in the glomeruli of control mice. 6 We have investigated the direct in vivo effect of AngII by systemic infusion into normal rats evaluating CTGF levels by RT-PCR. AngII-infused rats for 3 days presented elevated renal CTGF mRNA levels (5.9-fold versus control, P < 0.05, n = 8, RT-PCR; Figure 1 ▶ ). We evaluated CTGF production in the kidney of these animals using immunohistochemistry (Figure 2) ▶ . Control animals did not present staining for CTGF. After 3 days of AngII infusion, CTGF immunostaining was observed in glomeruli (located mainly in mesangial area), tubuli (mainly in distal tubular epithelial cells), and renal arteries (mainly in α-smooth muscle actin-positive cells). In AngII-infused rats for 7 days, CTGF overexpression remained elevated in the same renal structures (Figure 2C) ▶ .
Figure 1.

Systemic infusion of AngII increases renal CTGF mRNA expression via AT1. AngII (50 ng/kg/min; n = 8) was infused into rats for 3 days, total cortex RNA was extracted and analyzed by RT-PCR. Some animals were also treated with the AT1 antagonist Losartan (10 mg/kg/day; n = 8), or the AT2 antagonist PD123319 (30 mg/kg/day; n = 4), starting 24 hours before AngII infusion. Representative RT-PCR experiment that shows three different animals of each group (bottom) and data of densitometric analysis expressed as arbitrary units of mean ± SEM of four to eight animals of each group analyzed by duplicate (top). * P < 0.05 vs. control; # P < 0.05 vs. AngII infusion.
Figure 2.
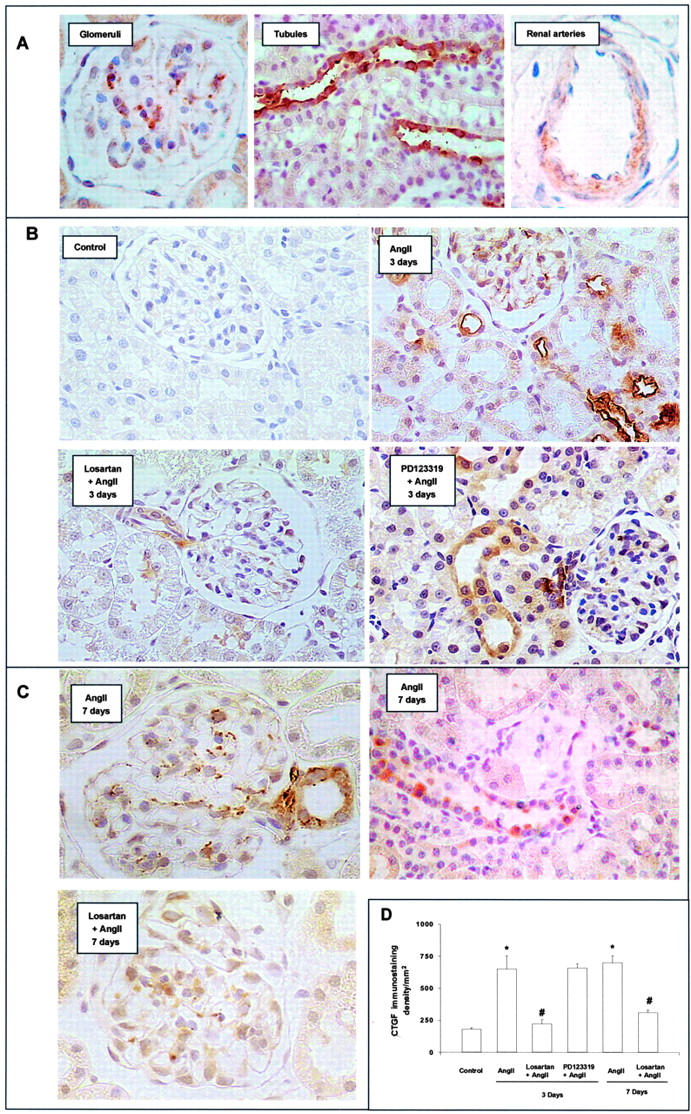
AngII infusion increases renal CTGF production via AT1. Kidney samples were analyzed by immunohistochemistry. A: In AngII-infused rats for 3 days, CTGF immunostaining was clearly shown in glomeruli (mesangial area), tubuli and renal arteries. B: Treatment with the AT1 antagonist Losartan, but not with the AT2 antagonist PD123319, diminished the AngII-induced CTGF production. C: In AngII-infused rats for 7 days, CTGF remained elevated in the same renal structures, and was diminished by Losartan. Figures show a representative of each group. Magnification: ×400. D: Computer analysis of CTGF staining score. Results are expressed in density per mm2 as mean ± SEM of data of four to eight animals per group. * P < 0.05 vs. control. # P < 0.05 vs. AngII infusion.
Systemic infusion of AngII causes morphological lesions in the kidney. After 3 days the majority of the glomeruli showed a normal appearance, and there was only slight and focal tubular injury (Table 1) ▶ . At day 7, animals presented mild mesangial matrix expansion and focal areas of tubular injury (Table 1) ▶ , as described. 28,30 To further study the implication of AngII on matrix accumulation, we determined fibronectin deposition by immunohistochemistry. In the normal kidney, fibronectin is present in the glomeruli, interstitium, and tubules along the basement membrane. During renal injury, fibronectin deposition is frequently increased. 31 Infusion of AngII for 3 days almost did not modify fibronectin deposition (not shown), but after 7 days fibronectin was markedly increased in glomerular and tubulointerstitial areas (Figure 3) ▶ . These data show that up-regulation of CTGF production occurs earlier than fibronectin deposition, suggesting that CTGF could be a mediator of AngII-induced renal fibrosis.
