

Abstract
The high rate of protein synthesis in β-cells renders them susceptible to endoplasmic reticulum (ER) stress, a condition that can be aggravated by additional imbalances in ER homeostasis and could potentially contribute to the pathogenesis of type-1 and type-2 diabetes. Islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) is an ER-resident protein that is specifically expressed in pancreatic β-cells and is a major target of diabetogenic CD8+ T cell responses in non-obese diabetic (NOD) mice. We produced transgenic mice expressing human IGRP (hIGRP) under the control of rat insulin promoter (RIP) to study epitopes in hIGRP capable of driving diabetogenic human leukocyte antigen (HLA)-restricted CD8+ T-cell responses in hIGRP/HLA transgenic NOD mice. Surprisingly, we found that 3 out of 14 lines expressing RIP-hIGRP in a non-T1D-prone genetic background developed a form of early-onset diabetes that was dissociated from autoimmune inflammation of pancreatic islets. We show that diabetes in these 3 lines resulted from increased rates of β-cell death because of ER stress. We hypothesize that IGRP compounds the viability of β-cells undergoing ER stress by generating unfolded proteins in the ER lumen, and that IGRP's location in the ER accounts, in part, for its exquisite immunogenicity in T1D-prone genetic backgrounds.
Keywords: IGRP, ER stress, beta-cell, type 1 diabetes mellitus
Introduction
Beta-cell death is a major contributor to the pathogenesis of both type 1 and type 2 diabetes [1]. Type 1 diabetes (T1D) is caused by a T cell-mediated autoimmune assault against pancreatic β-cells, leading to progressive β-cell depletion, insulin deficiency and hyperglycemia [2]. Although type 2 diabetes is largely caused by insulin resistance and β-cell insufficiency, recent studies have provided evidence for increased rates of β-cell death [3].
ER stress is a condition in which cellular insults like oxidative stress, aberrant Ca2+ regulation or protein overexpression promote inappropriate folding of proteins in the ER. This leads to a cellular response, coined as the unfolded protein response (UPR), which involves both the upregulation of ER chaperones as a way to enhance the protein folding capacity of the ER and an attenuation of new protein synthesis. This response increases the 'ER-associated degradation' (ERAD) of misfolded proteins and promotes cell death [4].
ER stress is a phenomenon that has been associated with different conditions leading to β-cell apoptosis and diabetes [5-7]. Given their role as insulin producers, β-cells have a very well developed ER capable of an extensive ER workload. This is reflected in increased expression of the ER chaperones calnexin and Bip and the ER stress transducer proteins Ire1α and PERK [8]. As a result, β-cells display increased sensitivity to insults like NO-mediated apoptosis [8]. In fact, β-cells are extremely sensitive to oxidative stress, a process that leads to NFκB activation, release of proinflammatory cytokines and chemokines and β-cell apoptosis or necrosis through poly ADP-ribose polymerase (PARP) activation [9]. Overexpression of unfolded or misfolded proteins has also been shown to induce ER stress in β-cells. For example, expression of a mutated form of insulin 2 in the Akita mouse fosters the accumulation of misfolded insulin 2 molecules in the ER of β-cells, leading to increased rates of β-cell loss [10].
Islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) is a member of G6Pase family that is specifically expressed in pancreatic islet cells, as an ER resident protein [11]. The role of this protein is unclear and its catalytic activity remains controversial. However, IGRP is a target of a very substantial population of islet-associated CD8+ T cells in NOD mice [12]. These CD8+ T cells - referred to as 8.3-like CD8+ T cells - express an invariant α chain characterized by Vα17-Jα42 recombination [13-16] and undergo a process of avidity maturation during the progression of islet inflammation to overt diabetes [17]. IGRP has also recently been shown to be a target of autoreactive CD8+ T cells in human diabetic individuals [18-20]. Here, we report on novel lines of transgenic mice that express a rat-insulin promoter (RIP)-driven human IGRP transgene. We show that overexpression of IGRP in pancreatic β-cells can induce a form of early onset non-immune-mediated diabetes that is associated with increased ER stress and β-cell death. Transgenic lines expressing equivalent amounts of endogenous mIGRP and hIGRP do not exhibit increased ER stress and should enable the mapping of epitopes in hIGRP that are capable of spontaneously eliciting diabetogenic CD8+ T cell responses in the context of T1D-associated HLA molecules in vivo.
