

Abstract
Transfer of collagen type II (CII)-specific monoclonal antibodies induces an acute form of arthritis (collagen type II antibody-induced arthritis, CAIA) in naïve mice. Arthritis was induced using a pair of monoclonal antibodies M2139 and CIIC1, binding to J1 and C1I epitopes of CII, respectively. Thereafter, lipopolysaccharide injection was used to increase the incidence and severity of the disease. This model was used to investigate the effect of genes, age, and sex as well as effector cells in the end-stage effector phase of arthritis pathogenesis. Injection of a single monoclonal antibody induced arthritis only after lipopolysaccharide stimulation. CAIA showed differences in disease penetration among the susceptible strains indicating the importance of non-major histocompatibility complex genes on the antibody effector pathway. B-cell-deficient mice were susceptible to CAIA and in some genetic backgrounds B-cell deficiency leads to enhanced arthritis. Histology of the affected paws revealed massive infiltrations of neutrophils along with bone and cartilage erosion, pannus formation, and fibrin deposition. Depletion of neutrophils significantly reduced the incidence and severity of the disease. CAIA susceptibility increased with age. Males were more susceptible than females and estrogen treatment decreased the development of arthritis. We conclude that CAIA is an acute arthritis triggered by antibody binding and neutrophils bypassing immune activation but with many characteristics in common with collagen-induced arthritis.
The most commonly used animal model for rheumatoid arthritis (RA) is the collagen-induced arthritis (CIA). Collagen type II (CII) is one of the major constituents of the articular cartilage matrix proteins and immunization with native CII in adjuvant induces autoimmune polyarthritis 1 by cross-reactive immune response to homologous collagen. As in RA, susceptibility to CIA is linked to the expression of certain class II major histocompatibility complex (MHC) alleles, 2 indicating the crucial role of T cells. The predominant role played by T cells in the initiation of CIA was demonstrated by using anti-CD4 3 or anti-TCRαβ 4 monoclonal antibodies (mAbs) and T-cell-deficient mice. 5 However, T-cell reactivity alone could not explain the disease pathology in CIA. Both the cellular and humoral immune mechanisms act in concert to mediate the progression of disease in CIA. 6
A requirement for the generation of CII-specific antibodies in the progression of CIA is well documented. B-cell-deficient mice with a highly CIA susceptible genetic background are resistant to the development of CIA. 7 A significant part of the inflammatory attack on the joints is mediated by pathogenic antibodies was emphasized using collagen-specific polyclonal sera both in rats 8,9 and mice. 10,11 Furthermore, purified mAbs were shown to induce arthritis in DBA/1 mice, however the arthritis was very mild. 12 The most likely reason for this is the use of single antibodies with too low concentration. Later, a mixture of anti-collagen mAbs purified from ascites 13,14 was shown to induce severe arthritis. However in these studies, the contribution of preformed immune complexes and other immune factors present in the immune sera or ascites, to the disease process could not be ruled out. Recent studies have shown that both polyclonal and monoclonal antibodies against the ubiquitously expressed self-antigen glucose-6-phosphate isomerase (GPI) could induce arthritis. 15,16 Collectively, these studies demonstrate that the autoantibodies are indeed directly pathogenic in vivo.
We have earlier demonstrated that CII-specific mAbs bind to normal joint cartilage surface in vivo, 17,18 indicating the direct accessibility of collagen epitopes on the articular cartilage. These antibodies in the form of immune complexes might play a central role in triggering the inflammation cascade either by activating the complement system 19 or via direct engagement of cells carrying Fc receptors. 20 It has been shown that both the complement-deficient 21 and Fcγ-chain-deficient 22 mice were resistant to CIA induction. Similarly, absolute abrogation of the GPI-specific antibody-transferred disease was observed in the neutrophil-depleted mice 23 and the importance of complement components, especially the alternative pathway and FcγRIII has been demonstrated to be involved in the disease process. 24,25 These studies propose that the antibody-mediated pathway is one of the major mechanisms of disease induction in arthritis. To facilitate the work, we have further characterized the induction of arthritis by CII-specific mAbs using purified antibodies from culture supernatants, ie, collagen antibody-induced arthritis, CAIA.
Materials and Methods
Animals
BALB/c, DBA/1, B10.Q, B10.RIII, C57BL/6, RIIIS/J, C3H.Q, NFR/N, NOD.Q, QD [(B10.Q X DBA/1)] F1, and QB [(BALB/c × B10.Q)] F1 mice at different ages were used in the present study. B10.Q and B10.RIII strains were originated from Professor Jan Klein (Tubingen, Germany) stock; C3H.Q was from DC Shreffler (St. Louis, MO); and BALB/c, C57Bl/6, NOD, DBA/1, RIII S/J, and Tlr4 (Toll-like receptor 4)-deficient mice were from Jackson Laboratories (Bar Harbor, ME). B-cell-deficient mice MT 26 were backcrossed (>10n) to different genetic backgrounds (B10.Q, B10.RIII, DBA/1, C3H.Q) and intercrossed to provide homozygous and heterozygous littermates. For transfer experiments, mice either from B-cell homozygous knockout breeding, in which wild-type animals were used as controls or from intercrosses were used. To select heterozygous mice during the backcross, DNA was analyzed by using the polymerase chain reaction technique with the 5′ sequence 5′CTATTCGGCTATGACTGG-3′(Neo3) and 5′CCCCACAACCATACTACC-3′(MT2) as the 3′sequence. All of the offsprings were investigated for absence of B cells by cytofluorometric analysis. Blood cells were stained with anti-CD45Ra (B220) antibody labeled with tricolor (Catalag, San Francisco, CA) before analysis. All mice were kept in a conventional but barrier animal facility (as defined in http://net.inflam.lu.se) with a climate-controlled environment having 12-hour light/dark cycles in polystyrene cages containing wood shavings, fed standard rodent chow and water ad libitum. Local animal welfare authorities permitted all of the animal experiments. Table 1 ▶ depicts the summary of the experiments done with the number of mice used in each experiment.
Table 1.
Summary of CAIA Experiments
| Mouse strains | Number of animals used with monoclonal antibody cocktail | CIA susceptibility | ||||
|---|---|---|---|---|---|---|
| Genetics* | Antibody titration | Age | Sex | PMNL depletion | ||
| BALB/c† | 46 | 22 | 15 | − | ||
| QB | 61 | 18 | 34 | 30 | 15 | +++ |
| C57BL/6 | 14 | +‡ | ||||
| DBA/1 | 19 | +++ | ||||
| B10.Q | 40 | ++ | ||||
| B10.Q (B−) | 14 | − | ||||
| QD | 34 | 32 | +++ | |||
| QD (B−) | 7 | − | ||||
| C3H.Q | 15 | +++ | ||||
| C3H.Q (B−) | 4 | − | ||||
| NFR/N | 6 | + (mild) | ||||
| NOD.Q | 11 | − | ||||
| B10.RIII | 36 | +++ | ||||
| B10.RIII (B−) | 17 | 82 | − | |||
| RIIIS/J | 7 | − | ||||
| Tlr4−/− | 10 | ND | ||||
*Cumulative number of mice used in all the experiments. (B−) indicates B-cell-deficient mice.
