

Abstract
Orientational constraints obtained from solid state NMR experiments on anisotropic samples are used here in molecular dynamics (MD) simulations for determining the structure and dynamics of several different membrane-bound molecules. The new MD technique is based on the inclusion of orientation dependent pseudo-forces in the COSMOS-NMR force field. These forces drive molecular rotations and re-orientations in the simulation, such that the motional time-averages of the tensorial NMR properties approach the experimentally measured parameters. The orientational-constraint-driven MD simulations are universally applicable to all NMR interaction tensors, such as chemical shifts, dipolar couplings and quadrupolar interactions. The strategy does not depend on the initial choice of coordinates, and is in principle suitable for any flexible molecule. To test the method on three systems of increasing complexity, we used as constraints some deuterium quadrupolar couplings from the literature on pyrene, cholesterol and an antimicrobial peptide embedded in oriented lipid bilayers. The MD simulations were able to reproduce the NMR parameters within experimental error. The alignment of the three membrane-bound molecules and some aspects of their conformation were thus derived from the NMR data, in good agreement with previous analyses. Furthermore, the new approach yielded for the first time the distribution of segmental orientations with respect to the membrane and the order parameter tensors of all three systems.
Keywords: Molecular dynamics simulations, Orientational NMR constraints, 2H-NMR, Oriented samples, Cholesterol, Pyrene, PGLa peptide, Order parameters, Force field calculations
Introduction
Using anisotropic media, such as partially oriented bicelles or macroscopically oriented membranes, NMR investigations can reveal a wealth of information about molecular properties, namely conformation, orientation and dynamics. In many solid state NMR studies of membrane-active peptides and transmembrane proteins, the samples are conveniently prepared with macroscopically oriented bilayers to obtain structural information. The NMR data analysis relies on a uniform alignment of all molecules with respect to the static magnetic field, as it makes use of the orientation dependence of the chemical shift, quadrupolar coupling or dipolar coupling interactions. In contrast to single crystal studies, where the molecules are immobilized in a unique conformation, in the case of lipid membranes and liquid crystalline systems one has to consider a wide distribution of molecular orientations and anisotropic motions. Here, we apply for the first time a new MD strategy to deduce such structural and dynamics information on three representative compounds in biomembranes with increasing complexity: (i) pyrene, (ii) cholesterol, and (iii) the antimicrobial peptide PGLa.
A convenient nucleus for solid state NMR investigations of oriented membranes is deuterium for several reasons (for reviews see e.g., Vold 1994; Davis 1983; Ulrich and Grage 1998). First, the influence of 2H substitutions on the molecular structure and dynamics is negligible. More importantly, in most biologically relevant cases the quadrupolar interaction tensor of a carbon-bound deuterium is to a good approximation aligned parallel to the 2H–C bond and directly reflects the local molecular orientation and dynamics of the labelled segment. Using 2H-NMR, the alignment and dynamic behaviour of lipids, small organic guest molecules and membrane-active peptides has been studied in numerous examples. Most previous strategies to extract the molecular orientation from NMR data, however, have been restricted to molecules with a rigid conformation, e.g. peptides assuming an ideal α-helix with fixed backbone torsion angles. In these studies, several individual labels are usually placed into strategic positions on the rigid molecular part. The measured quadrupolar splittings are then compared in a least squares RMSD analysis with the predicted values upon systematically varying the molecular alignment. The best-fit molecular orientation is determined from a 3D error plot as the one yielding minimal differences between the observed and calculated NMR parameters. However, this RMSD (root mean square deviation) analysis can only give preliminary results for flexible molecules such as peptides or proteins with a high degree of internal mobility.
A proper way to account for motional averaging of the NMR parameters caused by molecular mobility is to run all-atom MD simulations. When applied to biomembranes, however, such simulations pose two closely connected problems: (i) large size of the system and (ii) long simulation times. In particular, water surrounding the lipid bilayer has to be included into the MD simulations, and time-spans have to be covered approaching the NMR time scale. Moreover, the volume of interest has to be surrounded by multiple copies of the central cell to avoid boundary effects. Nevertheless, such simulations have been demonstrated to be feasible for moderately sized membrane segments hosting medium size molecules (see e.g. Tieleman et al. 2001). In the present contribution, we propose an alternative strategy, in which the oriented medium is not explicitly considered, but instead the ordering membrane environment is replaced in the MD simulations by pseudo-forces derived from the measured NMR parameters. This way sufficiently long MD runs become possible and allow to calculate the motionally averaged parameters observed in the NMR experiments. As a result, the full information about molecular orientation, order, segmental motions and even aspects of the molecular conformation could be revealed.
There is one remarkable advantage in the calculations involving orientational constraints. Since an experimentally observed NMR value usually represents a time average over the motions of the molecules, knowledge of the geometry and strength of these molecular motions is a prerequisite for properly interpreting the NMR data in terms of structural parameters. The order parameter, which is typically used to describe the effect of molecular motion on the NMR spectra, however, is often not known a priori. In the present approach, the molecular motions are intrinsically included and the order parameters are obtained as part of the simulation result.
Theory
NMR interaction tensors and coordinate transformations
The orientation dependence of the observed frequencies or splittings of an NMR property can be described by a 2nd rank tensor P, whose components are specified in the context of a coordinate system. On the one hand, a description in a molecule-fixed frame of reference is required to follow changes in orientation, but on the other hand the NMR observables are extracted from the interaction tensor in the laboratory frame related to the static magnetic field. We will define the molecule-fixed frames in the following way from the coordinates of the three bonded atoms A–B–C (see Fig. 1):
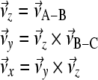 |
1 |
Fig. 1.
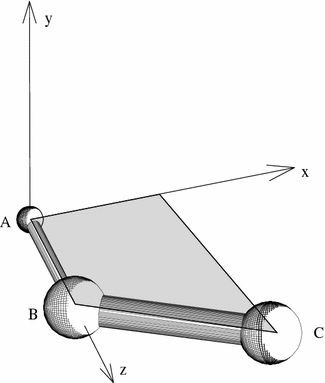
Local coordinate system used to define a tensorial NMR property on atom A. The bond from A to atom B is taken as the local z-direction, the plane spanned by A, B and C defines the x-direction in-plane, and the y-direction is perpendicular to that plane
Atom A is selected as the position of the nucleus of interest (for instance 2H), whose property PA is to be calculated. In case there are several possible ways of selecting atoms B and C, they will be defined such that the valence of bond B–C is a maximum (the valence can be estimated from the bond length, see O’Keefe and Brese 1991). This selection ensures that the y-axis will be perpendicular to a π-system if it exists, and the x-axis will be positioned within the π-plane.
Once we have selected a coordinate system that is fixed to the molecule, we can calculate the columns of the transformation matrix D from the unit vectors along 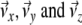 of this frame, given in coordinates of the laboratory frame.
of this frame, given in coordinates of the laboratory frame.
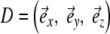 |
2 |
Matrix D can be used to transform the tensorial properties from the molecular to the laboratory frame. The tensor components of P in the molecular frame are thus transformed to yield P′ in the laboratory frame according to
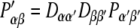 |
3 |
where D is the transformation matrix as given in Eq. 2 (the Einstein sum convention is used for Greek subscripts and α, β = {x, y, z}, the second index indicating the unit vector).
For most deuterium quadrupolar couplings, as well as vicinal dipolar couplings like 15N–1H and 13C–1H the tensor PA will be to a good approximation diagonal in this coordinate system, hence we can easily assign the principal values of PA to the three axes. In cases where the chemical shift tensor is to be analysed, the same molecular coordinate system can be used as in those calculations based on the bond polarization theory (BPT, see Sternberg 1988). Therefore, all these types of NMR interactions can be treated within the same formalism.
In static NMR experiments the tensor components are observed in a coordinate system of reference whose z-axis is aligned with B0. In this frame the measured frequency or splitting is given by the zz-component of the tensor P. However, in the approach presented here we can incorporate further tensor components in the MD simulations, which are accessible for example by measuring the sample at further tilt angles.