Material and methods
Production of transgenic mice
hIGRP cDNA was released from pPCR-Script Amp SK(+) (a gift from Dr. T. Dilorenzo, Albert Einstein College of Medicine, New York, NY) with HindIII and XhoI and ligated downstream of the rat insulin promoter (RIP) in a PBKS+ II plasmid. The RIP-hIGRP insert was then released with NotI, purified and injected into fertilized (B6xSJL) F2 oocytes. The 14 transgene-positive founders that were obtained were crossed with NOD mice. Mice of the F1 and N1 generation were used for this study.
Diabetes
Mice of the F1 and N1 generations were followed for diabetes development by measuring urine glucose levels. Animals with two consecutive readings of 3+ were considered diabetic (Diastix Reagent Strips for Urinalysis, Bayer Inc., Toronto, Ontario, Canada).
Immunohistochemistry and TUNEL staining
TUNEL assay was performed on formalin-fixed paraffin-embedded pancreatic tissue according to the manufacturer's protocol (ApopTag Plus peroxidase in situ Apoptosis Detection Kit, Chemicon International Inc., Temecula, CA). For insulin and CHOP (GADD34, a C/EBP transcription factor induced during the late stages of ER stress) staining, sections were stained with a guinea pig anti-insulin polyclonal Ab (Zymed/Invitrogen, San Francisco, CA) at a 1/100 dilution overnight at 4°C or with a rabbit anti-CHOP (GADD 153) polyclonal Ab (Santa Cruz Biotechnology Inc, Santa Cruz, CA), also at a 1/100 dilution for 60 minutes at room temperature, followed by a goat anti-rabbit secondary Ab (Vector Laboratories Inc., Burlington, Ontario, Canada).
Electron microscopy
Freshly isolated islets were fixed in 2.5% glutaraldehyde in 0.1M cacodylate buffer (pH = 7.4) for 2 hours. The supernatant was discarded and 2% warm agar was added to the pellet. The agar-embedded pellet was then cut into pieces, washed 3 times with the same buffer, post-fixed in 1% osmium tetroxide in cacodylate buffer for 1 hour and dehydrated through a graded series of ethanols and embedded in Spurr's resin. Ultrathin sections were cut on a Reichet-Jung Ultracut E microtome using a diamond knife and stained with uranyl acetate and lead citrate. The sections were examined in a Hitachy H 7000 transmission electron microscope at 75kV.
RT-PCR analysis
RNA was purified from freshly isolated pancreatic islets using RNeasy Mini Kit (Qiagen, Mississauga, Ontario, Canada). Real-time RT-PCR analysis was performed in an ABI Prism 7000 system using TaqMan One-step RT-PCR Master Mix Reagents (Applied Biosystems, Foster City, CA) using the following primers: mIGRP sense: GCTTGGGTTTCGCCATCA; mIGRP anti-sense: CGGCTTGGTGCCATTTTCT; mIGRP probe: 6FAM-CTCAGAAATGTTCCTTCGGAGCTG-TAMRA; hIGRP sense GCTTCTTAGGGTGCTCAACATTG; hIGRP anti-sense: GGGTTAGCACACCACTTTTTGG and hIGRP probe 6FAM-CCTGCTGTGGTCCGTGCCCAT-TAMRA. TaqMan rodent GAPDH control primers and probe (Applied Biosystems) were used to normalize the mRNA levels.
Results
Diabetes development versus hIGRP expression in islets of RIP-hIGR transgenic mice
Fourteen lines of RIP-hIGRP transgenic mice were generated. Unexpectedly, transgene-positive animals from 3 of these lines spontaneously developed diabetes at an early age. As shown in Figure 1A, line #1107: 3 out of 11 mice with an average age at onset of 73 ± 17 days; line #1108: 10 out of 21 mice with an average age at onset of 31 ± 2 days and line #1110: 12 out of 17 mice, with an average age at onset of 41 ± 2 days, developed diabetes.
Figure 1. Diabetes incidence and hIGRP expression levels in RIP-hIGRP transgenic lines.

Diabetes incidence in F1 and N1 RIP-hIGRP transgenic mice (A). Real-time PCR analysis of hIGRP mRNA expression in pancreatic islets of RIP-hIGRP-transgenic lines normalized to GAPDH (B and C) and mIGRP (D) mRNA levels.