†Twelve BALB/c mice received single monoclonal antibodies and 10 mice received isotype antibody control.
‡Strong adjuvant is needed. ND, not done; +, susceptible; ++, moderately susceptible; +++, highly susceptible.
Antibodies
The rat IgG2b monoclonal antibody (mAb) clone RB6-8C5 kindly provided by Dr. Coffman (DNAX Research Institute, Palo Alto, CA) was cultured and the antibodies were purified using affinity matrix. Biotinylation of antibodies was done as described earlier. 12
Generation and Characterization of CII-Specific mAbs
The CII-specific hybridomas were generated and characterized as described in detail elsewhere. 12,27,28 From the panel of mAbs generated, a combination of an IgG2b antibody of the clone M2139 binding to the J1 epitope (MP*GERGAAGIAGPK-P* indicates hydroxy proline) and an IgG2a antibody of the clone CIIC1 binding to the C1I epitope (GARGLT) was found to be more arthritogenic than the single mAb and used through out this study. Another CII-specific mAb, CIIF4 was found to inhibit the antibody-induced disease, 29 which might be because of the blocking of arthritogenic antibodies binding to CII via steric hindrance.
Purification of mAbs
CII-specific mAbs were generated in large scale as culture supernatants using Integra cell line 1000 flasks (Integra Biosciences, Walliselen, Switzerland) and the minibioreactor cellmax (GTF, Sweden). Antibodies were purified using γ-bind plus affinity gel matrix (Pharmacia, Sweden). Briefly, culture supernatants were centrifuged at 12,500 rpm for 30 minutes, filtered, and degassed before applying to the gel matrix. Antibodies were eluted using acetic acid buffer at pH 3.0 and neutralized with 1 mol/L of Tris-HCl, pH 9.0. The peak fractions were pooled and dialyzed extensively against phosphate-buffered saline (PBS), pH 7.0, with or without azide. The IgG content was determined either spectrophotometrically or by freeze-drying. The antibody solutions were filter-sterilized using 0.2-μm syringe filters (Dynagard; Spectrum Laboratories, CA), aliquoted, and stored at −70°C until used. Amount of endotoxin content in the antibody solutions prepared was found to be in the range of 0.02 to 0.08 EU/mg of protein as analyzed by the limulus amebocyte lysate (Pyrochrome) method (Cape Cod Inc., East Falmouth, MA).
Passive Transfer of Antibodies
The cocktail of M2139 and CIIC1 mAbs was prepared by mixing equal concentrations of each of the sterile-filtered antibody solutions to get a final amount of 9 mg. Mice were injected intravenously with 0.25 to 0.4 ml volumes of antibody solutions twice with a minimum of 3-hour intervals. As internal controls, mice received equal volumes of PBS. On day 5 or 10, lipopolysaccharide (LPS) (50 μg/mice) was injected intraperitoneally to all of the mice. A pair of irrelevant antibodies of same subclass [mouse anti-human HLA-DRα, IgG2a (L243) and mouse anti-human parathyroid epithelial cells, IgG2b (G11)] was injected in the most susceptible strain, BALB/c as controls.
Clinical Evaluation of Arthritis
Mice were examined daily for the arthritis development before and after LPS treatment for a minimum of 21 days or until the inflammation disappeared. Scoring of animals was done blindly using a scoring system based on the number of inflamed joints in each paw, inflammation being defined by swelling and redness. In this scoring system each inflamed toe or knuckle gave 1 point, whereas an inflamed wrist or ankle gave 5 points, resulting in a score of 0 to 15 (five toes + five knuckles + one wrist/ankle) for each paw and 0 to 60 points for each mouse.
Histological Preparations
Paws were fixed in 4% paraformaldehyde solution for 24 hours, decalcified for 3 to 4 weeks in an ethylenediaminetetraacetic acid solution, dehydrated, and embedded in paraffin. Sections of 6 μm were stained with either fast green and safranin O or hematoxylin and eosin. Mast cells and newly formed cartilage were visible after safranin O staining. For immunohistochemistry, paws were immediately dissected and frozen in OCT compound using isopentane on dry ice. The samples were stored at −70°C until cryosectioned at 10 μm at −30°C. Rat anti-mouse CD11b (M1/70) and rat anti-mouse Ly6G (RB6-8C5) antibodies were used as primary reagents. Either streptavidin or goat anti-mouse peroxidase was used for detection. Diaminobenzidine staining was performed according to established procedures.
Depletion of Neutrophils
RB6-8C5 antibody used in this study did not bind to mature macrophages, monocytes, or splenic B and T lymphocytes and has no significant effect on these populations. 30,31 Hence, to deplete the neutrophils in vivo, 0.5 mg of purified RB6 antibodies in 0.5 ml volume were injected intraperitoneally to naive mice once in 3 days starting from a day before the antibody transfer. In previous studies, this dose of antibody has been shown to completely eliminate the neutrophils in vivo. 23,32 However, CD8+ T cells have been shown to be moderately affected by this antibody treatment, but the effect on CD8+ cells occurs several days after the neutrophils are depleted 33 and as such will not affect this model because T-cell-deficient B10.Q mice were equally susceptible to the disease (Nandakumar et al, unpublished). Control mice received either IgG purified from pooled rat serum or PBS. Neutrophil depletion was monitored by fluorescence-activated cell sorting analysis using biotinylated RB6-8C5 and streptavidin-cychrome. All of the mice received arthritogenic mAb cocktail on day 0 and LPS on day 10 in this experiment. Arthritis was scored as described earlier.
Castration and Hormone Treatment
Both female and male mice were castrated under avertin anesthesia. The ovariectomy or vasectomy was done after a single incision through the peritoneum. After the castration, the mice were rested for 3 weeks before starting the hormone treatment. The hormone treatment was started 5 days before antibody transfer and given twice a week with subcutaneous injections of 3.2 μg of E2 (17β-estradiol-bensoate; Sigma, St. Louis, MO) in a volume of 100 μl of miglyol. The control groups were treated in a similar way with subcutaneous injections of miglyol only.
Statistical Analyses
All of the mice were included for calculation of arthritis susceptibility and severity. The severity of arthritis was analyzed by Mann-Whitney U-test or Kruskal-Wallis and the incidence by chi-square or Fisher’s exact test using the Statview 5.0.1 version. Significance was considered when P ≤ 0.05.