Constraints and calculation of molecular properties
To include experimental constraints in the MD simulations, pseudo-energy terms are added to the molecular energy provided by the force field. These pseudo-energies are defined as functions of the difference between the experimental and a calculated tensor property, Pi:
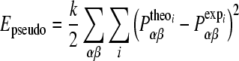 |
4 |
where k is a force constant which is chosen to adjust the size and unit of the energy. The first sum runs over all observed tensor components and does not need to include all possible elements. In a 1D NMR measurement of a single label in an oriented or monocrystalline sample, only the zz-value of the tensor of this site can be experimentally determined. However, any additional tensor component introduced into the calculations will improve the characterization of the orientation of the system. Because of the nature of the tensor transform (Eq. 3) each principal axis of the tensor can be inverted without changing the observed NMR frequency. Therefore, eight discrete tensor orientations are equivalent, even if all elements of the observed tensor are used as constraints. If, however, only the three principal values of the tensor are known, then in addition to this eightfold degeneracy a continuous set of orientations (instead of discrete possibilities) will be compatible with the experimental data from a single label. Therefore, a larger number of orientational constraints from several labels are necessary to determine the orientation of a rigid molecular segment in an unambiguous way. Obviously, if a molecule is intrinsically flexible, an even higher number of constraints will be required.
The experimental NMR values represent an ensemble average over a large number of molecules as well as a time average. The relevant time scale for averaging is given by the inverse of the frequency range that results from the anisotropy of the relevant interaction. When motions are faster than this time scale, a time average of the corresponding NMR parameters is observed. Motions slower than this time scale will result in a distribution of the respective NMR property. In all-atom MD simulations of larger molecules we can only perform calculations for a single molecular system and for time spans on the order of a Larmor period. Assuming that the NMR interactions are already completely averaged within this time, averaged properties can be simulated. This is likely to be the case for motions within the molecule and for the rotational diffusion of small molecules in oriented media such as the lipid bilayer considered here, which are on the time scale of 30 ps to 100 ns (see Brüschweiler 1994). We thus can assume that the time averages over the shorter period accessible for MD simulations do reflect the longer time averages observed by the NMR measurement. For the average of Ptheo we obtain using Eq. 3:
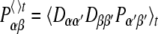 |
5 |
If the NMR property does not change with time, we have to perform the time averaging only over the products of the transformation matrix elements 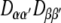 . For most deuterium quadrupolar couplings and vicinal dipolar couplings, any conformational influence on the NMR interaction can be neglected. In the case of chemical shift tensors, however, the change of the tensors in the course of the MD simulation has to be considered.
. For most deuterium quadrupolar couplings and vicinal dipolar couplings, any conformational influence on the NMR interaction can be neglected. In the case of chemical shift tensors, however, the change of the tensors in the course of the MD simulation has to be considered.
Since MD simulations are started with random velocities, we have to run many time steps before a thermal equilibrium is reached, and before the kinetic energy is redistributed properly between the different degrees of freedom of our system. During this equilibration phase of the MD the pseudo-energies will be large and unrealistic, since the proper mean values for the interaction tensors are not yet reached. Therefore the time averaging is performed here using an exponential memory function (Torda and van Gunsteren 1991):
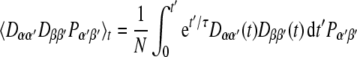 |
6 |
The memory time constant is denoted with τ, and N is the norm of the integral. This memory function serves to scale down the contributions from the beginning of the MD calculation exponentially. By proper selection of τ it can be ensured that we average mainly over the late, relevant time span of interest. Since in conventional MD simulations the equations of motion are integrated in finite time steps Δt, the integral in Eq. 6 is converted into a discrete sum S. During the MD simulation, the sum Sn+1 of the time step n + 1 is calculated from the sum of the previous time step Sn in a recursive manner:
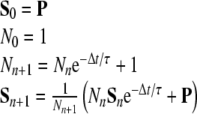 |
7 |
In the course of the MD simulation, S will contain the exponentially weighted time average of the property P, and in the case of a tensor the time averaging will be performed with all of its components.
Calculation of pseudo-forces
To include the NMR constraints into the equations of motion, pseudo-forces 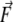 have to be calculated from the respective pseudo-energies (Eq. 4). They are obtained as the derivatives of the energies with respect to the coordinates of the atoms. In the case of orientational pseudo-forces, we have to derive the transformation matrices D with respect to the coordinates of the atoms that were used in their definition (see Eq. 1). The pseudo-forces in the Cartesian directions x, y and z (denoted with the Greek index γ) acting on the atoms A, B, and C (denoted with j, see Eq. 1) are then given by
have to be calculated from the respective pseudo-energies (Eq. 4). They are obtained as the derivatives of the energies with respect to the coordinates of the atoms. In the case of orientational pseudo-forces, we have to derive the transformation matrices D with respect to the coordinates of the atoms that were used in their definition (see Eq. 1). The pseudo-forces in the Cartesian directions x, y and z (denoted with the Greek index γ) acting on the atoms A, B, and C (denoted with j, see Eq. 1) are then given by
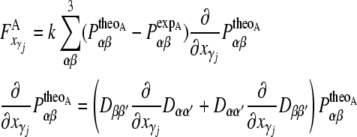 |
8 |
The calculation of the orientational pseudo-forces is thus reduced to determining the derivatives of the elements of the transformation matrices D with respect to the Cartesian coordinates of atoms A, B, and C.
In the following expressions we will use a short notation for the derivatives, omitting all indices:
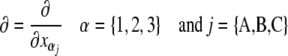 |
9 |
Using the definitions of Eqs. 1 and 9, these derivatives can be calculated in the following way:
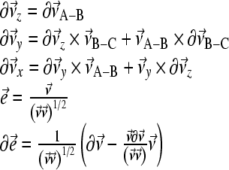 |
10 |
The derivatives of the unit vectors 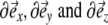 can be used to construct the derivatives of the elements of the transformation matrices
can be used to construct the derivatives of the elements of the transformation matrices 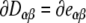 .
.
Because of the time dependence of the transformation matrices, the derivatives were calculated continuously during the MD simulation at each time step. The time average is only calculated for the NMR property.
As evident from Eq. 8, the pseudo-forces depend linearly on the difference between theory and experiment. To avoid too high pseudo-forces in the beginning of the MD simulation, when the deviation of the simulated and experimental values is still large, the contributions of the pseudo-forces are scaled down. Another possibility to avoid too large pseudo-forces is achieved by multiplying the pseudo-forces by weighting factors 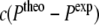 , which lead to nearly constant forces if
, which lead to nearly constant forces if 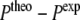 exceeds a threshold ΔP (see Witter et al. 2001):
exceeds a threshold ΔP (see Witter et al. 2001):
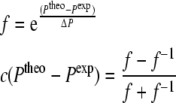 |
11 |
For 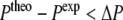 this factor behaves similar to the derivative of the original energy expression (4). It is possible to choose different force constants k (Eq. 8) and width parameters ΔP for each type of NMR property of interest.
this factor behaves similar to the derivative of the original energy expression (4). It is possible to choose different force constants k (Eq. 8) and width parameters ΔP for each type of NMR property of interest.
Order parameter calculation
Besides determining the structure of a molecule, it is equally important to describe its dynamic behaviour in an oriented medium such as a lipid bilayer. MD simulations provide a unique opportunity to obtain information on the molecular dynamics, which is difficult to determine experimentally. The traditional way to account for motional averages in NMR investigation is to introduce order parameters. To this aim, the time average of the transformation matrices D (Eq. 5) can be used to characterize the influence of restricted molecular motions on the anisotropic properties of a molecule. In a similar way, Saupe (1964) defined the so called order tensor to describe the average orientation of a molecule:
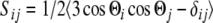 |
12 |
where 〈 〉 denotes the mean value and cosΘi are the z components of the unit vectors of a molecule-fixed frame within the laboratory system of reference. To describe the dynamic properties of flexible molecules, however, the Saupe approach is not sufficient, since molecular groups like peptide side chains may exhibit a larger mobility than the rest of the molecule. For this purpose Limmer (1989) introduced a segmental or site-specific order tensor W, which is the tensorial product of the Saupe order tensor and a conformational order tensor.