Table 1.
Renal Damage after Infusion of AngII
| Control | 3 Days | 7 Days | ||||
|---|---|---|---|---|---|---|
| AngII | Losartan + AngII | PD123319 + AngII | AngII | Losartan + AngII | ||
| Mesangial matrix expansion | − | − | − | − | +/++* | +/−† |
| Tubular injury score | − | + | − | + | ++/+++* | +/−† |
Quantification of mesangial matrix expansion and tubular injury. Results are expressed as semiquantitative score, as described in Materials and Methods. Four to eight animals were studied per group;
*P < 0.05 vs. control;
†P < 0.05 vs. AngII infusion.
Figure 3.

AngII infusion causes fibronectin overexpression via AT1. In control animals, slight fibronectin staining was observed in tubulointerstitial areas. In AngII-infused rats for 7 days there was a marked increase in fibronectin deposition in glomerular and tubulointerstitial areas, which diminished with the AT1 antagonist Losartan. A: A representative animal from each group. Magnification: ×200. B: Computer analysis of fibronectin staining score. Results are expressed in density per mm2 as mean ± SEM of four to eight animals per group. * P < 0.05 vs. control. # P < 0.05 vs. AngII infusion.
Pharmacological blockade of AngII receptors, using specific AT1 and AT2 antagonists, has contributed to defining their role in AngII-induced pathophysiological processes, such as regulation of fibrosis via AT1 and renal inflammatory cell infiltration through AT2. 21,28 In AngII-infused rats for 3 days, only treatment with the AT1 antagonist Losartan (n = 8), but not the AT2 antagonist PD123319 (n = 4), diminished CTGF mRNA expression and protein production (Figures 1 and 2) ▶ , suggesting that AngII-induced CTGF overexpression was mediated by the AT1 receptor. In AngII-infused rats for 7 days, the AT1 antagonist normalized blood pressure (101 ± 3 vs. 150 ± 12 mmHg in AngII-infused alone, n = 8, P < 0.05), decreased renal CTGF (Figure 2) ▶ and fibronectin protein deposition (Figure 3) ▶ and ameliorated glomerular sclerosis and tubular damage (Table 1) ▶ . All these results show that AngII via AT1 regulates CTGF, ECM proteins overexpression and fibrosis.
RAS Blockade Diminished CTGF Overexpression in an Experimental Model of Normotensive Immune Complex Nephritis
In human and experimental nephritis, renal CTGF overexpression has been described. 2,3 Our second aim was to determine whether RAS blockade could modulate CTGF during kidney injury. In a normotensive model of immune complex nephritis in rats, characterized by elevated renal AngII production and mRNA overexpression of TGF-β and ECM proteins, associated with fibrosis, 26 we have investigated whether RAS blockade did modulate renal CTGF expression. In nephritic animals, CTGF mRNA was increased compared to controls (Figure 4 ▶ ; 8.2-fold RT-PCR, n = 8, P < 0.05 vs. control). The ACE inhibitor quinapril diminished CTGF overexpression (Figure 4) ▶ , as well as the gene expression of TGF-β and ECM proteins, and ameliorated proteinuria and fibrosis. 26 These data show that during renal injury, blockade of renal AngII generation, without modification of blood pressure, diminished CTGF up-regulation and fibrosis.
Figure 4.

ACE inhibition diminishes renal CTGF overexpression in a normotensive model of immune complex nephritis in rats. Total cortex RNA was analyzed by RT-PCR. Some animals were treated with the ACE inhibitor quinapril (10 mg/kg/day; n = 8). Figure shows a representative RT-PCR experiment of two different animals of each group (bottom) and data of densitometric analysis expressed as arbitrary units of mean ± SEM of eight rats from each group (top). *P < 0.05 vs. control; # P < 0.05 vs. nephritis.
AngII Increases CTGF via AT1 in Cultured Renal Cells
To investigate whether AngII could regulate CTGF in renal tissue, independently of hemodynamic changes, we studied the direct effect of AngII on CTGF expression in two renal cell types, in which we have observed up-regulation of CTGF after AngII infusion in vivo: mesangial cells (MC) and tubuloepithelial cells (MCT).
By Northern blot we have observed that growth-arrested mesangial cells expressed low CTGF mRNA levels, as shown as a band of 2.4 kb, that increased in AngII-treated cells (Figure 5A) ▶ . This effect was seen within 1 hour, and CTGF mRNA levels remained elevated after 24 hours. The PKC activator PMA was used as positive control. Similar results were seen by RT-PCR (not shown).
Figure 5.

AngII increases CTGF mRNA expression in cultured mesangial cells. Growth-arrested mesangial cells were stimulated with 10−7 mol/L AngII from 1 to 24 hours and CTGF gene expression was determined by Northern blot. 10−7mol/L PMA was used as CTGF activator. The amount of CTGF mRNA was determined densitometrically and expressed as ratio CTGF/G3PDH of n-fold over control. A: A representative Northern blot; G3PDH was used as internal control. B: Values of mean ± SEM of eight experiments done. * P < 0.05 vs. control.
To investigate whether AngII also increased CTGF protein production, we used an anti-CTGF antibody that recognizes the 247–260 CTGF amino acids. In growth-arrested mesangial cells, the cell-associated fraction expressed a band of around 38–42 kd corresponding to the apparent molecular weight of CTGF. 4 AngII increased total cellular CTGF levels after 18 hours, and remained elevated until 48 hours (Figure 6) ▶ .
Figure 6.