Results
CII-Specific mAbs Induce a Severe Acute Arthritis (CAIA) in Mice
A mAb cocktail containing IgG2b antibody from the clone M2139 binding to J1 epitope and IgG2a antibody from the clone CIIC1 binding to C1I epitope was found to be arthritogenic in BALB/c and (BALB/c X B10.Q)F1 (= QB) mice. A dose titration showed that the cocktail induced arthritis at 3 mg but with the most efficient dose at 9 mg (Table 2) ▶ . Higher doses of antibodies had no further enhancing effect on arthritis (data not shown). Therefore, 9 mg was used as the standard dose. Single mAb injection induced arthritis only after LPS stimulation (Table 3) ▶ , whereas the mAb cocktail induced arthritis without LPS injection in many strains of mice (Figure 1, A and B) ▶ . Moreover, the duration of the disease induced by the cocktail (21 to 35 days) was always significantly longer than the single mAb alone (3 to 12 days). A typical arthritis disease curve induced by the cocktail of antibodies in BALB/c mice is depicted in Figure 2a ▶ . Compared to C1, 9 mg of M2139 antibodies induced significantly more severe arthritis with a higher incidence (Table 3) ▶ . However, 4.5 mg of neither M2139 nor CIIC1 induced arthritis in 6-month-old BALB/c mice. A control mAb cocktail of same antibody isotypes did not induce arthritis. Furthermore, there is no difference between the clinical disease induced by these mAbs before or after LPS injection (Figure 2, b and c) ▶ . Also Tlr4-deficient (LPS nonresponder) mice in the BALB/c background were highly susceptible to arthritis induced with the CII antibody cocktail, excluding a crucial role for endotoxin in the antibody preparation (Figure 1, A and B ▶ , and Figure 2c ▶ ).
Table 2.
Titration of Antibody Concentrations to Induce CAIA in BALB/c and QB Mice
| Mouse strain | Antibodies (mg) | Incidence | Max score (mean ± SEM) | ||
|---|---|---|---|---|---|
| A | L | A | L | ||
| BALB/c | 3 | 2/6 | 4/6 | 1.3 ± 0.8 | 11.5 ± 6.4 |
| 6 | 2/4 | 3/4 | 3.0 ± 2.3 | 21.3 ± 7.6 | |
| 9 | 7/7 | 7/7 | 5.7 ± 1.3 | 31.4 ± 3.2 | |
| 18 | 5/5 | 5/5 | 5.0 ± 0 | 30.2 ± 4.3 | |
| 3 | 0/3 | 1/3 | 0 | 1.6 ± 1.6 | |
| QB | 6 | 1/3 | 1/3 | 1.6 ± 1.6 | 5.0 ± 5.0 |
| 9 | 7/12 | 11/12 | 3.3 ± 1.5 | 19.1 ± 4.0 | |
Groups of 4 to 6 months-old naïve BALB/c and QB mice were injected with different amounts of the cocktail of monoclonal antibodies on day 0. All the mice received LPS i.p. on day 5. A, arthritis susceptibility at day 5 (before LPS injection). L, maximal arthritis after LPS injection.
Table 3.
Age Dependence of Monoclonal Antibody-Induced Arthritis
| Mouse strain | Age (months) | Incidence | Max score (mean ± SEM) | ||
|---|---|---|---|---|---|
| A | L | A | L | ||
| BALB/c | 6I | 0/5 | 5/5* | 0 | 13.4 ± 2.1 |
| 6II | 0/5 | 2/5 | 0 | 5.0 ± 3.9 | |
| QB | 2 | 0/6 | 1/6 | 0 | 3 ± 3 |
| 4 | 5/9† | 8/9‡ | 1.5 ± 0.9 | 17.4 ± 6.4† | |
| 8 | 5/6 | 6/6 | 7.5 ± 1.7§ | 19.6 ± 3.7 | |
| 10 | 5/10 | 10/10 | 4.2 ± 2.1 | 22.2 ± 4.5 | |
| 14 | 0/3 | 3/3 | 0 | 15.3 ± 7.5 | |
| QD | 2 | 0/8 | 0/8 | 0 | 0 |
| 6 | 9/24* | 22/24† | 5.1 ± 1.5 | 15.5 ± 1.5¶ | |
Five 6-month-old naïve BALB/c mice were injected with 9 mg of either I) M2139 or II) C1 single antibodies on day 0. Similarly, two other mice strains (QB and QD) were transferred with the cocktail of monoclonal antibodies at different ages at day 0 as described in Materials and Methods. All the mice received LPS on day 5. Mice were scored for arthritis for a minimum of 21 days. Statistically significant results compared between different groups were given.
†, 4 months: 2 months P ≤ 0.05;
‡, 4 months: 2 months P ≤ 0.005;
§, 8 months: 4 months P < 0.05;
¶, 8 months: 4 months P ≤ 0.05. A, indicates arthritis susceptibility at day 5 (before LPS injection) and L, indicates maximal arthritis after LPS injection.
*P ≤ 0.05; ¶ represents P ≤ 0.0001.
Figure 1.

Genetic influence on CAIA susceptibility. A group of normal and B cell-deficient (B−) mice from several different strains were injected with two mAbs on day 0 and all of the mice received LPS on day 5. A: Incidence of antibody-induced arthritis. B: Arthritis severity was indicated as mean max score ± SEM. BALB/c (n = 36), B10.RIII (n = 36), B10.RIII (B−) (n = 17), QB (n = 61), QD (n = 34), QD (B−) (n = 7), B10.Q (n = 40), B10.Q (B−) (n = 14), C3H.Q (n = 15), C3H.Q (B−) (n = 4), DBA/1 (n = 19), DBA/1 (B−) (n = 5), C57BL/6 (n = 14), NFR/N (n = 6), RIIIS/J (n = 7), and NOD.Q (n = 11) mice were used in this experiment. Tlr4-deficient (n = 10) and BALB/c (n = 10) mice were injected with two mAbs on day 0. n indicates the number of mice in each group.
Figure 2.

a: A typical CAIA disease curve in BALB/c mice. A number of mice (n = 12) were injected with two mAb cocktails (M2139 + CIIC1) intravenously on day 0. As an internal control an equal number of mice were injected with PBS. For isotype antibody control a pair of irrelevant antibodies (L243 + G11) were injected into a group of mice (n = 10) of the same age. All of the mice received 50 μg of LPS injection on day 5 intraperitoneally. Arthritis was scored as described in Materials and Methods for 30 days. None of the control mice developed arthritis. Macroscopic arthritis on day 10 was shown after antibody transfer in B10.RIII mice (b: left, normal paw; right, arthritis paw), after antibody transfer and LPS injection in BALB/c mice (c: left, arthritis paw; right, normal paw), and after antibody transfer in Tlr4-deficient mice (d: left, arthritis paw; right, normal paw).