By calculating the NMR properties in the course of the MD simulation it is possible to obtain the segmental order tensor WA of the vicinity of atom A. The principal axis system of the tensor property is used to represent the instantaneous segmental orientation, which is then related to the average tensor P determined by Eq. 5. The transformation matrices DPAS, which link the average P with the principal axis system of P at each time step of the simulation, can then be used to determine W (〈 〉t denotes the time average):
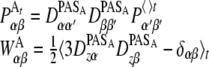 |
13 |
This average is calculated in every step of the MD simulation for all sites A for which the tensorial NMR properties are calculated. In general the complete order tensor has 81 elements, which reduce to nine components if we are interested only in the zz component of a property (Eq. 13). Since the order tensor W (and S as well) is symmetric and traceless, we have to consider only five independent elements. The transformation matrices DPAS in Eq. 13 can be calculated from the eigenvectors corresponding to the principal values of PA. After a full-length MD simulation, the order tensors WA will contain the information describing the time averaged orientation of all sites A.
To calculate the molecular Saupe order tensor, a coordinate system has to be fixed to the molecule. In a perfectly rigid molecule any set of three atoms could be selected at will. However, as most molecules are intrinsically flexible, it does not make sense to define the coordinate system that way, as the Saupe order tensor would then depend on this selection. We therefore decided to use the principal axis system of the tensor of inertia to represent the orientation of the entire molecule (see Burnell and De Lange 1980). This way we can calculate a molecular order tensor that is independent of any particular choice of coordinates:
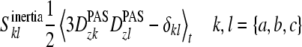 |
14 |
The axes of inertia are denoted with a, b and c, corresponding to the moments of inertia in increasing order. For rod-like molecules the long axis will thus be the a-axis of least inertia.
In many cases it is convenient to diagonalize the order tensor to introduce a principal frame of order (Low 2002). Within this frame there are only two independent parameters: the order parameter S and a biaxality parameter ξ:
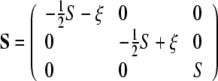 |
15 |
By ordering the principal components as 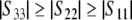 , one gets
, one gets 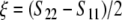 . If the biaxiality ξ is zero, the orientational distribution of the molecule is defined by a single axis, which is called director. In the biaxial case with a non-zero ξ, there exists a second distinct direction orthogonal to the director, and the molecular motions are characterized by two axes. A perfectly axially symmetric molecule that is oriented parallel to the membrane normal will lead to the following order tensor:
. If the biaxiality ξ is zero, the orientational distribution of the molecule is defined by a single axis, which is called director. In the biaxial case with a non-zero ξ, there exists a second distinct direction orthogonal to the director, and the molecular motions are characterized by two axes. A perfectly axially symmetric molecule that is oriented parallel to the membrane normal will lead to the following order tensor:
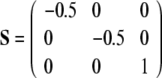 |
16 |
When the molecular Saupe tensor is used to interpret the NMR observables, the conformational tensors CA have to be known as well. These tensors mediate between the orientation and order of a site A and the molecular coordinate system. If the transformation matrices leading from the molecular coordinate system to the individual tensor systems of the sites A are known, CA can be calculated the same way as the order tensor (see Limmer 1989). Only in the case of rigid molecules, simple expressions for CA can be derived from the molecular geometry. Since in our MD simulations the segmental order tensors WA and the molecular Saupe order tensor are calculated directly, the conformational order tensors become obsolete.
Molecular dynamics simulation
The integration of the equations of motions is based on Verlet’s algorithm (Verlet 1967), and time steps of 0.5 fs were employed to sample all high frequency hydrogen atom vibrations. In constrained MD simulations it is generally necessary to control the temperature during the simulation time. This is accomplished by coupling the molecular system to a heat bath which dissipates the heat generated by the pseudo-forces. All prevailing differences between the constraints and their calculated values are sources of heat. To obtain an NTV assemble (with conserved particle number N, temperature T, and volume V), we introduced a proper thermostating procedure (see Evans and Morriss 1990). The coupling to the thermostat is controlled by a coupling time constant η which should be much larger than the time step. This time span η allows an adjustment of the range of thermal fluctuations in the simulated molecular system.
To prevent too large pseudo-forces at the start of the MD simulation the pseudo-forces were gradually increased towards their final values during the MD simulation. To this aim we introduced time dependent scaling factors 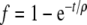 , which approach the value 1.0 in an exponential fashion. The time constant was set in most cases to 200 ps, leading to a relatively smooth course of the temperature.
, which approach the value 1.0 in an exponential fashion. The time constant was set in most cases to 200 ps, leading to a relatively smooth course of the temperature.
When applying the NMR orientational constraints during an MD run, the resulting pseudo-forces will “heat up” the system and enhance its rotational degrees of freedoms. Because the averaging procedure depends on the molecular re-orientations caused by the NMR constraints, some net rotational motion will prevail up to the end of the simulation. In standard MD simulations any overall molecular rotations and translations are subtracted from the velocities, since these external degrees of freedom are not of interest. In the present orientationally constrained calculations, however, only the net translations of the systems are removed.
Parametrization
For NMR spectroscopic considerations it is convenient to introduce the quadrupolar coupling tensor CQ in frequency units:
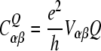 |
17 |
where Q is the quadrupolar moment of the deuterium nucleus, and V the electric field gradient tensor at the nuclear position. The trace of CQ is zero because of 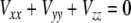 , and in the case of carbon-bonded deuterons the quadrupolar coupling tensor can be considered to a good approximation as diagonal in the coordinate system aligned with the C–2H bond. If the electron distribution is axially symmetric there is only one linearly independent tensor component, otherwise there are two. In the general high temperature case we observe two quadrupolar resonances with frequencies symmetric to zero:
, and in the case of carbon-bonded deuterons the quadrupolar coupling tensor can be considered to a good approximation as diagonal in the coordinate system aligned with the C–2H bond. If the electron distribution is axially symmetric there is only one linearly independent tensor component, otherwise there are two. In the general high temperature case we observe two quadrupolar resonances with frequencies symmetric to zero:
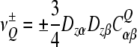 |
18 |
If the z-axis of the local electric field gradient points into the direction of the magnetic field, the corresponding quadrupolar splitting Δ ν is
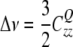 |
19 |
In the case of a C–2H bond the deuterium quadrupolar coupling depends only weakly on the molecular surrounding, hence representative values from static solid state NMR measurements of characteristic substances can be used as quadrupolar constants (Table 1). In this selection we have to keep in mind that the quadrupolar coupling constants depend strongly on the hybridization of the carbon, and to some extent also on the polarization of the C–2H bonds by partial charges caused by electronegative groups.
Table 1.
Deuterium quadrupolar coupling tensors
| Group | CQzz/kHz | CQyy/kHz | CQxx/kHz | Source |
|---|---|---|---|---|
| C(sp2)–2H | 193 | −102.29 | −90.71 | Benzene, naphthalene (Rinné and Depireux 1974) |
| C(sp3)–2H | 175 | −87.5 | −87.5 | Acenaphthene bridge (Rinné and Depireux 1974) |
| –CD3 | 58.33 | −29.17 | −29.17 | C(sp3)–2Ha |
a The values of the quadrupolar coupling constant are scaled by 1/3 to take into account the free methyl rotation
The coordinate system for the local tensors is defined in Fig. 1. The zz-component is aligned with the C–2H bond direction, and the yy-component is perpendicular to any π system. For an aromatic C–2H (sp2) bond an asymmetry parameter of 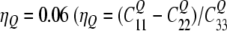 with
with 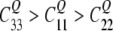 ) was assumed, and the tensor axes were assigned according to calculations of Bailey (1998). The value for the –CD3 group is obtained from the C(sp3)–2H value scaled by 1/3, assuming rapid rotation around the C–CD3 axis.
) was assumed, and the tensor axes were assigned according to calculations of Bailey (1998). The value for the –CD3 group is obtained from the C(sp3)–2H value scaled by 1/3, assuming rapid rotation around the C–CD3 axis.
Program implementation
The routines to apply the proposed orientational constraints are included into the COSMOS-NMR force field, which has been used in a number of previous applications, but which had so far been restricted to distance and chemical shift constraints (see e.g. Sternberg et al. 2003; Witter et al. 2002), and for the force field (Möllhoff and Sternberg 2001; Sternberg et al. 2001). These two types of NMR constraints can be combined with the new orientational constraints as will be demonstrated in the example of PGLa below. The authors provide the backend version of the full COSMOS program, containing all computational procedures without the graphics and modelling interface (GUI) (see http://www.cosmos-software.de). The COSMOS-backend (C++) was compiled for several operation systems including Windows, Unix and Linux.
