

Abstract
Fgfr4 has been shown to be important for appropriate muscle development in chick limb buds, however, Fgfr4 null mice show no phenotype. Here, we show that staged induction of muscle regeneration in Fgfr4 null mice becomes highly abnormal at the time point when Fgfr4 is normally expressed. By 7 days of regeneration, differentiation of myotubes became poorly coordinated and delayed by both histology and embryonic myosin heavy chain staining. By 14 days, much of the muscle was replaced by fat and calcifications. To begin to dissect the molecular pathways involving Fgfr4, we queried the promoter sequences for transcriptional factor binding sites, and tested candidate regulators in a 27 time point regeneration series. The Fgfr4 promoter region contained a Tead protein binding site (M-CAT 5′-CATTCCT-3′), and Tead2 showed induction during regeneration commensurate with Fgfr4 regulation. Co-transfection of Tead2 and Fgfr4 promoter reporter constructs into C2C12 myotubes showed Tead2 to activate Fgfr4, and mutation of the M-CAT motif in the Fgfr4 promoter abolished these effects. Immunostaining for Tead2 showed timed expression in myotube nuclei consistent with the mRNA data. Query of the expression timing and genomic sequences of Tead2 suggested direct regulation by MyoD, and, consistent with this, MyoD directly bound to two strong E-boxes in the first intron of Tead2 by chromatin immunoprecipitation assay. Moreover, co-transfection of MyoD and Tead2 intron reporter constructs into 10T1/2 cells activated reporter activity in a dose dependent manner. This activation was greatly reduced when the two E-boxes were mutated. Our data suggest a novel MyoD-Tead2-Fgfr4 pathway important for effective muscle regeneration.
Keywords: Muscle regeneration, Tead, TEF, Fgfr, MyoD, Microarray
Fibroblast growth factors (FGFs)1 and their receptors (FGFRs) are critical for the development of most cell types. There are at least 22 distinct FGF ligands and four receptors (FGFR1-4), and different ligand/receptor pairs regulate cell growth in either a positive or negative manner, depending on the cell type and stage of development (1,2). Gain-of-function mutations of FGFR1, FGFR2, and FGFR3 cause a series of important human disorders of bone development, most notably achondroplasia (dwarfism) (FGFR3), and different types of craniosynostosis (FGFR1, FGFR2, FGFR3) (3-7).
We and others have shown that the major receptor expressed in muscle is FGFR4, while the major ligand is FGF6 (8-17). Consistent with an important role for FGFR4 in muscle development, previous studies have shown that inhibition of FGFR4 leads to the arrest of muscle progenitor differentiation in chick embryo, with reduced expression of Myf5, MyoD, and embryonic myosin heavy chain and dramatic loss of limb muscles (18). This effect is independent of myoblast proliferation (18). Sp1 is shown as one of the factors to control the transcription of FGFR4 in skeletal muscle cells (19,20).
Despite the well-documented importance of Fgfr4 in muscle development in the chick, Fgfr4 null mice develop normally, with no evident muscle defects (21). The only phenotype shown to date for Fgfr4 null mice is increased liver injury and fibrosis induced by carbon tetrachloride, and increased cholesterol metabolism and bile acid synthesis (22-24).
In our studies of staged muscle regeneration in vivo (17,25,26), we had observed that Fgfr4 was strongly yet transiently induced at a key day 3 time point post-injection in muscle regeneration. This time point is commensurate with the myoblast-myotube transition, where proliferating myoblasts leave the cell cycle, fuse with neighboring cells, and terminally differentiate into multi-nucleated myotubes.
We hypothesized that Fgfr4 may be required for effective muscle regeneration, and tested this using staged degeneration/regeneration in Fgfr4 null mice. Regeneration in Fgfr4 was highly abnormal, with poorly differentiated myotubes at day 7, and extensive replacement of muscle by fat and calcifications by day 14. We then built a transcriptional pathway upstream of Fgfr4. We show that one of the TEA domain transcriptional factors, Tead2, is induced at day 3 during regeneration, whereupon it regulates the Fgfr4 promoter through the M-CAT motif (CATTCCT). Moreover, we show that MyoD directly binds to and activates Tead2 first intron. Thus, our data defines a MyoD-Tead2-Fgfr4 pathway important for effective muscle regeneration.