Genetic Heterogeneity
A series of inbred mouse strains were tested for CAIA susceptibility (Figure 1, a and b) ▶ . Many of the strains developed arthritis before the injection of LPS on day 5, starting from 24 hours after the transfer of antibodies. Clearly, BALB/c and B10.RIII are the most susceptible strains. DBA/1 develop severe arthritis but only after LPS injection as observed earlier. 13 A strong non-MHC genetic effect is apparent comparing strains with the same MHC haplotype. Surprisingly the B10.Q strain had a low susceptibility, as compared to B10.RIII, which would argue for a role of MHC, because these mice are MHC q and r congenic strains, respectively. However, we have found that these strains show genetic differences also at chromosome 10. 34 To confirm that the arthritis induction with antibodies was independent of cellular immune system we tested B-cell-deficient mice, which were as susceptible as their wild-type control. Interestingly, B-cell-deficient B10.Q mice were more susceptible than wild-type B10.Q indicating that the B-cell-deficient mice interact with the genetic difference observed between B10.RIII and B10.Q (Figure 1, a and b) ▶ . In fact, B-cell-deficient B10.Q mice were equally susceptible as that of B10.RIII mice arguing against a direct role for MHC.
Histological Evaluation of the Inflamed Paws
Safranin or hematoxylin staining of paraffin sections of the inflamed paws from antibody-transferred and LPS-injected mice with different genetic backgrounds showed variations in the microscopical inflammation, which correlated with clinical observations (Table 4 ▶ and Figure 3 ▶ ; a to d). Synovitis, infiltration of immune cells in the articular cavity, pannus formation, and bone and cartilage erosions were observed. Immunohistochemical staining of the cryosections of the arthritis paws injected with antibodies showed massive infiltrations of neutrophils with very few macrophages (Figure 3, h and l) ▶ . On the other hand, antibody-injected inflamed paws after LPS stimulation showed massive infiltration of both neutrophils and macrophages (Figure 3, f and j) ▶ . In both the phases of the clinical disease (before and after LPS stimulation) negligible numbers of T, B, and dendritic cells were observed among the infiltrating cell populations (data not shown).
Table 4.
Histological Evaluation of Inflamed Paws in Antibody-Induced Arthritis
| Mouse strain | Treatment | Max score (mean ± SEM) | Histological score |
|---|---|---|---|
| BALB/c | PBS + LPS | 0 | N |
| — | 0 | N | |
| mAbs + LPS | 55.3 ± 4.2 | A3 | |
| DBA/1 | PBS + LPS | 0 | N |
| — | 0 | N | |
| mAbs + LPS | 19.3 ± 5.3 | H1 | |
| B10.RIII | PBS + LPS | 0 | N |
| — | 0 | N | |
| mAbs + LPS | 26.3 ± 4.3 | A2 | |
| QD | PBS + LPS | 0 | N |
| — | 0 | N | |
| mAbs + LPS | 20 ± 2.5 | A2 | |
| QB | PBS + LPS | 0 | N |
| — | 0 | N | |
| mAbs + LPS | 27 ± 4.7 | A3 | |
| B10.Q | PBS + LPS | 0 | N |
| — | 0 | N | |
| mAbs + LPS | 20 ± 2.6 | A1 | |
| C3H.Q | PBS + LPS | 0 | N |
| — | 0 | N | |
| mAbs + LPS | 10 ± 5 | H1 |
Histological scores were evaluated as active score (A) from 1 to 3 and healing score (H) from 1 to 3. Active score 1 (A1) is mild synovitis; score 2 (A2), moderate synovitis with pannus formation; score 3 (A3), severe pannus formation, erosion of bone with more fibrin deposition. Healing score 1 (H1), new cartilage formation; score 2 (H2), cartilage and bone formation, score 3 (H3), cartilage and bone formation with ankylosis. N stands for normal. Three to six paws were examined for each group. Paws collected on day 11 after antibody injection (day 5 after LPS stimulation) were used for histological analysis. —, no treatment and paws were used from age-matched naïve mice.
Figure 3.
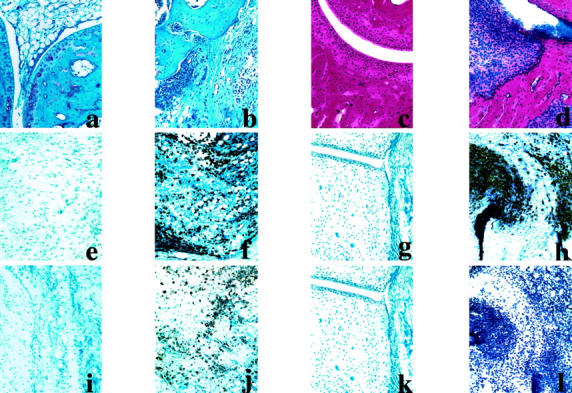
Histology of normal and arthritis joint sections and the infiltrated tissue surrounding the joints in CAIA. Top: Safranin staining of normal (a) and inflamed paw (b) of BALB/c mice on day 11 after antibody transfer (5 days after LPS injection) and hematoxylin staining of normal (c) and arthritis paw (d) of B10.RIII mice on day 17. Middle: Stained for RB6+ cells in normal and inflamed paws of BALB/c (e and f) and B10.RIII (g and h) mice, respectively. Bottom: Stained for Mac1+ cells in normal and inflamed paws of BALB/c (i and j) and B10.RIII (k and l) mice. e, f, h, i, j, and l: Surrounding tissues of the joints showing the nature of infiltrating immune cells in the inflamed paw. Results shown are representative of those obtained from three to four mice in each group. Original magnifications, ×20.
Effect of Neutrophil Depletion on Antibody-Induced Disease
To investigate the role of neutrophils in the antibody transfer model, we used the depleting mAb (RB6-8C5) specific for the neutrophil-restricted surface marker (Ly-6G), which has been used widely to deplete neutrophils in vivo. Both BALB/c and QB (BALB/c × B10.Q) F1 mice were treated intraperitoneally for 2 weeks (500 μg/injection) with the purified RB6-8C5 rat mAb, purified rat IgG or PBS once in 3 days starting from day −1 and arthritogenic mAb cocktail was injected intravenously on day 0. LPS (50 μg/mice) was injected intraperitoneally on day 10. Clinical score was monitored for 21 days after the mAb cocktail transfer, and the efficacy of neutrophil depletion in peripheral blood was monitored by flow cytometry. Incidence and severity of arthritis in mice treated with RB6-8C5 was significantly low, before and after LPS treatment. We found no observable difference between groups treated with either rat IgG or PBS in incidence and clinical scores (Table 5) ▶ . This data indicates that RB6-8C5-positive neutrophils have an important role in initiating CAIA.
Table 5.