Real-time RT-PCR analysis revealed variable degrees of hIGRP mRNA expression in islets isolated from different lines (Figure 1B). For all but 3 of the 14 lines, inter-strain differences in hIGRP transgene expression were consistent in independent experiments (Figure 1C). Comparison of the expression levels of hIGRP versus those of endogenous mouse IGRP (mIGRP) revealed that all but one of the 14 lines (#1114) had high hIGRP/mIGRP ratios (ranging from ~8 to ~33) (Figure 1D). Line #1114 expressed nearly identical levels of hIGRP and mIGRP (hIGRP/mIGRP = 1.4) (Figure 1D). Interestingly, these differences in mRNA expression did not correlate with differences in the diabetes proclivity of the different lines. For example, whereas the diabetes-prone lines #1107, #1108 and #1110 demonstrated moderate levels of expression, the two lines expressing the highest levels of hIGRP (#1105 and #1115 also displaying very high hIGRP/mIGRP ratios) were not diabetes susceptible.
Diabetes in RIP-hIGRP transgenic lines is associated with increased β-cell death
Because the levels of expression of the hIGRP transgene were assessed in purified islets, we considered the possibility that the lack of association between hIGRP mRNA levels and diabetes susceptibility might be due to effects of hIGRP expression on the number and/or function of β-cells within islets. That is, diabetes-prone lines might contain dysfunctional and/or fewer β-cells, as compared to their diabetes-resistant counterparts.
Hematoxilin and eosin staining of pancreatic tissue sections of mice from the diabetes-prone line #1108 did not show any evidence of islet inflammation, indicating that diabetes in these mice was not immune-mediated (Figure 2). We also compared the levels of insulin expression in islets of non-transgenic mice versus mice from non-diabetes-prone line #1115 and diabetes-prone line #1108. Compared to non-transgenic mice (Figure 3A), pancreata from line #1115 exhibited a decrease in insulin staining without displaying a significant reduction in the number of insulin-producing cells (Figure 3B), suggesting that hIGRP overexpression somehow inhibits insulin production by β-cells. In contrast, pancreata from line #1108 displayed both a clear decrease in the number of insulin-producing cells as well as in insulin levels (Figure 3C). This suggested that, in the β-cells of at least some lines, hIGRP overexpression not only downregulated insulin production but also caused β-cell death.
Figure 2. H and E staining of pancreatic islets.
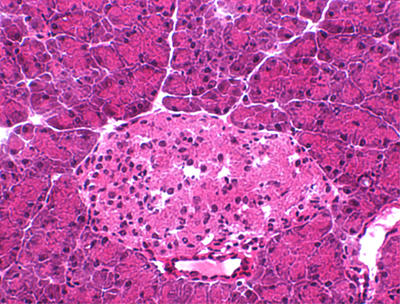
A representative H and E staining of pancreatic islets from diabetes-prone RIP-hIGRP transgenic line #1108 is shown.
Figure 3. Insulin immunohistochemistry of pancreatic islets.

The figure shows representative stains corresponding to pancreata from non-transgenic mice (A), RIP-hIGRP trangenic mice from the non-diabetes-prone line #1115 (B) and the diabetes-prone line #1108 (C).
hIGRP overexpression triggers β-cell necrosis through ER stress
To investigate in more detail the mechanisms underlying the depletion of β-cells in diabetes-prone RIP-hIGRP lines, we carried out electron microscopy analyses of RIP-hIGRP islets. Non-transgenic islets contained β-cells with characteristic insulin-containing granules and an ER with normal luminal space (Figures 4A and 4B). hIGRP overexpression in non-diabetes-prone lines was associated with a reduction in the number of insulin-containing granules in most β-cells. Some β-cells demonstrated signs of ER stress [21, 22], namely thickened nuclear membrane, increased ER luminal space and numerous cytoplasmic vacuoles (Figure 4C). Islets of diabetes-prone lines showed a significant reduction in the number of β-cells and a relative increase in the number of α and δ cells (Figure 4D). The remaining β-cells had significantly fewer insulin granules, pronounced signs of ER stress (Figure 4E) and loss of plasma membrane integrity (Figure 4F), a characteristic feature of necrotic cell death. Dead cells did not show signs of apoptosis such as nuclear fragmentation or formation of apoptotic bodies. In agreement with this interpretation of the data, islets of the diabetes-prone line #1108 did not contain TUNEL+ cells (Figure 5).
Figure 4. Electron micrographs of pancreatic islets.

Representative electron micrographs corresponding to pancreatic islets of non-transgenic mice (A and B) and RIP-hIGRP transgenic mice from non-diabetes-prone line #1115 (C) and diabetes-prone line #1108 (D-F). Note that, as compared to insulin granules, glucagon granules are round, fairly uniform in size and bear a membrane that is closely apposed to the granules' electron-dense content. Arrows indicate dilated ER or nuclear membrane. V: vacuoles. SG: secretory granules. ER: endoplasmic reticulum.