AngII up-regulates CTGF protein production in cultured mesangial cells. Growth-arrested mesangial cells were stimulated with 10−7 mol/L AngII from 3 to 48 hours, and total cellular proteins were analyzed. A band of ∼38 to 42 kd, corresponding to the predicted CTGF size, was detected. A: A representative Western blot of CTGF and α-tubuline was used as a loading control. B: Results of total CTGF production obtained from densitometric analysis, and expressed as ratio CTGF/α-tubuline of n-fold over control. Figure shows values of mean ± SEM of six experiments. * P < 0.05 vs. control.
In mouse proximal tubular epithelial cells (MCT), AngII stimulated CTGF mRNA expression, with a very rapid onset, increasing at 1 hour (Figure 7) ▶ . CTGF gene was also increased after at 24 hours, showing a similar behavior to that of mesangial cells. This up-regulation was accompanied by increased CTGF protein synthesis after 18 hours, prolonged for at least 48 hours (Figure 7) ▶ .
Figure 7.
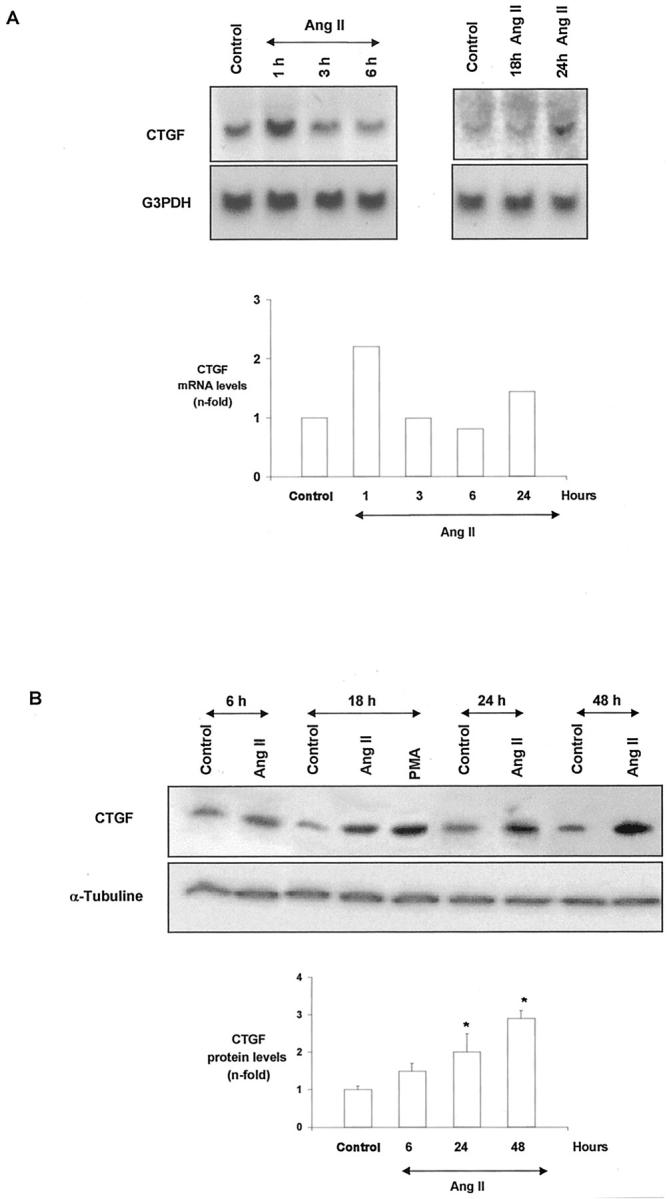
AngII increases CTGF mRNA expression and protein in mouse proximal tubular epithelial cells. Growth-arrested tubular cells were stimulated with 10−7 mol/L AngII from increasing times, CTGF gene expression was determined by Northern blot and protein levels by Western blot. A: A representative Northern blot. Top: Mean of two experiments done with identical results. B: A representative Western blot. Bottom: values of mean ± SEM of five experiments. * P < 0.05 vs. control.
Mesangial cells express both AT1 and AT2 receptors, 28 but ECM accumulation is mediated by AT1 pathway. 20,24 By specific receptor antagonists we examined the receptor subtype involved in CTGF regulation in MC. The AT1 antagonist Losartan (10−6 mol/L) caused a significant diminution in AngII-induced CTGF at both mRNA and protein levels. However, the AT2 antagonist PD123319 (10−5 mol/L) had no effect (Figure 8) ▶ . These results suggest that AngII-induced CTGF up-regulation was mediated by AT1 receptors and confirm the in vivo data.
Figure 8.