EXPERIMENTAL PROCEDURES
Staged muscle regeneration and expression profiling—Fgfr4 null mice were originally generated by Dr. Chu-Xia Deng (NIDDK, NIH; 21), and maintained by Dr. Wallace McKeehan at Texas A&M University.
Staged muscle degeneration/regeneration was done in both 8 week old wild-type and Fgfr4 null mice by intramuscular injection of 100 μl of 10 μM cardiotoxin (CTX) using a 10-needle manifold, as we have previously described (25,26). Both gastrocnemii of three mice per time point were injected and studied.
H+E staining—Each muscle was examined histologically in the belly (center) of the gastrocnemius muscle. Cryosections (8μm) were cut using an IEC Minotome® cryostat, collected on Superfrost Plus slides (Fisher Scientific), and stained with hematoxylin and eosin, or used for immunostaining and immunolocalizations.
Quantitative multiplex fluorescent PCR—Quantitative multiplex fluorescent PCR (QMF-PCR) was done as previously described using a LI-COR DNA analyzer (25,27). Briefly, 1μg of total RNA was used to synthesize cDNA using oligo(dT) primer (Roche) in a 20μl reaction. 0.5μl of cDNA was then used for RT-PCR in a 12.5μl reaction. PCR was done at 94°C for 30s, 55°C for 30s, and 72°C for 30s for 18 to 20 cycles, and then a 72°C extension for 10 min. Primers used for RT-PCR are Tead2 (NM_011565) forward: 5′ GGAGTGAGCAGCCAGTATGAGAG 3′, Tead2 reverse: 5′ TACACAAAGCGCCCGTCCTC 3′. Primers for control genes (NIPI-like protein, Cytidine monophospho-N-acetylneuraminic acid synthetase) are NIPI-like protein (U67328) forward: 5′ AACAGGGAACCTATGGTGGC 3′, reverse: 5′ CTGTCCACAGGGTGACTGAAG 3′; Cytidine monophospho-N-acetylneuraminic acid synthetase (Cmas, AJ006215) forward: 5′ GACCTAGTCTTGCTCCGACCTC 3′, reverse: 5′ CAGGGGTGTCTTACCAGACTC 3′. Forward primers were labeled with an infrared fluorescent dye (IRDye 700, LI-COR). PCR products are 135bp (Tead2), 142bp (NIPI-like protein), and 128bp (Cmas). PCR products were quantitated using Gene ImagIR 3.56 (LI-COR). Expression of tested gene was normalized to that of NIPI-like protein (control1) and Cmas (control2) by using the following formula: (test/control1 + test/control2)/2.
Real time RT-PCR—Real time RT-PCR was done using ABI Prism 7900HT Sequence Detection System. Tead2 and Cmas primers are the same as these used in QMF-PCR. Primers for Vgl2 (NM_153786.1) are forward: 5′ TCGGTCTGGCTGGGTGGTACAG 3′, reverse: 5′ CCATTCATCCTAGCCACACACCG 3′. 0.5μl of cDNA was used for RT-PCR in a 25μl reaction with SYBR® Green fluorescence labeling. PCR was done at 95°C for 15s, 57°C for 30s, and 72°C for 30s for 40 cycles followed by a dissociation step to check the specificity of the products.
Antibody production, western blotting and immunostaining—Rabbit polyclonal antibodies were produced against Tead2 using a single peptide showing no homology with other Tead family members (WTGSEEGSEEGTGGS). Immune sera were affinity purified using the peptide. Antibody is available from the authors by request.