Effect of Neutrophil Depletion on the Antibody-Induced Disease
| Mouse strain | Treatment | Incidence | Max score (mean ± SEM) | ||
|---|---|---|---|---|---|
| A | L | A | L | ||
| BALB/c | RB6* | 1/5 | 2/5 | 1.0 ± 1.0 | 2.0 ± 2.0NS |
| IgG | 4/5 | 4/5 | 15.0 ± 5.4 | 16.0 ± 5.3 | |
| PBS | 4/5 | 5/5 | 11.2 ± 3.9 | 20.2 ± 9.1 | |
| QB | RB6* | 1/5 | 2/5 | 1.0 ± 1.0 | 1.6 ± 1.0 |
| IgG | 4/5 | 5/5 | 13.6 ± 5.0 | 17.2 ± 4.6 | |
| PBS | 4/5 | 5/5 | 14.0 ± 3.3 | 15.0 ± 5.2 | |
Groups of naïve BALB/c (n = 15) and QB mice (n = 15) were treated six times with 0.5 mg of either RB6 or affinity-purified normal rat IgG in 0.5 ml volume or PBS alone i.p. once in every 3 days starting from 1 day before to the anti-collagen antibody transfer. LPS was injected on day 10. Mice were scored for arthritis development up to day 19. A, indicates arthritis susceptibility at day 5 (before LPS injection) and L, indicates maximal arthritis after LPS injection.
*, P ≤ 0.05 when RB6 group was compared to IgG and PBS groups together. NS, not significant.
Age Influence on CAIA
Increasing age of mice has earlier been shown to increase susceptibility to CIA. 35 CAIA shows a similar age-dependent effect as shown in Table 3 ▶ . As the age progresses, the incidence and disease severity were found to be increased in both the strains of mice (QB and QD), when cocktail of mAbs were used to induce the disease.
Gender Influence on CAIA
Male mice are more susceptible to CIA than females 36 and estrogen has been shown to exert a suppressive effect on arthritis. 37 CAIA showed partially the same gender and hormone influence as CIA. As depicted in Table 6 ▶ , before LPS injection an increased incidence of arthritis in the male mice compared to female mice was observed. However, such difference in arthritis susceptibility between sexes was abolished after LPS injection. In B-cell-deficient mice, male mice have enhanced susceptibility compared to females both before and after LPS injection (Table 7) ▶ .
Table 6.
Antibody-Induced Arthritis in Male and Female QB (BALB/c × B10.Q)F1 Mice
| Sex | Age (months) | Incidence | Max score (mean ± SEM) | ||
|---|---|---|---|---|---|
| A | L | A | L | ||
| Male | 4 | 3/5* | 4/5 | 2.4 ± 1.4 | 7.6 ± 3.3 |
| Female | 4 | 0/5 | 4/5 | 0 | 10.4 ± 5.1 |
| Male | 10 | 5/10† | 10/10 | 4.6 ± 2.1 | 23.1 ± 4.8 |
| Female | 10 | 0/10 | 7/10 | 0 | 15.1 ± 4.6 |
Each group of animals (n = 5 to 10) was injected with a cocktail of two monoclonal antibodies (CI + M2139) i.v. on day 0 and on day 5, all the animals received 50 μg of LPS i.p. Equal number of animals injected with PBS on day 0 and LPS on day 5 served as controls. Arthritis was scored for 24 days. None of the control animals developed arthritis. A, indicates arthritis susceptibility at day 5 (before LPS injection) and L, indicates maximal arthritis after LPS injection.
*, P ≤ 0.05;
† P ≤ 0.01.
Table 7.
Amelioration of Antibody-Induced Disease by Estrogen in B-Cell-Deficient Mice
| Sex | Operation | Treatment | Incidence | Max score (mean ± SEM) | ||
|---|---|---|---|---|---|---|
| A | L | A | L | |||
| Male | — | — | 2/11 | 8/11 | 15.0 ± 5.0 | 16.5 ± 2.3 |
| Female | — | — | 0/11 | 3/11* | 0 | 12.7 ± 2.4 |
| Male | Sham | — | 0/4 | 3/4 | 0 | 38.0 ± 8.7 |
| Male | Castrated | — | 1/6 | 6/6 | 1 ± 0 | 42.3 ± 5.0 |
| Male | — | Miglyol | 5/8 | 6/8 | 15.8 ± 2.6 | 27.8 ± 3.0 |
| Male | — | E2 | 3/7 | 3/7 | 8.7 ± 1.9 | 9.3 ± 1.7* |
| Female | Sham | — | 3/12 | 3/12 | 10.7 ± 5.2 | 16.6 ± 4.0 |
| Female | Castrated | — | 3/10 | 8/10 | 15.0 ± 8.7 | 20.3 ± 5.0 |
| Female | Castrated | Miglyol | 3/7 | 7/7 | 8.7 ± 1.9 | 19.4 ± 5.1 |
| Female | Castrated | E2 | 0/7 | 1/7* | 0 | 15.0 ± 0 |
Groups of B-cell-deficient mice (n = 6 to 12) of both sexes in B10.RIII background were used in these experiments. Anti-CII antibody cocktail was injected i.v. on day 0, followed by i.p. injection of LPS on day 5. Animals were scored for a minimum of 21 days. A, indicates arthritis susceptibility at day 5 (before LPS injection) and L, indicates maximal arthritis after LPS injection.
*, P ≤ 0.05.
To clarify the influence of female sex hormones in CAIA, male and castrated female μMTBR−/− mice were treated with 17β-estradiol (E2) solubilized in a synthetic oil (miglyol) or with miglyol only as a control, before and after injection with antibodies. The E2-treated castrated females had a clearly decreased incidence compared with the miglyol-treated mice. In the noncastrated male mice, the E2 treatment led to less severe disease but with no differences in the arthritis incidence (Table 7) ▶ .
Discussion
The role for antibodies in the pathogenesis of arthritis has recently been highlighted and the CIA model has been, and will be, of crucial importance in such studies. Although the arthritogenic potential of antibodies to CII is well known, it is of importance to have well-characterized mAbs as tools for these studies. We have here characterized a pair of such antibodies and describe several new characteristics of the collagen antibody-induced arthritis (CAIA).