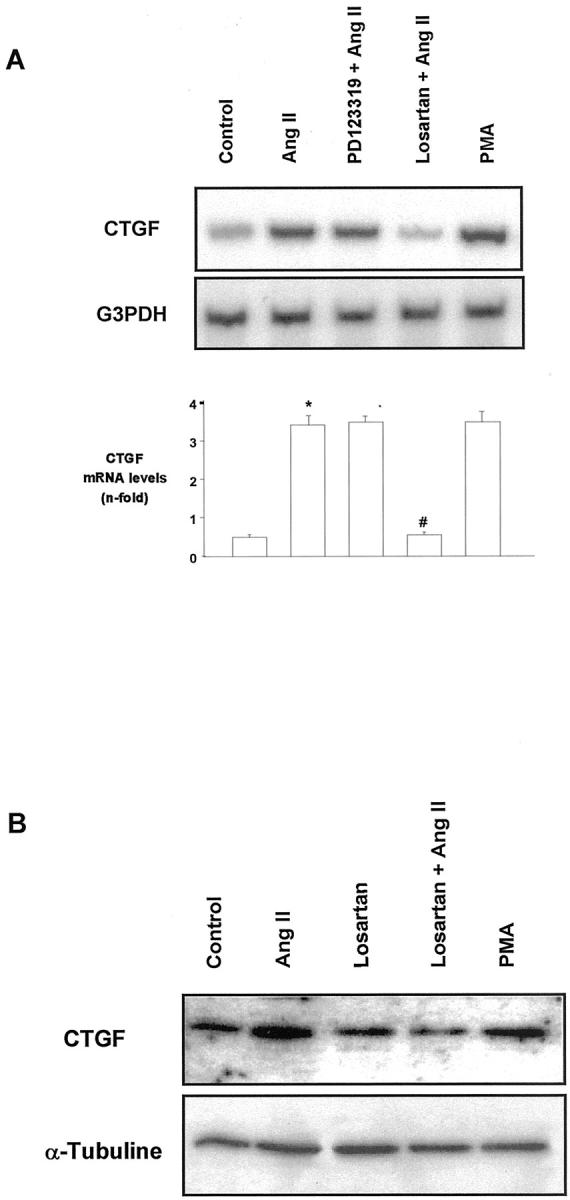
CTGF is regulated by the AT1 receptor in cultured mesangial cells. Cells were pretreated for 30 minutes with 10−6 mol/L of Losartan (AT1 antagonist) or 10−5 mol/L PD123319 (AT2 antagonist), and then stimulated with 10−7 mol/L AngII for additional 24 hours. A: A representative RT-PCR. Bottom: values of mean ± SEM of five experiments. * P < 0.05 vs. control. # P < 0.05 vs. AngII. B: Western blot of six experiments.
TGF-β Is Involved in AngII-Induced CTGF Regulation
TGF-β is the most important regulator of ECM, since it increases matrix production, inhibits matrix degradation, and modulates integrin receptors. 25 Previous studies have demonstrated that TGF-β is a direct stimulus for CTGF gene transcription. 8,11 In human fibroblasts, TGF-β presents a rapid and maintained CTGF gene up-regulation, 11,15,33 similar to that observed in mesangial cells treated with AngII. In contrast, platelet-derived growth factor (PDGF), epidermal growth factor, fibroblast growth factor, serotonin, and lysophosphatidic acid provoked only a transient and weak response. 33-35 We, and others, have previously demonstrated that the blockade of TGF-β, with antibodies that neutralize active TGF-β, blocked AngII-induced ECM production in renal cells. 23,25 For this reason, we evaluated whether TGF-β could be involved in CTGF regulation in response to AngII. The presence of a TGF-β neutralizing antibody diminished AngII-induced CTGF mRNA induction after 24 hours and protein production after 48 hours (Figure 9) ▶ , suggesting that CTGF production caused by AngII is, at least in part, mediated by endogenous TGF-β synthesis.
Figure 9.
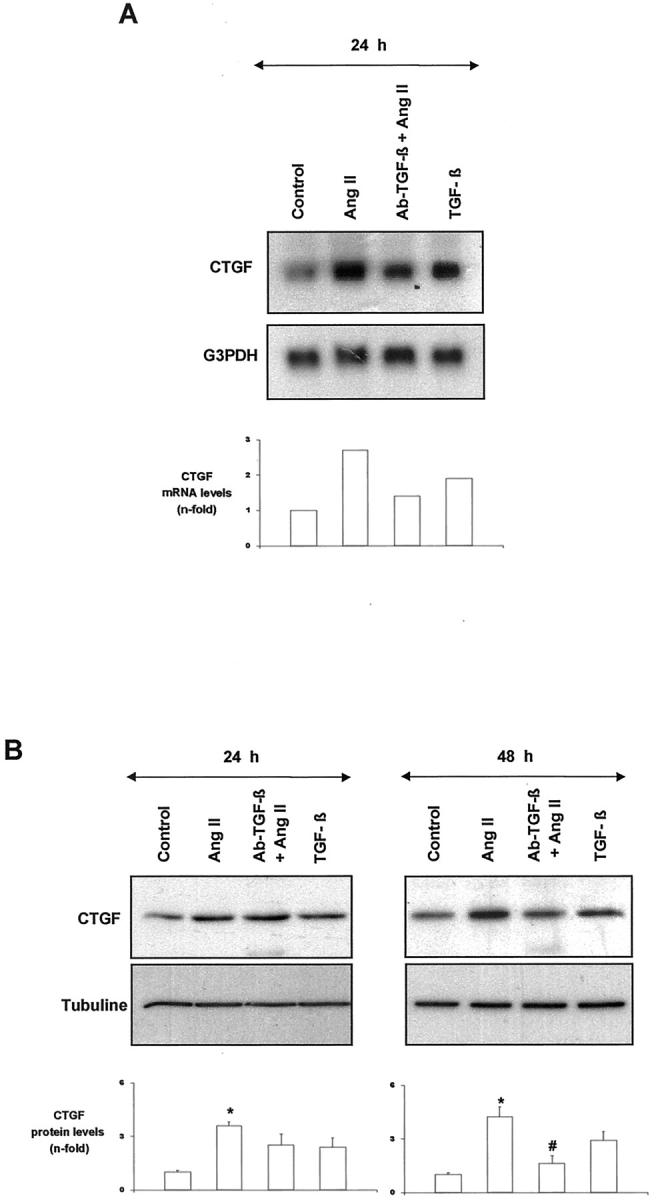
TGF-β is involved in AngII-induced CTGF regulation. In mesangial cells, TGF-β (1 ng/ml) increased CTGF mRNA expression (A) and synthesis (B). Cells were treated with AngII (10−7 mol/L) in the presence of a TGF-β neutralizing antibody (10 μg/ml) for different times. A: Top shows a representative Northern blot; bottom shows values of mean of two experiments. B: Top shows a representative Western blot; bottom shows values of mean ± SEM of four experiments. * P < 0.05 vs. control. # P < 0.05 vs. AngII infusion.