Applications
The use of NMR orientational constraints is particularly well suited to gain insight into the alignment and dynamics of molecules embedded in biomembranes. In the three examples presented here, we will first demonstrate our new MD approach on pyrene as a simple model compound dissolved in lipid bilayers, then we will apply it to cholesterol as an intrinsic membrane lipid, and finally to the antimicrobial peptide PGLa, which forms an amphiphilic α-helix in membranes.
Pyrene
Pyrene was used as a first example, not only because of its relatively simple and rigid structure, but also because a full set of data is available from a recent 2H-NMR analysis in oriented lipid bilayers (Hoff et al. 2005). The same publication included an all-atom MD simulation of four pyrene molecules in a POPC (1-palmitoyl-2-oleoyl-phosphatedylcholine) membrane. Those previous results will thus allow a direct comparison of our new strategy with established simulation techniques. The present orientational constraint-driven simulation utilizes the experimental 2H-NMR data of deuterium-labelled pyrene (see Fig. 2) embedded at 4 mol% in POPC bilayers. Spectra had been acquired at two discrete sample orientations, namely with the membrane normal aligned parallel and perpendicular to the B0 field. In both orientations two pairs of narrow lines were observed (corresponding to the sites 2H1 and 2H2 + 2H3, respectively), whose splittings changed by a factor of 1/2. This situation can only originate from axially symmetric averaged quadrupolar tensors that are aligned with the membrane normal. This means that the pyrene molecules reorient quickly (on the time scale of the NMR interaction) about the membrane normal. Since the principal axis system of each averaged quadrupolar deuterium coupling tensor coincides with the membrane-fixed coordinate system, the absolute values of all 6 or 5 linearly independent components of the averaged coupling tensor are known and can be included in the present MD simulation. In this case, we could even make use of the signs of the principal quadrupolar components, as they had been deduced from the previous MD simulations mentioned above (Hoff et al. 2005).
Fig. 2.
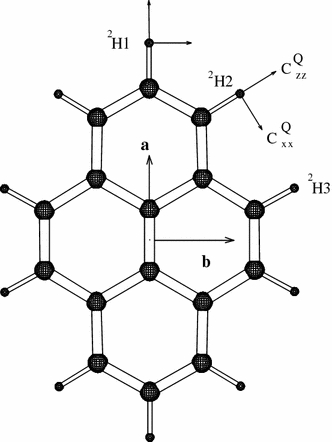
Fully deuterated pyrene molecule with three kinds of labelled sites (2H1, 2H2 and 2H3), illustrating the principal axes of the local deuterium quadrupolar tensors and of the principal axes of inertia a, b and c
Using the 2H-NMR data from the literature, we performed two MD simulations on pyrene constrained by the complete tensors. In the first run, only the contributions to the pseudo-energy arising from the three diagonal elements (first sum in Eq. 4) were considered, and in a second run also the three off diagonal values (in this case 0.0) were included. In the first run, 11.4 ns were simulated, with the MD parameters set as given in Table 2. To study the time behaviour of the pseudo-forces they were not scaled up exponentially as in the other two case studies below. The width parameter was set equal to the experimental error as ΔP = 1 kHz, and the memory time was τ = 200 ps. The time development of the simulated tensor components for the first nanosecond can be seen in Fig. 3. The trajectories are shown for the 2H2 site, which possesses an experimental splitting of 40.5 kHz. After an initial time period of about 250 ps, the final average coupling tensor components are reached and their values fluctuate only weakly. The residual fluctuations depend on four parameters: (i) the thermal fluctuations controlled by the coupling to the heat bath, (ii) the pseudo-energy, (iii) the width parameter ΔP given in Eq. 11, and (iv) the memory time τ of Eq. 6. In the simulated 2H-NMR spectra (Fig. 4) these prevailing small fluctuations translate into line broadening, and the resulting line shapes are indeed very similar to the experimentally observed spectrum (Hoff et al. 2005).
Table 2.
General parameters for the MD simulations with orientational constraints
Fig. 3.
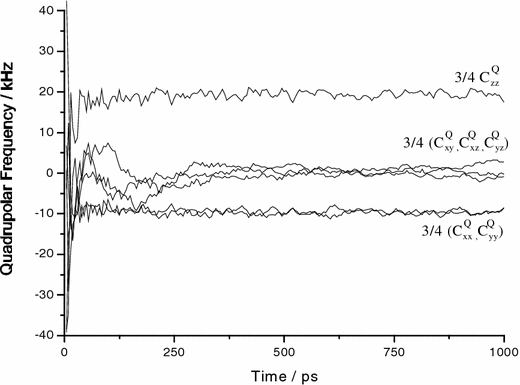
Trajectory of the time development of the averaging for the deuterium tensor components for 2H2 of pyrene. Under the influence of orientational pseudo-forces all components approach their final values after a period of about 250 ps. The component at about 20 kHz is experimentally observable in an oriented sample with the membrane normal parallel to B0. The component at −10 kHz would correspond to an experiment with a perpendicular sample alignment
Fig. 4.
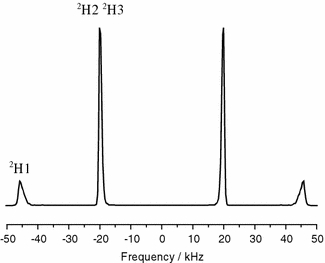
2H-NMR spectrum from the MD simulation of deuterated pyrene, using orientational constraints from an NMR measurement in oriented POPC membranes (Hoff et al. 2005)
In this first simulation we had constrained only the three principal components of the quadrupolar tensor. It turned out that in this case small off-diagonal values did not approach zero but remain in the range of −1.5 to 2 kHz up to the end of the simulation (see Fig. 3). At the beginning of the simulation a high pseudo-energy of 31,000 kJ/mol was encountered, which dropped to 0.8 kJ/mol by the end of the simulation. Because of the large pseudo-energies at the start, we realized that it is preferable to first run a preliminary MD simulation (1–4 ns) with much smaller pseudo-forces, and then to step up the pseudo- forces after some initial averaging has been performed. In the next two cases studies the pseudo-forces were switched on exponentially to avoid long preliminary equilibration periods.
In a second MD simulation of pyrene over 7.8 ns, we also constrained the three off-diagonal tensor components Cxy, Cxz, and Cyz to zero, which are averaged by rotations of the molecule about the membrane normal. The corresponding values calculated from the MD run yielded off-diagonal elements with absolute values smaller than 0.23 kHz. The pseudo-energy increased by a factor of two, as it now contains contributions from both the diagonal and off-diagonal tensor components. In an attempt to compensate for the higher pseudo-energy, we set the memory time to a higher value of τ = 1,000 ps (see Eq. 6). Altogether we obtained smaller fluctuations of the coupling tensor components and much narrower lines in simulated spectra, which can be most likely attributed to the damping effect of the memory function outweighing the higher pseudo-energies.
Table 3 summarizes the calculated quadrupolar splittings corresponding to the results of the second simulation, in which all tensor components were constrained. The values were averaged for any sites on pyrene with mutually parallel C–2H bond directions, so that the 2H1 splitting is a mean value of two and the others are mean values of four sites each. The MD simulation is seen to reflect the experimental 2H-NMR data very well in several aspects. First of all, the calculated quadrupolar splittings reproduce the experimental values very well. The marginally lower values found for the simulated splittings might be explained by a slightly elevated temperature encountered during the simulation. Namely, the mean temperature of this simulation was 302 K and the final temperature 299 K. Since the target temperature of the simulation was 290 K, the NMR constraints produced a constant temperature increase of about 10 K. This difference would explain the slightly smaller simulated splittings compared to the experiment. In full agreement with the experimental data, no significant difference was found for the sites 2H2 and 2H3. In the simulations, the difference between their splittings was within the scatter of the individual sites, and this difference could neither be resolved experimentally. Furthermore, also the averaging of the pyrene molecule in the membrane is reflected correctly in the simulation. The values for the tensor components perpendicular to the B0 field direction are negative and half of the values of Table 3, indicating that the simulation does indeed lead to an averaged axially symmetric tensor aligned parallel to the membrane normal.
Table 3.