Several groups have reported transfer of arthritis using anti-CII antibodies with LPS. 13,14,38-45 In the present study, we could transfer arthritis with pure IgG mAbs in different strains of mice without any other stimulants, which open up the field to understand the molecular parameters of the antibodies responsible for their arthritogenicity with reduced complexity. The presence of secondary stimulus, LPS increases the disease incidence and severity. LPS has been previously shown to exacerbate arthritis both in CIA 46,47 and the antibody transfer model. 13 In the later model, LPS was shown to decrease the threshold level of antibodies required to induce arthritis apart from bypassing their epitope specificity. 13 The difference in the disease penetration in different strains of mice could be because of the genetic variation in the non-MHC gene regions on the antibody effector pathway. Genetic studies involving different mouse crosses are underway to address this question directly. It is of interest to note that the non-MHC genes do have an influence on several different immunological parameters in the inbred mouse strains used to study the autoimmune diseases 48 and in the immune complex-mediated diseases contribution of complement or FcγRs could be varied depending on the genetic background of the mice. 49
Antibodies as a constituent of immune complexes play a pivotal role in triggering inflammation in a number of autoimmune diseases. It is most likely that the first step in the initial triggering event in this transfer model is the formation of collagen-IgG immune complexes on the cartilage surface or in the synovium. Collagen epitopes are located in a repetitive structure formed on the cartilage surface, hence it is possible that the two different antibodies can form multimeric complexes more effectively on joint surfaces favoring arthritogenicity either by optimal complement activation or binding to FcgR-bearing cells. Earlier it has also been demonstrated that the autoreactive antibodies to CII initiate inflammation by binding to articular cartilage and thus causing activation of complement, C3 deposition, 10 and eventual cleavage of C5. 50 FcRs on mast cells can act as a link between the immune complexes and the downstream inflammatory mechanisms. 51
Complement fragments binding to immune complexes, tissue damage, and/or FcγR cross linking can activate local mononuclear cells that in turn could release proinflammatory cytokines in or near the joints inducing neutrophil and macrophage recruitment. These phagocytes can further get activated and create a proinflammatory cytokine milieu that could affect the activities of resident cell populations present in the joint. Using anti-CII antibody transfer, it has been shown that interleukin-1 and tumor necrosis factor-α but not interleukin-6 is important in this model, 41 similar to anti-GPI antibody-induced disease. 52 Release of granules containing many tissue-degrading enzymes by macrophages and neutrophils can amplify their responses. Earlier it has been reported that neutrophil elastase can increase the availability of CII epitopes for antibody binding by disrupting the outer layer of the articular cartilage surface. 53 Moreover, it has been demonstrated that mice deficient for neutrophil-derived protease dipeptidyl peptidase I were protected from anti-CII antibody-induced arthritis. 54 Figure 4 ▶ describes the possible scenario of antibody-induced disease in the inflamed joint.
Figure 4.

Schematic diagram of possible interactions of immune effector molecules in the joint after antibody transfer. IC, immune complex; N, neutrophils; M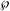 , macrophages; M, mast cells; C1q, C3, C3a, C5a, and B, complement components; FcγR, Fcγ receptor; IL-1, interleukin 1; TNF, tumor necrosis factor; TGF, transforming growth factor; LTB4, leukotriene B4; MIP-1α, macrophage inflammatory protein.
, macrophages; M, mast cells; C1q, C3, C3a, C5a, and B, complement components; FcγR, Fcγ receptor; IL-1, interleukin 1; TNF, tumor necrosis factor; TGF, transforming growth factor; LTB4, leukotriene B4; MIP-1α, macrophage inflammatory protein.
We have earlier shown that CIA is modulated by female sex hormones. Castrated female mice develop a more severe disease compared to normal mice. Estrogen treatment of castrated mice immunized with CII strongly modulates the disease by delaying the onset of arthritis, decrease in incidence and less severe disease in a dose-dependent manner. 55 In CIA, estrogen has been shown to decrease CII-specific antibody response, while increasing the polyclonal antibody production. 37 In the present study we observed that the E2 treatment significantly reduced the arthritis incidence indicating that estrogen could also have an important role in the antibody effector mechanisms either by down-regulating the proinflammatory mediators 56 or affecting the neutrophil-mediated inflammation. 57
In RA patients, activated B cells and plasma cells are present in the inflamed synovium that differentiate locally into antibody-producing plasma cells and in the established disease, lymphoid follicles with germinal centers may be present as well. This results in high levels of local Ig production and the deposition of immune complexes (which may include IgG and IgM rheumatoid factors) in the synovium and articular cartilage. 58,59 Immune complexes in RA have been proposed to play a role in the generation of invasive pannus and in irreversible cartilage matrix degeneration. Indeed, B cells and immune complexes are often located adjacent to and within sites of tissue destruction in RA joints. Antibodies sequestered within cartilage layers have been detected in more than 80% of cartilage samples from RA patients. 60,61 Intravenous injection of antibodies to CII, purified from the plasma of a RA patient, induced arthritis in normal mice. 62 These data suggest that the threshold level, epitope specificity, affinity, and isotype of the accumulated antibodies on the joint cartilage surface and the presence of inducers of proinflammatory cytokines such as LPS from intestinal flora might contribute to the ultimate pathogenicity of the antibodies.
In conclusion CIA, the most common animal model for RA is a very complex disease, which consists of two distinct phases; immune response and the inflammatory process. Previous studies on CIA induction have shown that results obtained from examination of the mechanisms leading to the onset of inflammation cannot be extrapolated to the analysis of established inflammatory responses. This is amply demonstrated by the example of anti-CD4 mAb treatment, which prevents disease when administered at the time of challenge with CII but is ineffective in altering the course of established disease. 3 It is often difficult to separate and study the inflammatory process from the immune response. However in the antibody-induced arthritis used in this study, inflammation occurs in the absence of a primary immune response, thereby allowing one to study the inflammatory processes more clearly avoiding complexity.
Acknowledgments
We thank Carlos Palestro for taking good care of animals and Margareta Svejme for staining histological sections.
Footnotes
Address reprint requests to Dr. Kutty Selva Nandakumar, Section for Medical Inflammation Research, I-11, BMC, 22184, Lund, Sweden. E-mail: nan@inflam.lu.se.
Supported by grants from the Swedish Rheumatism Association; the Swedish Research Council; King Gustaf V’s 80-year; and the Crafoord, Kock, and Österlund Foundations.