CTGF Is a Downstream Mediator of AngII-Induced Fibrosis
We investigated whether CTGF was involved in AngII-induced fibrosis, evaluating its effect on soluble fibronectin production by Western blot. We analyzed the effects of endogenous CTGF, blocking its production using an antisense CTGF oligodeoxynucleotide. In cultured mesangial cells, AngII significantly increased fibronectin release into the medium after 48 and 72 hours of incubation. Coincubation with the CTGF antisense oligonucleotide diminished AngII-induced fibronectin production at both times (Figure 10) ▶ , suggesting that CTGF has a crucial role in the profibrotic action of AngII in renal cells.
Figure 10.

CTGF is involved in AngII-induced fibronectin production. Mesangial cells were treated with AngII (10−7 mol/L) for 48 and 72 hours, and soluble fibronectin production was analyzed by Western blot. To neutralize CTGF actions, cells were co-treated with a CTGF antisense oligonucleotide (ODN). As negative control a CTGF sense oligonucleotide was used, and as positive control of fibronectin production TGF-β (1 ng/ml) was used. A band of ∼220 kd, corresponding to the molecular weight of soluble fibronectin, is shown (top). Figure shows a representative Western blot of five experiments done. Values of media ± SEM are shown (bottom). * P < 0.05 vs. control. # P < 0.05 vs. AngII.
Discussion
Our results clearly demonstrate that AngII in vivo and in vitro regulates CTGF and suggest that CTGF could be a mediator of fibrogenic effects of AngII in the kidney.
In human normal kidney, CTGF mRNA was mainly expressed in epithelial cells and some interstitial cells, 8 as observed in rats. 9 In human and experimental models of renal injury, such as crescentic glomerulonephritis, IgA nephropathy, focal and segmental glomerulosclerosis, lupus nephritis, chronic transplant rejection, focal glomerulosclerosis, membranoproliferative glomerulonephritis, 5/6 nephrectomy and diabetic nephropathy, CTGF was strongly up-regulated. 3,6,8-10 In contrast, CTGF expression did not appear to be increased in minimal change nephrotic syndrome and membranous nephropathy, glomerular diseases characterized by non-inflammatory lesions and proteinuria. 3 We have observed a marked renal CTGF overexpression in two different models of renal injury, AngII-induced renal damage and immune complex nephritis. These findings clearly support the hypothesis that CTGF is a common growth factor involved in renal damage.
In several models of kidney damage, as unilateral ureteral obstruction, anti-Thy-1.1 nephritis and experimental diabetic glomerulosclerosis, TGF-β1 and CTGF were coordinately up-regulated from the early stage of interstitial fibrosis followed by marked induction of ECM proteins. 8-10 In diabetic nephropathy, CTGF overexpression was observed when mesangial expansion was mild, and interstitial disease and proteinuria were absent. 6,8 In AngII-induced renal damage, CTGF up-regulation was observed after 3 days, preceding elevated fibronectin deposition, which was found after 7 days, when tubular damage was also observed. In rats with immune complex nephritis, renal CTGF mRNA expression was strongly up-regulated coincidentally with overexpression of TGF-β and ECM proteins. In this phase of the disease, animals also present renal damage, fibrosis, and severe proteinuria. 26 In a model of skin fibrosis, CTGF mRNA levels remained elevated in areas of persistent fibrosis, 36 as we observed after 7 days of AngII infusion. In addition, injection of CTGF into the skin induces the formation of fibrous tissue. 12 These data strongly suggest that CTGF could be a mediator of AngII-induced fibrosis in the kidney.
Pharmacological blockade of the RAS has demonstrated beneficial effects in several kidney diseases. In experimental models of renal damage, ACE inhibitors and AT1 antagonists reduced gene up-regulation of TGF-β and ECM proteins as well as fibrosis. 25-27 We have observed that an ACE inhibitor decreased CTGF overexpression in rats with immune nephritic and an AT1 antagonist diminished AngII-induced CTGF up-regulation, as well as renal fibrosis. These data show that the beneficial effects of RAS blockers could be due to the modulation of growth factors such as CTGF. Our data demonstrate that drugs controlling AngII are probably some of the best ways of avoiding fibrosis in progressive renal diseases.
Hypertension causes structural changes in the kidney, including cellular hypertrophy and ECM accumulation, which have been attributed to hemodynamic changes associated to mechanical stress and to factors like AngII and TGF-β. Mechanical strains, such as stretch, shear stress, and static pressure, are possible regulatory elements in CTGF expression. 6,37 In mesangial cells, high static pressure and cyclic mechanical stretching up-regulated CTGF and ECM mRNA. 37-39 These effects were reversed by a CTGF antisense oligonucleotide. 39 In contrast, Riser et al 6,8 did not find any increase in CTGF protein after 4 to 72 hours of cyclin stress either under serum or serum-depleted conditions. Systemic infusion of AngII into normal rats caused a slight blood pressure elevation; however, several data suggest that the cellular actions of AngII are responsible for CTGF up-regulation. In a rat model of cyclosporine A and high sodium diet-induced hypertension, an AT1 antagonist normalized increased cardiac CTGF expression, while it only partially diminished blood pressure elevation. 40 In rats with immune complex nephritis, renal CTGF mRNA expression was strongly up-regulated in the absence of systemic hypertension, and blockade of renal AngII generation by ACE inhibition diminished CTGF mRNA expression. In AngII-infused rats, the AT1 antagonist Losartan diminished renal CTGF up-regulation. Moreover, in cultured mesangial cells the AT1 antagonist also decreased CTGF gene expression and synthesis. All these in vivo evidence, together with our in vitro data, suggest that CTGF is up-regulated by a direct, non-pressure-mediated, AngII-effect.