Figure 5. TUNEL assay.

Representative TUNEL staining of pancreatic islets from RIP-hIGRP transgenic mice from the diabetes-prone line #1108.
Protein overexpression has been shown to trigger cell death by inducing ER stress [22-24]. CHOP (GADD34) is a member of the C/EBP family of transcription stress factors, which is strongly induced during late stages of ER stress [4, 8, 23]. We evaluated the presence of CHOP in islets of RIP-hIGRP transgenic mice. Although islets from non-transgenic mice (Figure 6A) and mice of the non-diabetes-prone line #1115 did not express CHOP (Figure 6B), islets of mice of the diabetes-prone line #1108 demonstrated clear staining for CHOP in a significant fraction of islet cells (Figure 6C). These data confirmed that, in at least some transgenic lines, overexpression of RIP-hIGRP caused ER stress and promoted β-cell death.
Figure 6. CHOP immunostaining of pancreatic islets.

Representative staining of pancreata from non-transgenic mice (A), and RIP-hIGRP-transgenic mice from non-diabetes-prone line #1115 (B) and diabetes-prone line #1108 (C). Arrows indicate CHOP staining.
Discussion
This study demonstrates that IGRP overexpression in pancreatic β -cells can activate an ER stress pathway that leads to β-cell death and a non-autoimmune form of early onset diabetes. We generated 14 RIP-hIGRP transgenic lines in a non-type 1 diabetes-prone genetic background and showed that they expressed variable levels of hIGRP mRNA. In these mice, hIGRP overexpression was associated with a significant reduction in the number of insulin granules, dilation of the ER lumen and CHOP expression. These signs of beta cell ER stress were accentuated in mice displaying diabetes proclivity and were accompanied by β-cell necrosis, leading to insulin deficiency and diabetes.
Since IGRP is a member of G6Pase family [11], it is possible that increased expression of this protein interferes with the process of glucose-stimulated insulin secretion through increased dephosphorylation of glucose-6-phosphate and subsequent reduction in the ATP/ADP ratio. However, we think that this is unlikely because IGRP overexpression in transfected cell lines induced no [11, 25] or little [26] glucose-6-phosphatase activity. That IGRP has only minor enzymatic activity has been recently confirmed in IGRP-deficient mice [27], which display subtle disturbances in glucose metabolism. Accordingly, our observation that RIP-hIGRP transgenic mice contain significantly reduced numbers of insulin granules is more consistent with a defect in insulin production than with a defect in insulin secretion.
Diabetes resulting from targeted overexpression of proteins in pancreatic β-cells is not unique to hIGRP and has been seen in other models. The underlying mechanisms, however, were unclear. Lo et al. reported that expression of major histocompatibility complex (MHC) class II molecules in β-cells resulted in a fully penetrant form of diabetes that was dissociated from lymphocytic infiltration of pancreatic islets [24]. In another report, RIP-driven expression of hen egg lysozyme (RIP-HEL) in H-2k-congenic NOD mice caused diabetes, but not insulitis, in 60% of male mice [22]. In a very similar way to RIP-hIGRP mice, diabetes in these animals was associated with a decreased number of insulin granules, reduced β-cell mass and dilation of the ER lumen. The authors suggested ER stress as the underlying cause of the diabetic phenotype of these animals. Unlike diabetes in RIP-HEL transgenic NOD.H-2k mice, which predominantly occurred in male mice, diabetes in RIP-hIGRP transgenic (SJLxB6) F2 x NOD F1 mice was not gender-biased. The mechanisms underlying this gender bias are unclear, but may have to do with differences in the genetic backgrounds of the mice.
The extensive secretory workload of β-cells renders them more susceptible to ER stress than other cell types. The fact that β-cells are the most affected cell type in mice lacking PERK (PKR-like ER kinase, a Ser/Thr kinase involved in cellular responses to ER stress) supports this concept [21]. These mice develop an early onset form of diabetes that is caused by ER stress and reduced β-cell survival. The Akita mouse, which carries a spontaneous mutation in the Ins2 gene, also develops an early-onset form of non-autoimmune-mediated diabetes. Misfolded proinsulin-2 accumulates in the ER of these mice, causing ER distention and expression of the ER chaperone BiP and the transcription factor CHOP [10].