References
- 1.Trentham DE, Townes AS, Kang AH: Autoimmunity to type II collagen an experimental model of arthritis. J Exp Med 1977, 146:857-868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wooley PH, Luthra HS, Stuart JM, David CS: Type II collagen-induced arthritis in mice. I. Major histocompatibility complex (I region) linkage and antibody correlates. J Exp Med 1981, 154:688-700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ranges GE, Sriram S, Cooper SM: Prevention of type II collagen-induced arthritis by in vivo treatment with anti-L3T4. J Exp Med 1985, 162:1105-1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moder KG, Luthra HS, Kubo R, Griffiths M, David CS: Prevention of collagen induced arthritis in mice by treatment with an antibody directed against the T cell receptor alpha beta framework. Autoimmunity 1992, 11:219-224 [DOI] [PubMed] [Google Scholar]
- 5.Corthay A, Johansson A, Vestberg M, Holmdahl R: Collagen-induced arthritis development requires alpha beta T cells but not gamma delta T cells: studies with T cell-deficient (TCR mutant) mice. Int Immunol 1999, 11:1065-1073 [DOI] [PubMed] [Google Scholar]
- 6.Seki N, Sudo Y, Yoshioka T, Sugihara S, Fujitsu T, Sakuma S, Ogawa T, Hamaoka T, Senoh H, Fujiwara H: Type II collagen-induced murine arthritis. I. Induction and perpetuation of arthritis require synergy between humoral and cell-mediated immunity. J Immunol 1988, 140:1477-1484 [PubMed] [Google Scholar]
- 7.Svensson L, Jirholt J, Holmdahl R, Jansson L: B cell-deficient mice do not develop type II collagen-induced arthritis (CIA). Clin Exp Immunol 1998, 111:521-526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stuart JM, Cremer MA, Townes AS, Kang AH: Type II collagen-induced arthritis in rats. Passive transfer with serum and evidence that IgG anticollagen antibodies can cause arthritis. J Exp Med 1982, 155:1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takagishi K, Kaibara N, Hotokebuchi T, Arita C, Morinaga M, Arai K: Serum transfer of collagen arthritis in congenitally athymic nude rats. J Immunol 1985, 134:3864-3867 [PubMed] [Google Scholar]
- 10.Stuart JM, Dixon FJ: Serum transfer of collagen-induced arthritis in mice. J Exp Med 1983, 158:378-392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watson WC, Brown PS, Pitcock JA, Townes AS: Passive transfer studies with type II collagen antibody in B10. D2/old and new line and C57Bl/6 normal and beige (Chediak-Higashi) strains: evidence of important roles for C5 and multiple inflammatory cell types in the development of erosive arthritis. Arthritis Rheum 1987, 30:460-465 [DOI] [PubMed] [Google Scholar]
- 12.Holmdahl R, Rubin K, Klareskog L, Larsson E, Wigzell H: Characterization of the antibody response in mice with type II collagen-induced arthritis, using monoclonal anti-type II collagen antibodies. Arthritis Rheum 1986, 29:400-410 [DOI] [PubMed] [Google Scholar]
- 13.Terato K, Harper DS, Griffiths MM, Hasty DL, Ye XJ, Cremer MA, Seyer JM: Collagen-induced arthritis in mice: synergistic effect of E. coli lipopolysaccharide bypasses epitope specificity in the induction of arthritis with monoclonal antibodies to type II collagen. Autoimmunity 1995, 22:137-147 [DOI] [PubMed] [Google Scholar]
- 14.Terato K, Hasty KA, Reife RA, Cremer MA, Kang AH, Stuart JM: Induction of arthritis with monoclonal antibodies to collagen. J Immunol 1992, 148:2103-2108 [PubMed] [Google Scholar]
- 15.Maccioni M, Zeder-Lutz G, Huang H, Ebel C, Gerber P, Hergueux J, Marchal P, Duchatelle V, Degott C, van Regenmortel M, Benoist C, Mathis D: Arthritogenic monoclonal antibodies from K/BxN mice. J Exp Med 2002, 195:1071-1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto I, Staub A, Benoist C, Mathis D: Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 1999, 286:1732-1735 [DOI] [PubMed] [Google Scholar]
- 17.Holmdahl R, Mo JA, Jonsson R, Karlstrom K, Scheynius A: Multiple epitopes on cartilage type II collagen are accessible for antibody binding in vivo. Autoimmunity 1991, 10:27-34 [DOI] [PubMed] [Google Scholar]
- 18.Mo JA, Scheynius A, Nilsson S, Holmdahl R: Germline-encoded IgG antibodies bind mouse cartilage in vivo: epitope- and idiotype-specific binding and inhibition. Scand J Immunol 1994, 39:122-130 [DOI] [PubMed] [Google Scholar]
- 19.Colten HR: Immunology. Drawing a double-edged sword. Nature 1994, 371:474-475 [DOI] [PubMed] [Google Scholar]
- 20.Ravetch JV, Clynes RA: Divergent roles for Fc receptors and complement in vivo. Annu Rev Immunol 1998, 16:421-432 [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA: A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J Immunol 2000, 164:4340-4347 [DOI] [PubMed] [Google Scholar]
- 22.Kleinau S, Martinsson P, Heyman B: Induction and suppression of collagen-induced arthritis is dependent on distinct Fc gamma receptors. J Exp Med 2000, 191:1611-1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wipke BT, Allen PM: Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol 2001, 167:1601-1608 [DOI] [PubMed] [Google Scholar]
- 24.Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, Ezekowitz A, Carroll MC, Brenner M, Weissleder R, Verbeek JS, Duchatelle V, Degott C, Benoist C, Mathis D: Arthritis critically dependent on innate immune system players. Immunity 2002, 16:157-168 [DOI] [PubMed] [Google Scholar]
- 25.Solomon S, Kolb C, Mohanty S, Jeisy-Walder E, Preyer R, Schollhorn V, Illges H: Transmission of antibody-induced arthritis is independent of complement component 4 (C4) and the complement receptors 1 and 2 (CD21/35). Eur J Immunol 2002, 32:644-651 [DOI] [PubMed] [Google Scholar]
- 26.Kitamura D, Roes J, Kuhn R, Rajewsky K: A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 1991, 350:423-426 [DOI] [PubMed] [Google Scholar]
- 27.Karlsson R, Mo JA, Holmdahl R: Binding of autoreactive mouse anti-type II collagen antibodies derived from the primary and the secondary immune response investigated with the biosensor technique. J Immunol Methods 1995, 188:63-71 [DOI] [PubMed] [Google Scholar]
- 28.Schulte S, Unger C, Mo JA, Wendler O, Bauer E, Frischholz S, von der Mark K, Kalden JR, Holmdahl R, Burkhardt H: Arthritis-related B cell epitopes in collagen II are conformation-dependent and sterically privileged in accessible sites of cartilage collagen fibrils. J Biol Chem 1998, 273:1551-1561 [DOI] [PubMed] [Google Scholar]
- 29.Burkhardt H, Koller T, Engstrom A, Nandakumar KS, Turnay J, Kraetsch HG, Kalden JR, Holmdahl R: Epitope-specific recognition of type II collagen by rheumatoid arthritis antibodies is shared with recognition by antibodies that are arthritogenic in collagen-induced arthritis in the mouse. Arthritis Rheum 2002, 46:2339-2348 [DOI] [PubMed] [Google Scholar]