Mesangial cells are the main cells involved in the development of glomerulosclerosis. In these cells, CTGF significantly increased fibronectin and collagen type I and IV production, induced its own expression, regulated mesangial cell migration, but did not increase cell proliferation. 6-8 CTGF is up-regulated by high extracellular glucose, TGF-β, and, as we have observed, AngII, but not by PDGF. 6-8 Other factors, including TNF-α, cAMP, and NO, suppress CTGF. 17,41,42 AngII, via AT1, regulates mesangial cell behavior, inducing cell proliferation or hypertrophy and increasing ECM accumulation. 20 These processes seem to be mediated by the endogenous production of several factors, including TGF-β 20,23,24 and CTGF. In this regard, we have observed that an antisense CTGF oligonucleotide diminished AngII-induced fibronectin production. In mesangial cells, CTGF is a downstream mediator of profibrogenic actions of TGF-β. 7,8 Using a neutralizing TGF-β antibody, CTGF induction caused by AngII was diminished, showing that TGF-β mediates, at least in part, CTGF production. In AngII-infused rats, CTGF immunostaining was elevated in the mesangial area, and preceded mesangial matrix expansion observed after 7 days. These data suggest that CTGF is a mediator of AngII-induced mesangial matrix accumulation.
The chronic accumulation of ECM in glomeruli and tubulointerstitial regions is critical in the progressive loss of function in renal diseases. 24 Tubulointerstitial fibrosis is considered to be a predictor of poor outcome in renal diseases. In human nephritis, the increase in the number of cells expressing CTGF mRNA was observed at sites of chronic tubulointerstitial damage, which correlated with the degree of damage. 3 We have found that AngII increases CTGF in tubular cells in vivo and in vitro. Moreover, AngII infusion for 7 days caused tubular damage. In murine proximal tubuloepithelial cells AngII via AT1 induces hypertrophy, production of TGF-β and ECM accumulation 22 and, as observed by us, CTGF. In these cells, other factors also up-regulate CTGF including TGF-β, calcium oxalate, and mechanical wounding, but not high glucose, 43-45 and CTGF increased fibronectin but not collagen type I and type III expression. 43 These data suggest that CTGF could also be involved in AngII-induced fibrotic responses in tubular cells.
Cell-derived CTGF may act on renal cells by an autocrine mode, contributing to the increased expansion of ECM proteins. The primary target cells for CTGF in the renal interstitium are myofibroblasts, which are responsible for providing ECM proteins in interstitial fibrosis. In this sense, in normal rat kidney (NRK) fibroblasts, CTGF increases ECM proteins. 14-17 In these cells AngII also increases cell proliferation, TGF-β, and ECM production. 23 CTGF is also involved in myofibroblast differentiation. 46 In the rat remnant kidney model, CTGF is highly expressed in interstitial fibroblasts, coinciding with damage, regeneration, and fibrosis. The description of CTGF mRNA expression in the proximity of regenerative epithelial cells and those transdifferentiating to myofibroblasts suggests that CTGF may modulate renal tubular epithelial differentiation. 15 These results indicate that CTGF has a crucial role in the profibrotic action of TGF-β and AngII in renal cells, providing a potential therapeutic target against tubulointerstitial fibrosis.
On the whole, we have presented information showing that AngII via CTGF is implicated in the process of renal fibrosis. Our results suggest that CTGF up-regulation is an important factor in the pathogenesis of AngII-induced renal fibrosis, regulating mesangial matrix accumulation, progressive glomerulosclerosis, and tubular damage, acting downstream of AngII. TGF-β is considered the main factor involved in matrix regulation. The blockade of TGF-β actions with neutralizing antibodies and decorin has demonstrated reduction of fibrosis in experimental models; however, the TGF-β knockout mice is lethal, presenting a hyperinflammatory phenotype, 47 suggesting that other more specific targets for antifibrotic therapy are necessary. Since there are no data available on the involvement of CTGF in modulation of inflammation and immune reactions, and this growth factor is a downstream mediator of TGF-β and, as shown here, of AngII-induced fibrosis, CTGF maybe a useful target for novel antifibrotic therapies.
Footnotes
Address reprint requests to Marta Ruiz-Ortega, Ph.D., Vascular and Renal Research Laboratory, Fundación Jiménez Díaz, Avenida Reyes Católicos, 228040 Madrid, Spain. E-mail: mruizo@fjd.es.
Supported by grants from Fondo de Investigación Sanitaria (01/3130, PI020513, and PI020822), Comunidad Autónoma de Madrid (08.4/0017/2000 and 08.4/0018/2001), Sociedad Española de Nefrología, Fundación Renal Iñigo Alvarez de Toledo, European grant of shared-cost Research and Technological Development actions (QLRT-200101215), Fondecyt 1000433, and Fondo Europeo de Desarrollo Regional.