IGRP is an autoantigen recognized by a substantial fraction of islet-associated CD8+ T cells in NOD mice, which we have referred to as '8.3-like' CD8+ T cells. Although 8.3-like CD8+ T cells recognize residues 206-214 of IGRP, multiple other subdominant epitopes are targeted by islet-associated CD8+ T cells. In fact, about 40% of all islet-associated CD8+ T cells are IGRP-reactive [28]. On the basis of the data presented here, it is tempting to speculate that IGRP's exquisite immunogenicity is somehow associated with its location in the ER. Its hydrophobicity may facilitate the generation of a relatively large number of misfolded IGRP molecules and defective ribosomal initiation products. Proteasomal degradation of these byproducts of IGRP synthesis could potentially fuel the generation, expression and recognition (by CD8+ T-cells) of diabetogenic levels of target peptide/MHC class I complexes on the β-cell surface.
It is even possible that IGRP contributes to β-cell death in certain circumstances such as, for example, when β-cells undergo ER stress in response to genetic and environmental factors or in patients who upregulate IGRP expression in response to endogenous or exogenous cues. In addition, by virtue of its localization to the ER membrane, β-cell death resulting from either necrosis or apoptosis would result in the segregation of IGRP into necrotic debris and apoptotic bodies, which are known to be enriched for ER components [29]. These necrotic debris and apoptotic bodies would serve as shuttle vehicles for IGRP to access the antigen cross-presentation pathway, which is responsible for activation and recruitment of diabetogenic CD8+ T cells.
Regardless of whether or not IGRP contributes to ER stress, some of the RIP-hIGRP lines described here should prove useful for investigating the biochemical and pathological consequences of ER stress in β-cells. Other lines (i.e. line #1114) should allow the precise definition of naturally processed epitopes in hIGRP that are capable of eliciting the activation and recruitment of autoreactive CD8+ T cells in humanized HLA transgenic NOD mice lacking endogenous mIGRP.
Acknowledgments
We thank Dr. T. DiLorenzo for providing the hIGRP cDNA and also M. DeCrom, H. Metselaar, T. Irvine, C. Gwozd and W. Dong for excellent technical assistance and animal care. This work was supported by grants from the Canadian Diabetes Association, the Canadian Institutes of Health Research and the Juvenile Diabetes Research Foundation. A.S. is supported by a studentship of the Alberta Heritage Foundation for Medical Research (AHFMR). P.S. is a Scientist of the AHFMR. The JMDRC is supported by the Diabetes Association (Foothills).
References
- 1.Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 2.Eizirik DL, Mandrup-Poulsen T. A choice of death - the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia. 2001;44:2115–2133. doi: 10.1007/s001250100021. [DOI] [PubMed] [Google Scholar]
- 3.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 4.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51(Suppl 3):S455–S461. doi: 10.2337/diabetes.51.2007.s455. [DOI] [PubMed] [Google Scholar]
- 6.Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345. doi: 10.1023/a:1016175429877. [DOI] [PubMed] [Google Scholar]
- 7.Lipson KL, Fonseca SG, Urano F. Endoplasmic reticulum stress-induced apoptosis and auto-immunity in diabetes. Curr Mol Med. 2006;6:71–77. doi: 10.2174/156652406775574613. [DOI] [PubMed] [Google Scholar]
- 8.Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, Akira S, Araki E, Mori M. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci U S A. 2001;98:10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bottino R, Balamurugan AN, Tse H, Thirunavukkarasu C, Ge X, Profozich J, Milton M, Ziegenfuss A, Trucco M, Piganelli JD. Response of human islets to isolation stress and the effect of antioxidant treatment. Diabetes. 2004;53:2559–2568. doi: 10.2337/diabetes.53.10.2559. [DOI] [PubMed] [Google Scholar]
- 10.Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arden SD, Zahn T, Steegers S, Webb S, Bergman B, O'Brien RM, Hutton JC. Molecular cloning of a pancreatic islet-specific glucose-6-phosphatase catalytic subunit-related protein. Diabetes. 1999;48:531–542. doi: 10.2337/diabetes.48.3.531. [DOI] [PubMed] [Google Scholar]