Calculated quadrupolar splittings in a 7.8 ns constrained MD simulation of pyrene, compared with the experimental values obtained from a 2H-NMR measurement of the deuterated pyrene in oriented POPC membranes
| Site | Sign of tensor componenta | Constrained MD splitting Δν (kHz) | NMR experimentalb splitting Δ ν (kHz) |
|---|---|---|---|
| 2H1 | + | 91.4 (91.39, 91.4)c | 93.0 |
| 2H2 | + | 39.9 (39.8–40.0)c | 40.5 |
| 2H3 | + | 39.7 (39.6–39.8)c | 40.5 |
a The sign is given for the orientation of the membrane normal in B0 field direction
b Hoff et al. (2005)
c Range of simulated values for two or four sites, respectively
The experimental 2H-NMR data only reflect dynamic averages of e.g. the C–2H bond orientation, but can access only little information on the dynamic processes leading to the observed splittings. Here, the constraint-driven MD simulation provides a way to obtain details on molecular motions that are compatible with the experimental data. In the example of pyrene, the symmetry of the molecule and its motions in the membrane allowed us to derive information on the dynamic behaviour of the molecule from its observed splittings. Since in our case the coupling tensors are diagonal within the bond coordinate system and the segmental order tensor is also diagonal, simple expressions for the quadrupolar splittings are obtained:
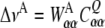 |
20 |
and for the case of an axially symmetric coupling tensor CQ we get
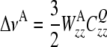 |
21 |
The latter expression is especially useful, since the quadrupolar splitting of a site A is directly related to a single segmental order parameter. Assuming that the order tensor is diagonal in the laboratory frame, Eq. 21 applies to dipolar splittings as well, since the static dipolar coupling tensor is always axially symmetric and traceless, and the same holds for a rapidly rotating CD3-group. However, since the coupling tensor CQ for the C(sp2)–2H bonds is not axially symmetric (see Table 1), the three principal elements of the order tensors WA contribute to the splitting as in Eq. 20. It is therefore not possible to extract the order parameter tensor components from a single experimental splitting. From the course of our MD simulation, on the other hand, all components of the orders tensors WA (Table 4) are available, and the terms of Eq. 20 can be readily analysed.
Table 4.
Simulated order parameters for pyrene in POPC
| Site | Wxx | Wyy | Wzz | | Wxy,Wxz,Wyz| |
|---|---|---|---|---|
| 2H1 | +0.021 | −0.342 | +0.321 | ≤0.03 |
| 2H2 | +0.181 | −0.310 | +0.129 | ≤0.07 |
| 2H3 | +0.165 | −0.294 | +0.129 | ≤0.03 |
| Sbb | Scc | Saa | |Sab, Sac, Sbc| | |
| Moleculea | +0.048 | −0.275 | +0.228 | ≤0.006 |
a The values for the molecular Saupe order tensor are ordered in the columns such that the axes of inertia fit to the corresponding coordinate axes of the 2H1 site (see Fig. 2)
For a perfectly rigid pyrene molecule the molecular Saupe order tensor S and the segmental order tensors WA are linked by fixed conformation tensors. With our choice of the molecular coordinate system, S and WA are identical for the site 2H1, and for the other two sites the tensors are related by simple geometric expressions. Comparing the molecular order tensor S (last row in Table 4) and the segmental order tensor W (e.g. of the site 2H1, which is expressed in the same coordinate system, first row in Table 4), it is noticed that most of the molecular S tensor components are slightly smaller than the corresponding values of the segmental order tensor W. This effect is caused by bond vibrations and molecular twists occurring during the MD simulation. However, as these contributions to the dynamics (manifested in the difference between segmental and molecular order) are small, the mobility of the pyrene in the membrane is well described by the molecular order tensor S. Further qualitative conclusions on the behaviour of the pyrene molecule in the membrane can thus be drawn from the order tensor. Because Saa constitutes the largest (signed value) component, it is obvious that the corresponding axis a (in the system of inertia), and with it the long molecular axis of pyrene, shows some preference for an orientation in the direction of the membrane normal. Since Scc and Sbb are different, the molecular motions are not axially symmetric, but display a preference for a second axis, expressed in a biaxiality parameter ξ = 0.11 (see Eq. 16). This pronounced biaxiality is a consequence of the restricted motions of the flat pyrene molecule within the lipid matrix. A further indication for such a restricted motion is the fact that the smallest component of the order tensor S is found along the c-axis direction perpendicular to the aromatic ring system. This direction has thus the smallest average component  along the membrane normal and is most conserved, as expected for the planar shape of the molecule. In their all-atom MD simulation including 128 lipid molecules and water, Hoff et al. (2005) obtained essentially the same results as in this work, with the exception that they found a much higher Saupe order component Szz = 0.42 (in this work denoted with Scc).
along the membrane normal and is most conserved, as expected for the planar shape of the molecule. In their all-atom MD simulation including 128 lipid molecules and water, Hoff et al. (2005) obtained essentially the same results as in this work, with the exception that they found a much higher Saupe order component Szz = 0.42 (in this work denoted with Scc).
Cholesterol
Cholesterol is a constituent of many biomembranes, and its orientation and motion within the lipid bilayer is of great interest to explain its influence on membrane properties. The behaviour of cholesterol in membranes has been studied extensively, especially by 2H-NMR in view of its sensitivity towards molecular alignment and dynamics (Smith and Oldfield 1984). We have based our MD analysis on a recent study by Marsan et al. (1999), who had measured and interpreted 2H-NMR spectra of partially deuterated cholesterol in oriented DMPC (dimyristoylphosphatidylcholine) bilayers. The labelled sites in the sterol ring system are indicated in Fig. 5.
Fig. 5.
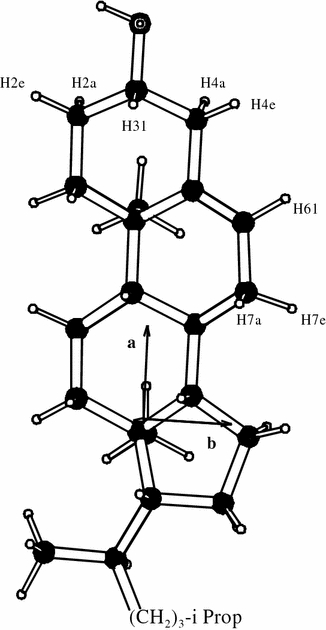
Cholesterol molecule with 2H-labelled sites indicated, for which experimental orientational constraints were available. From the MD simulation quadrupolar splittings were calculated for all deuterons in the ring system
The authors examined six different models of cholesterol structures proposed in the literature using an RMSD analysis to assign all observed lines in the 2H-NMR spectrum. In this analysis the quadrupolar splittings were calculated from the respective rigid molecular models, treating the molecular orientation and the Saupe tensor as free variables. It turned out that the results depended crucially on the selection of the molecular coordinates, and only the data from a neutron diffraction analysis (McMullan et al. 1992) of a cholesterol derivative (20-CH3-methylpregnene-3,20-diol) produced convincing results. This strong dependence on the exact coordinates is regarded as one of the main drawbacks of RMSD analyses of rigid molecular models.
In the analysis presented here, we performed MD simulations for both methylpregnenediol and cholesterol, having modelled the latter with COSMOS (see Fig. 5). As constraints for the MD simulations we used the 2H-NMR splittings of the eight experimentally observed deuterium labels of Marsan et al. (1999), together with their best assignment (see Table 5). In addition, from the MD simulations we predicted the putative quadrupolar splittings of all other protonated sites of the sterol ring system. This new data from our MD simulations can thus be used in the future to assign further 2H-NMR spectra without the need for any special labelling schemes or particular molecular coordinates. As outlined above, we again used the full quadrupolar tensors as constraints. Two short preliminary MD runs were performed to determine the signs of the coupling constants, since this information is required for the full tensorial constraints. All quadrupolar splittings of cholesterol which had been measured in oriented samples with the membrane normal parallel to B0 yielded negative coupling constants, (giving correspondingly positive values for the perpendicular orientation, see Table 5). As in the example of pyrene, MD simulations over 1,000 ps were sufficient to reproduce all features of molecular motion. To avoid too large pseudo-forces at the start of the simulation, the pseudo-forces were exponentially increased to their final values during the MD run. The parameters used in the simulations are summarized in Table 2.
Table 5.