M. R., O. L., and V. E. are fellows of Fondo de Investigación Sanitaria.
References
- 1.Lau LF, Lam SCT: The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res 1999, 248:44-57 [DOI] [PubMed] [Google Scholar]
- 2.Gupta S, Clarkson MR, Duggan J, Brady HR: Connective tissue growth factor: potential role in glomerulosclerosis and tubulointerstitial fibrosis. Kidney Int 2000, 58:1389-1399 [DOI] [PubMed] [Google Scholar]
- 3.Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, Goldschmeding R: Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 1998, 53:853-861 [DOI] [PubMed] [Google Scholar]
- 4.Oemar BS, Luscher TF: Connective tissue growth factor: friend or foe? Arterioscler Thromb Vasc Biol 1997, 17:1483-1489 [DOI] [PubMed] [Google Scholar]
- 5.Murphy M, Godson C, Cannon S, Kato S, Mackenzie HS, Martin F, Brady HR: Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J Biol Chem 1999, 26:5830-5804 [DOI] [PubMed] [Google Scholar]
- 6.Riser BL, Denichillo M, Cortes P, Baker C, Grodin JM, Yee J, Narins RG: Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis J Am Soc Nephrol 2000, 11:25-38 [DOI] [PubMed] [Google Scholar]
- 7.Blom IE, van Dijk AJ, Wieten L, Duran K, Ito Y, Kleij L, deNichilo M, Rabelink TJ, Weening JJ, Aten J, Goldschmeding R: In vitro evidence for differential involvement of CTGF, TGF-β, and PDGF-BB in mesangial response to injury. Nephrol Dial Transplant 2001, 16:1139-1148 [DOI] [PubMed] [Google Scholar]
- 8.Riser BL, Cortes P: Connective tissue growth factor and its regulation: a new element in diabetic glomerulosclerosis. Ren Fail 2001, 23:459-470 [DOI] [PubMed] [Google Scholar]
- 9.Ito Y, Goldschmeding R, Bende R, Claessen N, Chand M, Kleij L, Rabelink T, Weening J, Aten J: Kinetics of connective tissue growth factor expression during experimental proliferative glomerulonephritis. J Am Soc Nephrol 2001, 12:472-484 [DOI] [PubMed] [Google Scholar]
- 10.Yokoi H, Sugawara A, Mukoyama M, Mori K, Makino H, Suganami T, Nagae T, Yahata K, Fujinaga Y, Tanaka I, Nakao K: Role of connective tissue growth factor in profibrotic action of transforming growth factor-β: a potential target for preventing renal fibrosis. Am J Kidney Dis 2001, 3:S134-S138 [DOI] [PubMed] [Google Scholar]
- 11.Kucich U, Rosenbloom JC, Herrick DJ, Abrams WR, Hamilton AD, Sebti SM, Rosenbloom J: Signaling events required for transforming growth factor-β stimulation of connective tissue growth factor expression by cultured human lung fibroblasts. Arch Biochem Biophys 2001, 395:103-112 [DOI] [PubMed] [Google Scholar]
- 12.Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR: Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol 1996, 107:404-411 [DOI] [PubMed] [Google Scholar]
- 13.Kothapalli D, Grotendorst GR: CTGF modulates cell cycle progression in cAMP-arrested NRK fibroblasts. J Cell Physiol 2000, 182:119-126 [DOI] [PubMed] [Google Scholar]
- 14.Clarkson MR, Gupta S, Murphy M, Martin F, Godson C, Brady HR: Connective tissue growth factor: a potential stimulus for glomerulosclerosis and tubulointerstitial fibrosis in progressive renal disease. Curr Opin Nephrol Hypertens 1999, 8:543-548 [DOI] [PubMed] [Google Scholar]
- 15.Frazier KS, Paredes A, Dube P, Styer E: Connective tissue growth factor expression in the rat remnant kidney model and association with tubular epithelial cells undergoing transdifferentiation. Vet Pathol 2000, 37:328-335 [DOI] [PubMed] [Google Scholar]
- 16.Grotendorst GR: Connective tissue growth factor: a mediator of TGF-β action on fibroblasts. Cytokine Growth Factor Rev 1997, 8:171-179 [DOI] [PubMed] [Google Scholar]
- 17.Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, Grotendorst GR: Connective tissue growth factor mediates transforming growth factor β-induced collagen synthesis: down-regulation by cAMP. EMBO J 1999, 13:1774-1778 [PubMed] [Google Scholar]
- 18.Hisikawa K, Nakaki T, Fujii T: Transforming growth factor-β1 induces apoptosis via connective tissue growth factor in human aortic smooth muscle cells. Eur J Pharmacol 1999, 385:287-290 [DOI] [PubMed] [Google Scholar]
- 19.Shimo T, Nakanishi T, Nishida T, Asano M, Kanyama M, Kuboki T, Tamatani T, Tezuka K, Takemura M, Matsumura T, Takigawa M: Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem 1999, 126:137-145 [DOI] [PubMed] [Google Scholar]
- 20.Egido J: Vasoactive hormones and renal sclerosis. Kidney Int 1996, 49:578-597 [DOI] [PubMed] [Google Scholar]
- 21.Matsubara H: Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ Res 1998, 83:1182-1191 [DOI] [PubMed] [Google Scholar]
- 22.Wolf G, Neilson EG: Angiotensin II as a renal growth factor. J Am Soc Nephrol 1993, 3:1531-1540 [DOI] [PubMed] [Google Scholar]
- 23.Ruiz-Ortega M, Egido J: Angiotensin II modulates cell growth-related events and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int 1997, 52:1497-1510 [DOI] [PubMed] [Google Scholar]
- 24.Mezzano S, Ruiz-Ortega M, Egido J: Angiotensin II and renal fibrosis. Hypertension 2001, 38:635-638 [DOI] [PubMed] [Google Scholar]
- 25.Border WA, Noble NA: Interactions of transforming growth factor-β and angiotensin II in renal fibrosis. Hypertension 1998, 31:181-188 [DOI] [PubMed] [Google Scholar]