- 12.Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, Serreze DV, Shabanowitz J, Hunt DF, Nathenson SG et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A. 2003;100:8384–8388. doi: 10.1073/pnas.0932778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santamaria P, Utsugi T, Park BJ, Averill N, Kawazu S, Yoon JW. Beta-cell-cytotoxic CD8+ T cells from nonobese diabetic mice use highly homologous T cell receptor alpha-chain CDR3 sequences. J Immunol. 1995;154:2494–2503. [PubMed] [Google Scholar]
- 14.Verdaguer J, Yoon JW, Anderson B, Averill N, Utsugi T, Park BJ, Santamaria P. Acceleration of spontaneous diabetes in TCR-beta-transgenic nonobese diabetic mice by beta-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-alpha chains. J Immunol. 1996;157:4726–4735. [PubMed] [Google Scholar]
- 15.Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med. 1997;186:1663–1676. doi: 10.1084/jem.186.10.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiLorenzo TP, Graser RT, Ono T, Christianson GJ, Chapman HD, Roopenian DC, Nathenson SG, Serreze DV. Major histocompatibility complex class I-restricted T cells are required for all but the end stages of diabetes development in nonobese diabetic mice and use a prevalent T cell receptor alpha chain gene rearrangement. Proc Natl Acad Sci U S A. 1998;95:12538–12543. doi: 10.1073/pnas.95.21.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amrani A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature. 2000;406:739–742. doi: 10.1038/35021081. [DOI] [PubMed] [Google Scholar]
- 18.Unger WW, Pinkse GG, Mulder-van der Kracht S, van der Slik AR, Kester M, Ossendorp F, Drijfhout JW, Serreze D, Roep BO. Human clonal CD8 autoreactivity to an IGRP islet epitope shared between mice and men. Ann N Y Acad Sci. 2007 doi: 10.1196/annals.1394.024. In Press. [DOI] [PubMed] [Google Scholar]
- 19.Mallone R, Martinuzzi E, Blancou P, Novelli G, Afonso G, Dolz M, Bruno G, Chaillous L, Chatenoud L, Bach JM et al. CD8+ T-cell responses identify beta-cell autoimmunity in human type 1 diabetes. Diabetes. 2007;56:613–621. doi: 10.2337/db06-1419. [DOI] [PubMed] [Google Scholar]
- 20.Ouyang Q, Standifer NE, Qin H, Gottlieb P, Verchere CB, Nepom GT, Tan R, Panagiotopoulos C. Recognition of HLA class I-restricted beta-cell epitopes in type 1 diabetes. Diabetes. 2006;55:3068–3074. doi: 10.2337/db06-0065. [DOI] [PubMed] [Google Scholar]
- 21.Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 22.Socha L, Silva D, Lesage S, Goodnow C, Petrovsky N. The role of endoplasmic reticulum stress in nonimmune diabetes: NOD.k iHEL, a novel model of beta cell death. Ann N Y Acad Sci. 2003;1005:178–183. doi: 10.1196/annals.1288.022. [DOI] [PubMed] [Google Scholar]
- 23.Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo D, Burkly LC, Widera G, Cowing C, Flavell RA, Palmiter RD, Brinster RL. Diabetes and tolerance in transgenic mice expressing class II MHC molecules in pancreatic beta cells. Cell. 1988;53:159–168. doi: 10.1016/0092-8674(88)90497-7. [DOI] [PubMed] [Google Scholar]
- 25.Martin CC, Bischof LJ, Bergman B, Hornbuckle LA, Hilliker C, Frigeri C, Wahl D, Svitek CA, Wong R, Goldman JK et al. Cloning and characterization of the human and rat islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) genes. J Biol Chem. 2001;276:25197–25207. doi: 10.1074/jbc.M101549200. [DOI] [PubMed] [Google Scholar]
- 26.Petrolonis AJ, Yang Q, Tummino PJ, Fish SM, Prack AE, Jain S, Parsons TF, Li P, Dales NA, Ge L et al. Enzymatic characterization of the pancreatic islet-specific glucose-6-phosphatase-related protein (IGRP) J Biol Chem. 2004;279:13976–13983. doi: 10.1074/jbc.M307756200. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Martin CC, Oeser JK, Sarkar S, McGuinness OP, Hutton JC, O'Brien RM. Deletion of the gene encoding the islet-specific glucose-6-phosphatase catalytic subunit-related protein autoantigen results in a mild metabolic phenotype. Diabetologia. 2007;50:774–778. doi: 10.1007/s00125-006-0564-1. [DOI] [PubMed] [Google Scholar]
- 28.Han B, Serra P, Amrani A, Yamanouchi J, Maree AF, Edelstein-Keshet L, Santamaria P. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat Med. 2005;11:645–652. doi: 10.1038/nm1250. [DOI] [PubMed] [Google Scholar]
- 29.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]