Calculated deuterium quadrupolar splittings from a constrained MD simulation compared with the experimental 2H-NMR data (Marsan et al. 1999)
| Site | Constrained MD splitting of methyl-pregnenediol (kHz) | Constrained MD splitting of cholesterol (kHz) | Experimental NMR splitting (kHz) |
|---|---|---|---|
| H2a | −101.5 | −101.4 | 101.68 |
| H2e | −66.7 | −67.7 | 67.86 |
| H3_1 | −107.1 | −106.7 | 107.30 |
| H4e | −63.2 | −62.3 | 62.68 |
| H4a | −94.3 | −93.0 | 94.98 |
| H6 | −7.29 | −6.69 | 6.44 |
| H7a | −92.9 | −95.4 | 96.12 |
| H7e | −91.6 | −91.3 | 91.48 |
| C18–CD3 | −33.0 | −33.8 | |
| C19–CD3 | −37.5 | −34.7 | |
| H1a | −104.2 | −99.2 | |
| H1e | −69.4 | −46.1 | |
| H8_1 | −107.0 | −100.3 | |
| H9_1 | −112.6 | −103.0 | |
| H11a | −106.4 | −102.7 | |
| H11e | −50.7 | −74.7 | |
| H12a | −46.4 | −13.1 | |
| H12e | −103.1 | −101.1 | |
| H14_1 | −103.4 | −100.3 | |
| H15_1 | −102.1 | −81.8 | |
| H15_2 | −93.5 | −88.9 | |
| H16_1 | −17.6 | −40.9 | |
| H16_2 | −23.1 | −27.6 | |
| H17_1 | −85.7 | −92.7 |
The final results for the quadrupolar splittings obtained from the constrained MD simulations of both sterols are summarized in Table 5. The corresponding simulated 2H-NMR spectrum of cholesterol is shown in Fig. 6, indicating the peak positions of all ring deuterons. This presentation can be helpful in the future for assigning the signals of fully deuterated cholesterol derivatives. Overall, the theoretical and experimental values agree very well within 1 kHz (except for 2H6). The only noteworthy deviation is seen for 2H7a, where the calculated splitting of methylpregnenediol differs by 3.2 kHz from the experiment. Overall, the simulated cholesterol quadrupolar couplings agree slightly better with the experimental values than the respective methylpregnenediol values. This finding is also reflected in the lower mean temperature reached in the simulation of cholesterol (323 K) than for methylpregnenediol (336 K). The higher mean temperature of the latter is probably also responsible for the slight differences between the calculated splittings of the two molecules in some of the other sites, too (see Table 5).
Fig. 6.
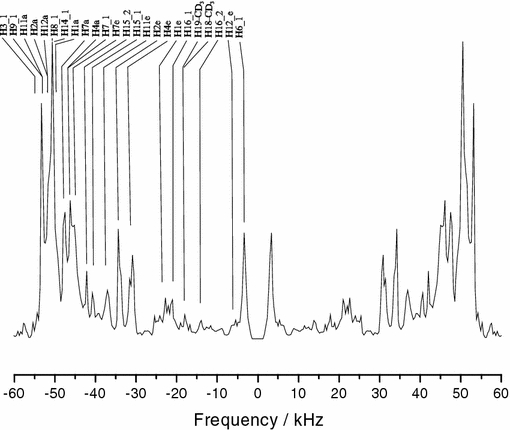
Calculated 2H-NMR spectrum from the MD simulation of deuterated cholesterol in an oriented membrane, with the membrane normal parallel to B0. Pseudo-forces were applied in the MD simulation to a sub-set of deuterons for which experimental data was available (see Fig. 5). The peak positions of all other deuterons connected to the ring skeleton are also predicted and indicated (see Table 5). The intensity of the CD3-signals has to be multiplied by a factor of three
The constrained MD simulation showed that both cholesterol derivatives essentially assume an upright orientation in the membrane and perform rotations mainly around their long molecular a-axis of inertia. This behaviour is reflected in the molecular Saupe order tensor (see Table 6), whose values are not far from the ideal axially symmetric case of Eq. 16 with a low biaxiality of ξ = 0.01. Previous calculations of the order tensor from the NMR data (Marsan et al. 1999) had given similar results as the present MD simulation. We find that the preferred orientation of the long a-axis of the molecule displays a small mean tilt angle Θ a of 13° with respect to the membrane normal. A similar result was obtained by Marsan et al. (1999), who reported a tilt of the long molecular axis (as defined in the paper of Dufourc et al. 1984) with respect to the membrane normal of 12° (provided that in their RMSD data analysis the restriction to a rotationally symmetric order tensor was dropped).
Table 6.
Molecular Saupe order tensor derived from the constrained MD simulation, compared with the values derived from the previous RMSD analysis (Marsan et al. 1999)
| Tensor component | MD simulation of methylpregnenediol | MD simulation of cholesterol | Static RMSD analysis |
|---|---|---|---|
| Saa | 0.87 | 0.88 | 0.94 |
| Sbb | −0.44 | −0.44 | −0.48 |
| Scc | −0.43 | −0.44 | −0.46 |
| Sab | 0.0 | −0.11 | |
| Sac | 0.0 | −0.12 | |
| Sbc | 0.0 | 0.0 |
We note that the axially symmetric averaging of cholesterol does not imply that its motion resembles a smooth rotation about a fixed axis. In the MD simulation we observed frequent jumps of the long axis by 20 to 30°, accompanied by a change of the conformation of the aliphatic tail of the molecule. This behaviour can be examined more closely by inspecting the trajectory of the angle Θ a between the a-axis of inertia and the membrane normal (z-axis, see Fig. 7). Under the influence of the full pseudo-forces after 150 ps, the long axis of cholesterol frequently jumps off the most preferred orientation of Θ a = 4°. As a consequence, the mean orientation and the most frequent orientation do not coincide. Methylpregnenediol behaves clearly different in the MD simulation, since it has only a short aliphatic tail that does not give rise to conformational changes.
Fig. 7.
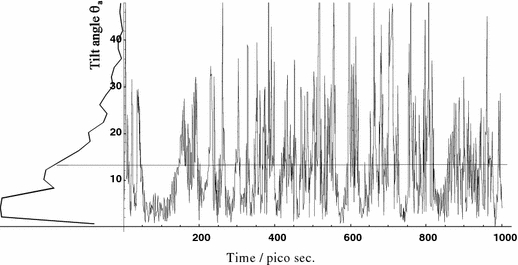
Trajectory of the time development of the angle Θa between the a-axis of inertia and the z-direction (membrane normal). The axis frequently jumps off its most preferred orientation due to conformational changes in the aliphatic tail of the molecule, hence the mean tilt (13°) and the most preferred tilt (4°) do not coincide
PGLa
In our third case study, the new MD approach was applied to a membrane bound peptide. PGLa is a 21-residue cationic peptide (GMASKAGAIAGKIAKVALKAL-NH2) from the magainin family of antibiotics present in frog skin, which folds into an amphiphilic α-helix when bound to lipid bilayers. The mechanism of antimicrobial activity has been addressed in numerous studies and is attributed to the perturbation of bacterial membranes. Solid state NMR has yielded much insight into its structure and dynamic behaviour in model membranes (Bechinger 1999). For our MD simulations we used the constraints from the 2H-NMR investigations of Strandberg et al. (2005), who had labelled four native alanine residues (positions 6, 8, 10 and 14) and two isoleucines (positions 9 and 13) one by one with 2H3-alanine. At a peptide-to-lipid ratio of 1:200 in DMPC, it was found from the quadrupolar splittings of these six CD3-groups that the α-helical PGLa is aligned flat in the plane of the membrane, and the peptide undergoes fast rotational diffusion about the membrane normal at 35°C. From the same data we now derived the full quadrupolar coupling tensors for the six 2H-labelled sites, including the sign of the tensor elements, and used these as constraints for the MD simulations. For the quadrupolar coupling tensors we used the parameters of the CD3-group (Table 1), both for the native Ala substitutions as well as for the nominal constraint along the Cα–Cβ segment of isoleucine.
It is known from previous NMR studies (Strandberg et al. 2005) that membrane-bound PGLa forms an α-helix in the range of the labelled stretch, and further evidence for an α-helical conformation between residues 6 and 21 is provided by (Bechinger 1999). We therefore started the MD simulations with an idealized α-helix, and for all backbone hydrogen bonds we introduced 18 additional distance constraints of 1.86 Å to keep the molecular model helical. At the end of the simulation, the RMSD of the backbone hydrogen bond lengths from their ideal values was only 0.2 Å, thus confirming that that the molecule stayed indeed mostly helical during the MD simulation. It also had to be taken into account that the four lysine side chains and N-terminus are positively charged. Since charged NH+3 groups have a strong tendency to form hydrogen bonds, we added a water molecule near each hydrogen atom of a charged group. This way any undesired hydrogen bonds of lysines to the backbone could be prevented, and indeed at the end of the simulation all 15 water molecules were still in contact with the peptide.