- 26.Ruiz-Ortega M, González S, Serón D, Condom E, Bustos C, Largo R, González E, Ortiz A, Egido J: ACE inhibition reduces proteinuria, glomerular lesions and extracellular matrix production in a normotensive rat model of immune complex nephritis. Kidney Int 1995, 48:1778-1791 [DOI] [PubMed] [Google Scholar]
- 27.Wu LL, Cox A, Roe CJ, Dziadek M, Cooper ME, Gilbert RE: Transforming growth factor-β1 and renal injury following subtotal nephrectomy in the rat: role of the renin-angiotensin system. Kidney Int 1997, 51:1555-1567 [DOI] [PubMed] [Google Scholar]
- 28.Ruiz-Ortega M, Lorenzo O, Ruperez M, Blanco J, Egido J: Systemic Infusion of angiotensin II into normal rats activates nuclear factor κB and AP-1 in the kidney: role of AT1 and AT2 receptors. Am J Pathol 2001, 158:1743-1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenzo O, Ruiz-Ortega M, Suzuki Y, Rupérez M, Esteban V, Sugaya T, Egido J: Angiotensin III activates nuclear transcription factor κB in cultured mesangial cells mainly via AT2 receptors: studies with AT1 receptor-knockout mice. J Am Soc Nephrol 2002, 13:1162-1171 [DOI] [PubMed] [Google Scholar]
- 30.Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Pritzl P, Floege J, Schwartz SM: Renal injury from angiotensin II-mediated hypertension. Hypertension 1992, 19:464-474 [DOI] [PubMed] [Google Scholar]
- 31.Adler S, Striker LJ, Sriker GE, Perkinson DT, Hibbert J, Couser WG: Studies of progressive glomerular sclerosis in the rat. Am J Pathol 1986, 123:553-562 [PMC free article] [PubMed] [Google Scholar]
- 32.Grotendorst GR, Okochi H, Hayashi N: A novel transforming growth factor-β response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ 1996, 7:469-480 [PubMed] [Google Scholar]
- 33.Igarashi A, Okochi H, Bradham DM, Grotendorst GR: Regulation of connective tissue growth factor expression in human skin fibroblast and during wound repair. Mol Cell Biol 1993, 4:637-645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hahn A, Heusinger-Ribeiro J, Lanz T, Zenkel S, Goppelt-Struebe M: Induction of connective tissue growth factor by activation of heptahelical receptors: modulation by rho proteins and the actin cytoskeleton. J Biol Chem 2000, 1:37429-37435 [DOI] [PubMed] [Google Scholar]
- 35.Heusinger-Ribeiro J, Eberlein M, Wahab NA, Goppelt-Struebe M: Expression of connective tissue growth factor in human renal fibroblasts: regulatory roles of rhoA and cAMP. J Am Soc Nephrol 2001, 12:1853-1861 [DOI] [PubMed] [Google Scholar]
- 36.Mori T, Kawara S, Shinozakai M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T, Takehara K: Role and interaction of connective tissue growth factor with transforming growth factor-β in persistent fibrosis: a mouse model of fibrosis. J Cell Physiol 1999, 181:153-159 [DOI] [PubMed] [Google Scholar]
- 37.Riser BL, Cortes P, Zhao X, Bernstein J, Dumler F, Narins RG: Intraglomerular pressure and mesangial stretching stimulate extracellular matrix formation in the rat. J Clin Invest 1992, 90:1932-1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riser BL, Cortes P, Yee J: Modelling the effects of vascular stress in mesangial cells. Curr Opin Nephrol Hypertens 2000, 9:43-47 [DOI] [PubMed] [Google Scholar]
- 39.Hishikawa K, Oemar BS, Nakaki T: Static pressure regulates connective tissue growth factor expression in human mesangial cells. J Biol Chem 2001, 18:16797-16803276 [DOI] [PubMed] [Google Scholar]
- 40.Finckenberg P, Lassila M, Inkinen K, Pere AK, Krogerus L, Lindgren L, Mervaala E, Vapaatalo H, Nurminen LM, Ahonen J: Cyclosporine induces myocardial connective tissue growth factor in spontaneously hypertensive rats on high-sodium diet. Transplantation 2001, 71:951-958 [DOI] [PubMed] [Google Scholar]
- 41.Rupprecht HD, Keil A, Hofer G, Schocklamann HO: Regulation of gene expression in cultured rat mesangial cells by NO: downregulation of connective tissue growth factor. J Am Soc Nephrol 1999, 10:579 [Google Scholar]
- 42.Abraham DJ, Shiwen X, Black CM, Sa S, Xu Y, Leask A: Tumor necrosis factor-α suppresses the induction of connective tissue growth factor by transforming growth factor-β in normal and scleroderma fibroblasts. J Biol Chem 2000, 275:15220-15225 [DOI] [PubMed] [Google Scholar]
- 43.Wang S, Denichilo M, Brubaker C, Hirschberg R: Connective tissue growth factor in tubulointerstitial injury of diabetic nephropathy. Kidney Int 2001, 60:96-105 [DOI] [PubMed] [Google Scholar]
- 44.Hammes MS, Lieske JC, Pawar S, Spargo BH, Toback FG: Calcium oxalate monohydrate crystals stimulate gene expression in renal epithelial cells. Kidney Int 1995, 48:501-509 [DOI] [PubMed] [Google Scholar]
- 45.Pawar S, Kartha S, Toback FG: Differential gene expression in migrating renal epithelial cells after wounding. J Cell Physiol 1995, 165:556-565 [DOI] [PubMed] [Google Scholar]
- 46.Folger PA, Zekaria D, Grotendorst G, Masur SK: Transforming growth factor-β-stimulated connective tissue growth factor expression during corneal myofibroblast differentiation. Invest Ophthalmol Vis Sci 2001, 42:2534-2541 [PubMed] [Google Scholar]
- 47.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S: Transforming growth factor-β1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA 1993, 15:770-774 [DOI] [PMC free article] [PubMed] [Google Scholar]