- 30.Conlan JW, North RJ: Neutrophils are essential for early anti-Listeria defense in the liver, but not in the spleen or peritoneal cavity, as revealed by a granulocyte-depleting monoclonal antibody. J Exp Med 1994, 179:259-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedrosa J, Saunders BM, Appelberg R, Orme IM, Silva MT, Cooper AM: Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect Immun 2000, 68:577-583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verdrengh M, Tarkowski A: Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect Immun 1997, 65:2517-2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Oca RM, Buendia AJ, Del Rio L, Sanchez J, Salinas J, Navarro JA: Polymorphonuclear neutrophils are necessary for the recruitment of CD8(+) T cells in the liver in a pregnant mouse model of Chlamydophila abortus (Chlamydia psittaci serotype 1) infection. Infect Immun 2000, 68:1746-1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karlsson J, Zhao X, Lonskaya I, Neptin M, Holmdahl R, Andersson A: Novel quantitative trait loci controlling development of experimental autoimmune encephalomyelitis and proportion of lymphocyte subpopulations. J Immunol 2003, 170:1019-1026 [DOI] [PubMed] [Google Scholar]
- 35.Holmdahl R, Jansson L, Gullberg D, Rubin K, Forsberg PO, Klareskog L: Incidence of arthritis and autoreactivity of anti-collagen antibodies after immunization of DBA/1 mice with heterologous and autologous collagen II. Clin Exp Immunol 1985, 62:639-646 [PMC free article] [PubMed] [Google Scholar]
- 36.Holmdahl R, Jansson L, Larsson E, Rubin K, Klareskog L: Homologous type II collagen induces chronic and progressive arthritis in mice. Arthritis Rheum 1986, 29:106-113 [DOI] [PubMed] [Google Scholar]
- 37.Holmdahl R, Jansson L, Meyerson B, Klareskog L: Oestrogen induced suppression of collagen arthritis: I. Long term oestradiol treatment of DBA/1 mice reduces severity and incidence of arthritis and decreases the anti type II collagen immune response. Clin Exp Immunol 1987, 70:372-378 [PMC free article] [PubMed] [Google Scholar]
- 38.Han Z, Chang L, Yamanishi Y, Karin M, Firestein GS: Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum 2002, 46:818-823 [DOI] [PubMed] [Google Scholar]
- 39.Itoh T, Matsuda H, Tanioka M, Kuwabara K, Itohara S, Suzuki R: The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J Immunol 2002, 169:2643-2647 [DOI] [PubMed] [Google Scholar]
- 40.Johansson AC, Hansson AS, Nandakumar KS, Backlund J, Holmdahl R: IL-10-deficient B10.Q mice develop more severe collagen-induced arthritis, but are protected from arthritis induced with anti-type II collagen antibodies. J Immunol 2001, 167:3505-3512 [DOI] [PubMed] [Google Scholar]
- 41.Kagari T, Doi H, Shimozato T: The importance of IL-1 beta and TNF-alpha, and the noninvolvement of IL-6, in the development of monoclonal antibody-induced arthritis. J Immunol 2002, 169:1459-1466 [DOI] [PubMed] [Google Scholar]
- 42.Kato H, Nishida K, Yoshida A, Takada I, McCown C, Matsuo M, Murakami T, Inoue H: Effect of NOS2 gene deficiency on the development of autoantibody mediated arthritis and subsequent articular cartilage degeneration. J Rheumatol 2003, 30:247-255 [PubMed] [Google Scholar]
- 43.Svensson L, Nandakumar KS, Johansson A, Jansson L, Holmdahl R: IL-4-deficient mice develop less acute but more chronic relapsing collagen-induced arthritis. Eur J Immunol 2002, 32:2944-2953 [DOI] [PubMed] [Google Scholar]
- 44.Wallace PM, MacMaster JF, Rouleau KA, Brown TJ, Loy JK, Donaldson KL, Wahl AF: Regulation of inflammatory responses by oncostatin M. J Immunol 1999, 162:5547-5555 [PubMed] [Google Scholar]
- 45.Yumoto K, Ishijima M, Rittling SR, Tsuji K, Tsuchiya Y, Kon S, Nifuji A, Uede T, Denhardt DT, Noda M: Osteopontin deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Proc Natl Acad Sci USA 2002, 99:4556-4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terato K, Ye XJ, Miyahara H, Cremer MA, Griffiths MM: Induction by chronic autoimmune arthritis in DBA/1 mice by oral administration of type II collagen and Escherichia coli lipopolysaccharide. Br J Rheumatol 1996, 35:828-838 [DOI] [PubMed] [Google Scholar]
- 47.Yoshino S, Sasatomi E, Ohsawa M: Bacterial lipopolysaccharide acts as an adjuvant to induce autoimmune arthritis in mice. Immunology 2000, 99:607-614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johansson AC, Vestberg M, Holmdahl R: Non-major histocompatibility complex dependent variations in lymphocyte activity between inbred mouse strains susceptible to various autoimmune diseases. Scand J Immunol 2000, 52:21-29 [DOI] [PubMed] [Google Scholar]
- 49.Heller T, Gessner JE, Schmidt RE, Klos A, Bautsch W, Kohl J: Cutting edge: Fc receptor type I for IgG on macrophages and complement mediate the inflammatory response in immune complex peritonitis. J Immunol 1999, 162:5657-5661 [PubMed] [Google Scholar]
- 50.Wang Y, Rollins SA, Madri JA, Matis LA: Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci USA 1995, 92:8955-8959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB: Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 2002, 297:1689-1692 [DOI] [PubMed] [Google Scholar]
- 52.Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, Gravallese E, Mathis D, Benoist C: Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med 2002, 196:77-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jasin HE, Taurog JD: Mechanisms of disruption of the articular cartilage surface in inflammation. Neutrophil elastase increases availability of collagen type II epitopes for binding with antibody on the surface of articular cartilage. J Clin Invest 1991, 87:1531-1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adkison AM, Raptis SZ, Kelley DG, Pham CT: Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest 2002, 109:363-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jansson L, Mattsson A, Mattsson R, Holmdahl R: Estrogen induced suppression of collagen arthritis. V: physiological level of estrogen in DBA/1 mice is therapeutic on established arthritis, suppresses anti-type II collagen T-cell dependent immunity and stimulates polyclonal B-cell activity. J Autoimmun 1990, 3:257-270 [DOI] [PubMed] [Google Scholar]
- 56.Salem ML, Hossain MS, Nomoto K: Mediation of the immunomodulatory effect of beta-estradiol on inflammatory responses by inhibition of recruitment and activation of inflammatory cells and their gene expression of TNF-alpha and IFN-gamma. Int Arch Allergy Immunol 2000, 121:235-245 [DOI] [PubMed] [Google Scholar]
- 57.Josefsson E, Tarkowski A, Carlsten H: Anti-inflammatory properties of estrogen. I. In vivo suppression of leukocyte production in bone marrow and redistribution of peripheral blood neutrophils. Cell Immunol 1992, 142:67-78 [DOI] [PubMed] [Google Scholar]
- 58.Kim HJ, Krenn V, Steinhauser G, Berek C: Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J Immunol 1999, 162:3053-3062 [PubMed] [Google Scholar]
- 59.Schroder AE, Greiner A, Seyfert C, Berek C: Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci USA 1996, 93:221-225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jasin HE: Autoantibody specificities of immune complexes sequestered in articular cartilage of patients with rheumatoid arthritis and osteoarthritis. Arthritis Rheum 1985, 28:241-248 [DOI] [PubMed] [Google Scholar]
- 61.Terato K, Shimozuru Y, Katayama K, Takemitsu Y, Yamashita I, Miyatsu M, Fujii K, Sagara M, Kobayashi S, Goto M, Nishioka K, Miyasaka N, Nagai Y: Specificity of antibodies to type II collagen in rheumatoid arthritis. Arthritis Rheum 1990, 33:1493-1500 [DOI] [PubMed] [Google Scholar]
- 62.Wooley PH, Luthra HS, Singh SK, Huse AR, Stuart JM, David CS: Passive transfer of arthritis to mice by injection of human anti-type II collagen antibody. Mayo Clin Proc 1984, 59:737-743 [DOI] [PubMed] [Google Scholar]