Compared to the two small-molecule case studies above, the NMR data on PGLa provide only few constraints on the large and rather flexible molecule, as the pseudo-forces act only on six amino acids out of 21. Therefore, unrealistically large-pseudo forces at the start of the MD simulation have to be avoided. As a first measure, the pseudo-forces were increased exponentially to their final values during the MD simulation with a time constant of ρ = 100 ps, like in the cholesterol simulation. From the NMR studies it was known that PGLa rotates rapidly (on the NMR time scale) about the membrane normal. This motion opens up a second possibility to scale the pseudo-forces properly. We calculated rotational averages of the quadrupolar tensors in every step of the MD. Only three tensor values rotated by 120° around the director axis (in this case the membrane normal oriented parallel to the B0 field directions) are necessary to obtain a mean tensor within the limits of a fast rotation. All off-diagonal values of the simulated tensors thus become zero and only the principal values of the tensors are left as constraints. The mean temperature at the end of the simulation was only 11 K above the target temperature of 293 K, indicating that exceedingly high pseudo-forces have been successfully avoided indeed, which would otherwise have lead to additional heat production.
As in the other two case studies above, the constraint-driven MD simulations were able to reproduce the experimental 2H-NMR data correctly, as evident from the comparison in Table 7. All calculated frequencies are well within the experimental error margins (as estimated from the experimental line widths). For some additional sites the quadrupolar frequencies were predicted from the MD simulation, and it would be of interest whether future measurements can confirm these values.
Table 7.
Calculated deuterium quadrupolar splittings from a constrained MD simulation of PGLa, compared with the experimental 2H-NMR data (Strandberg et al. 2005) in DMPC
| Site | MD (kHz) | NMR (kHz) |
|---|---|---|
| Ala3 | −22.2 | |
| Ala6 | +15.7 | +15.6 |
| Ala8 | +17.1 | +17.2 |
| Ala10 | −15.0 | −15.0 |
| Ala14 | −26.8 | −26.6 |
| Ala17 | +17.1 | |
| Ala20 | −25.5 | |
| Ile9 | −4.9 | −5.2 |
| Ile13 | +26.2 | 26.4 |
The major aim of the present NMR data analysis was the determination of the time- and ensemble averaged alignment of the helical PGLa backbone in the membrane environment. As illustrated in Fig. 8, the principal axes of inertia offer a suitable molecule-fixed frame of reference to describe the peptide orientation. In the PGLa the a-axis of least inertia points along the helix, and the c-axis of highest inertia is located between the hydrophilic lysine-rich face and the hydrophobic face of the molecule formed by alanines, valines, leucines and isoleucines. Intuitively, we would expect the helix to be in contact with biomembranes just like it is displayed in this view, namely with the intermediate b-axis collinear with the membrane normal.
Fig. 8.
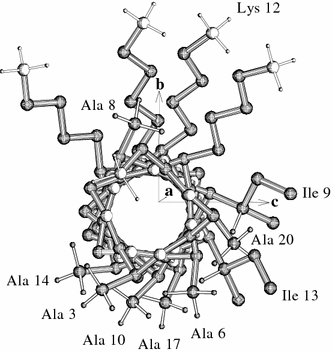
View along the long axis of PGLa modelled as an idealized α-helix with only the lysine, alanine and isoleucine side chains displayed. The latter two types were labelled by 2H3-alanine, and their 2H-NMR splittings were included into the MD simulation. The principal a-axis of inertia, points along the helix, and the b-axis of inertia points radially through Cα of lysine 12
The outcome of the MD simulation is illustrated most appropriately in Fig. 9, which visualizes the alignment and dynamic behaviour of PGLa in DMPC. Here, the peptide orientations during the MD simulation are presented as a scatter plot of the inertia axes a, b and c on the surface of a unit sphere. A narrow range of orientations is seen for the a-axis representing the peptide helix. In contrast, the broad scatter for b and c indicates that a high degree of motional freedom exists for motions around the helix axis. The time-averaged tilt angle of the a-axis with respect to the membrane normal in found to be 99°, with a mean deviation of about ±4°. This value is in very good agreement with the tilt angle τ ≈ 98° of PGLa that had been deduced from an RMSD plot of the 2H-NMR by Strandberg et al. (2005) assuming a rigid idealized α-helix. We note, however, that such helix axis definition is not exactly comparable to the a-axis of inertia used in our MD calculations. The authors further described the azimuthal alignment of the amphiphilic peptide by defining an angle ρ between an axis pointing from the centre of the helix radially through the Cα-atom of lysine 12 and the membrane plane. In our case, the b-axis of inertia also happens to be directed to Cα of lysine 12 (see Fig. 8), but the centre of inertia does not exactly coincide with the helix axis. As can be seen from Fig. 10, the angle Θ b between the b-axis and the membrane normal oscillates between 10° and 70°, with a mean value of 33°. This average value corresponds to an angle ρ of approximately 123°, which compares well with ρ ≈ 115° extracted from the previous analysis (Strandberg et al. 2005). In this comparison we have to keep in mind that in our simulations the PGLa molecule was found to be far from rigid and possesses a high degree of mobility around the helix long axis. The lysine side chains are found to spread out much more than in the initial model, and the PGLa helix itself exhibits a small bend.
Fig. 9.
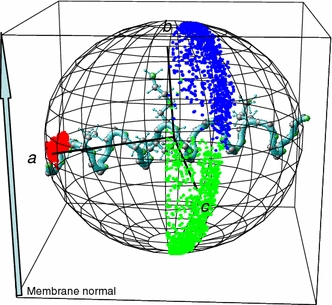
Visualization of the alignment and motional behaviour of the α-helical peptide PGLa (only lysine side chains shown) in a DMPC membrane, as determined by a 1 ns MD simulation using experimental 2H-NMR constraints (Strandberg et al. 2005) The instantaneous orientation of the three axes of inertia a, b and c is displayed as a scatter plot on a sphere, representing snapshots of these axes orientations every picosecond. The poles of the sphere are oriented parallel to the membrane normal. The helix long axis displays only a small scatter with a mean tilt angle of 99°, while the two other axes oscillate about this direction producing a large scatter
Fig. 10.
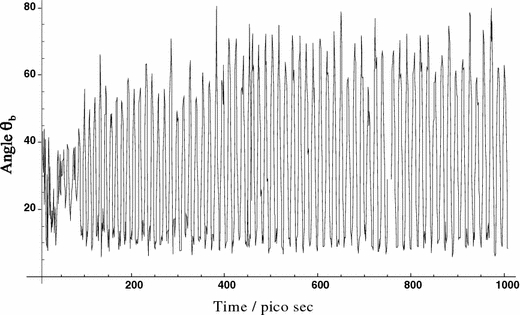
Molecular dynamics trajectory of the angle Θb between the intermediate b-axis of inertia and the membrane normal
Similarly important as the alignment of the peptide are its dynamic properties, which also enter into the observed 2H-NMR data. Table 8 lists the components of the molecular Saupe tensor, which is again used to describe the average alignment a molecule fixed frame (i.e. the principal axes of inertia) with respect to the laboratory frame. However, in contrast to the two previous case studies, this Saupe tensor does not represents the fast reorientation of the molecule around the membrane normal. No pseudo-forces were driving this motion in the simulation, since a rotational mean value was introduced. Here, the order tensor represents only the wobble of the long a-axis and the pronounced oscillations about this molecular axis (see Fig. 10). As expected from the distribution of polar and hydrophobic residues on the different faces of the helix, it is seen that the Saupe tensor is not diagonal Table 8). The biaxiality of ξ = 0.09 (S = 0.76, see Eq. 15) is clear evidence that the molecule does not behave like a rod-shaped object, and rotation about its long axis is restricted. In fact, oscillations of about 60° around the long axis, but no complete rotation events were observed in the MD simulation (see Fig. 10). These oscillations start after about 100 ps when the pseudo-forces approach their full strength.
Table 8.
Molecular Saupe order tensor from a constrained MD simulation of PGLa
| Tensor component | MD |
|---|---|
| Saa | +0.49 |
| Sbb | −0.28 |
| Scc | −0.46 |
| Sab | −0.46 |
| Sac | +0.06 |
| Sbc | −0.08 |
At a first glance the rather regular oscillations of PGLa in Fig. 10, or the rotations of cholesterol (Fig. 7), may not seem to reflect reality. However, we have to keep in mind that our MD simulations are performed with single molecules in vacuum, and the temperature is controlled by a continuously acting NTV “thermostat” adjustment. In nature, heat is transferred by stochastic interactions with other molecules, thereby introducing a stochastic behaviour of molecular rotations and re-orientations. This stochastic aspect is not present in our MD simulations, but nonetheless the amplitudes, velocities and directions of the motions adopt realistic values as the NMR constraints have to be satisfied. The molecule in the simulation cannot stop in one preferred orientation, because then no averaging would be performed anymore.
Conclusions
Solid state NMR is a valuable technique to gain insight into the behaviour of peptides and proteins in oriented media, provided the data can be interpreted in terms of molecular structure and dynamics. In this contribution we developed a new strategy in which all-atom MD simulations and NMR data obtained from oriented samples are combined to obtain such structural and motional information. To this aim, a molecular mechanics force field (in this case COSMOS-NMR) was extended to include pseudo-forces, which drive the molecular dynamics to meet the NMR constraints. They “heat up” molecular rotations or re-orientations, leading to proper averaging of the calculated tensor values such that the calculated tensor values agree with the corresponding experimental observations. The orientational constraints can be further combined with intramolecular constraints such as distances or chemical shifts. This way, similar results can be obtained as in full membrane MD simulations, but without the computational burden of having to perform a detailed simulation of the lipids and surrounding water molecules. Because they are performed in vacuum, the constrained MD simulations can be completed in relatively short simulation times (≤1 ns), still reaching a complete averaging of the NMR observables.
In three case studies, ranging from a small rigid compound to a 21-residue membrane-active peptide, the MD simulations with orientational NMR constraints succeeded to produce a detailed picture of the molecular motions and orientations in oriented membranes. It could be demonstrated that this new method is not limited to rigid molecules and does not depend on the choice of the initial coordinates. Deuterium quadrupolar splittings from 2H-labelled pyrene, cholesterol and PGLa in oriented lipid bilayers have been used as constraints in the present examples, but the general formalism presented will be applicable to all kinds of tensorial NMR properties.
Acknowledgement
Many helpful discussions with Dr. Stephan Grage are gratefully acknowledged.
Contributor Information
Ulrich Sternberg, Phone: +49-7247-826950, FAX: +49-7247-824842, Email: Ulrich.Sternberg@ibg.fzk.de.
Anne S. Ulrich, Email: anne.ulrich@ibg.fzk.de
References
- Bailey WC (1998) B3LYP calculation of deuterium quadrupole coupling constants in molecules. J Molec Spec 190:318–323 [DOI] [PubMed]
- Bechinger B (1999) The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochem Biophys Acta 1462:157–183 [DOI] [PubMed]
- Brüschweiler R (1994) Connections between NMR relaxation measurements and theoretical models of structural dynamics of biopolymers in solution. In: Tycko R (ed) Nuclear magnetic resonance probes molecular dynamics. Kluwer, pp 301–334
- Burnell EE, De Lange CA (1980) Effects of Interaction between molecular internal motion and reorientation on NMR of anisotropic liquids. J Magn Reson 39:461–480
- Davis JH (1983) The description of membrane lipid conformation, order and dynamics by 2H-NMR. Biochim Biophys Acta 737:117–171 [DOI] [PubMed]
- Dufourc EJ, Parish EJ, Chitrakorn S, Smith ICP (1984) Structural and dynamical details of cholesterol-lipid interactions as revealed by deuterium NMR. Biochemistry 23:6062–6071 [DOI]
- Evans DJ, Morriss GP (1990) Statistical mechanics of nonequilibrium liquids. Academic Press, London
- Hoff B, Strandberg E, Ulrich AS, Tieleman DP, Posten C (2005) 2H-NMR study and molecular dynamics simulation of the location, alignment, and mobility of pyrene in POPC bilayers. Biophys J 88:1818–1827 [DOI] [PMC free article] [PubMed]
- Limmer S (1989) Physical principles underlying the experimental methods for studying the orientational order of liquid crystals. Fortschritte der Physik 37:879–931
- Low RJ (2002) Measuring order and biaxiality. Eur J Phys 23:111–117 [DOI]
- Marsan MP, Muller I, Ramos C, Rodriguez F, Dufourc EJ, Czaplicki J, Milon A (1999) Cholesterol orientation and dynamics in dimyristoylphosphatidylcholine bilayers: A solid state deuterium NMR analysis. Biophys J 76:351–359 [DOI] [PMC free article] [PubMed]
- McMullan RK, Koetzle TF, Fronckowiak MD (1992) Structure of [20-CH3],[20-CD3]-methylpregnene-3,20-diol methanolate from neutron diffraction at 123 K. Acta Cryst C48:1509–1512
- Möllhoff M, Sternberg U (2001) Molecular mechanics with fluctuating atomic charges—a new force field with a semi-empirical charge calculation. J Mol Model 7:90–102
- O’Keefe M, Brese NE (1991) Atom sizes and bond lengths in molecules and crystals. J Am Chem Soc 113:3226–3229 [DOI]
- Rinné M, Depireux J (1974) Nuclear quadrupole coupling constant of deuterium bound to carbon in organic molecules. In: Smith JAS (ed) Advances in nuclear quadrupole resonance, vol 1. Heyden & Son Ltd, London, pp 345–357
- Saupe A (1964) Kernresonanzen in kristallinen Flüssigkeiten und kristallinflüssigen Lösungen. Z f Naturforschung 19a:161–171
- Smith RL, Oldfield E (1984) Dynamic structure of membranes by deuterium NMR. Science 255:280–288 [DOI] [PubMed]
- Sternberg U (1988) Theory of the influence of the second co-ordination sphere on the chemical shift. Mol Phys 63:249–267 [DOI]
- Sternberg U, Koch F-Th, Bräuer M, Kunert M, Anders E (2001) Molecular mechanics for zinc complexes with fluctuating atomic charges. J Mol Model 7:54–64
- Sternberg U, Witter R, Ulrich AS (2003) 3D structure elucidation using NMR chemical shifts. Annu Rep NMR Spectroscopy 52:53–103 [DOI]
- Strandberg E, Wadhwani P, Tremouilhac P, Dürr UHN, Ulrich AS (2006) Solid-state NMR analysis of the PGLa peptide orientation in DMPC bilayers: Structural fidelity of 2H-labels versus high sensitivity of 19F-NMR. Biophys J 90:1676–1686 [DOI] [PMC free article] [PubMed]
- Tieleman DP, Berendsen HCJ, Sansom MSP (2001) Voltage-dependent insertion of alamethicin at phospholipid/water and octane/water interfaces. Biophys J 80:331–346 [DOI] [PMC free article] [PubMed]
- Torda AE, van Gunsteren WF (1991) The refinement of NMR structures by molecular dynamics simulation. Comput Phys Commun 62:289–296 [DOI]
- Ulrich AS, Grage SL (1998) Solid state 2H-NMR. In: Ando I, Asakura T (eds) Solid state NMR of polymers. Elsevier, Amsterdam, pp 190–211
- Verlet L (1967) Computer “experiments” on classical fluids. Phys Rev 159:98–103 [DOI]
- Vold RR (1994) Deuterium NMR studies of dynamics in solids and liquid crystals. In: Tycko R (ed) Nuclear magnetic resonance probes of molecular dynamics. Kluwer Academic Publishers, pp 27–112
- Witter R, Prieß W, Sternberg U (2001) Chemical shift driven geometry optimization. J Comp Chem 23:298–305 [DOI] [PubMed]
- Witter R, Seyfart L, Greiner G, Reissmann S, Weston J, Anders E, Sternberg U (2002) Structure determination of a pseudotripeptide zinc complex with the COSMOS-NMR force field and DFT methods. J Biomol NMR 24:277–289 [DOI] [PubMed]