Whole protein extraction from muscles was quantitated with Bradford protein assay (BioRad) and 10μg of proteins for each sample were loaded onto NuPAGE® Novex Bis-Tris gels (Invitrogen). Proteins were transferred to nitrocellulose membrane, incubated with Tead2 antibodies for 2 hours, and then with HRP-bovine-anti-rabbit IgG (Santa Cruz) for 1 hour. Samples were detected with ECL Western blotting detection reagents (Amersham pharmacia biotech) and exposed to film.
Immunostaining was done as previously described (28). R-phycoerythrin-conjugated rat anti-mouse CD16/CD32 monoclonal antibodies (1:40 dilution) were purchased from BD Biosciences Pharmingen. Rat anti-mouse Laminin a2 monoclonal antibodies (1:50 dilution) were purchased from Alexis Biochemicals. Monoclonal antibodies against embryonic myosin heavy chain were obtained from Developmental Studies Hybridoma Bank. Secondary antibodies Cy3-conjugated donkey anti-rabbit IgG (1:500 dilution), Cy3-conjugated donkey anti-mouse IgG (1:500 dilution) and Cy2-conjugated goat anti-rat IgG (1:100 dilution) were purchased from Jackson ImmunoResearch Laboratories.
Quantitation of anti-CD16/CD32 fluorescence was done using Image-Pro® Plus imaging software (Media Cybernetics). Muscle areas were measured using ImageJ (http://rsb.info.nih.gov/ij/). Fluorescence was normalized to the area of each muscle.
Mineral Analysis—Injected muscles were cryosectioned and either stained by the von Kossa technique to demonstrate the presence of phosphate containing mineral or placed on barium fluoride spectroscopic windows and analyzed by Fourier transform infrared imaging (29) using a Perkin Elmer Spotlight Imaging system (PerkinElmer Instruments). The spatial resolution was ∼7μm. Individual spectra were extracted from the images where mineral was visible (based on the increased intensity of the phosphate band at 900-1200 wavenumbers), and compared to spectra of bone.
PCR amplification of the Fgfr4 promoter, Tead2 first intron, subcloning and mutagenesis—A 226bp Fgfr4 genomic region containing the M-CAT site was PCR amplified from genomic DNA isolated from C2C12 skeletal muscle cells. A MluI site was added to the forward primers and an XhoI site was added to the reverse primers. Primer sequences are- forward: 5′ CAGTACGCGTAACGACTGAGACTGGGCGATCC 3′; reverse: 5′ TAGTCTCGAGACACTCACCCGCCCGGAGCTC 3′. After PCR amplification, the Fgfr4 genomic products were first subcloned into pCR-II-TOPO vector (Invitrogen), and then digested with MluI and XhoI. The Fgfr4 restricted fragments were subcloned in the pGL2 luciferase reporter vectors pGL2-Basic and pGL2-Promoter (Promega) digested with MluI and XhoI. Six consecutive point mutations were introduced in the M-CAT site (CATTCCT to CCGGAAA) of the FGFR4 promoter using the QuikChange XL site-directed mutagenesis kit (Stratagene). Both the wild type sequence and mutations were confirmed by DNA sequencing of the Fgfr4 constructs.
A 213bp Tead2 first intron region containing two E-boxes was amplified from C2C12 cell genomic DNA. A KpnI site was added to the forward primers and an XhoI site was added to the reverse primers. Primer sequences are- forward: 5′ TCAGGGTACCATGCCCCCTTTTGCTGTGTCG 3′ reverse: 5′ AGTCCTCGAGAGTGCCTGAGGCTGTGTTTGG 3′. PCR products were digested with KpnI and XhoI and subcloned into pGL3-Promoter vector (Promega). Point mutations were introduced in the E-boxes (CAGCTGCTGCCCTTCTCTGAGCACCTG to CAGCGACTGCCCTTCTCTGAGTCCCTG) using the QuikChange XL site-directed mutagenesis kit (Stratagene). Both wild type and mutant constructs were confirmed by DNA sequencing.
Cells, Transfections and Luciferase Assay—C2C12 skeletal muscle cells (ATCC) were cultured in growth medium, GM (DMEM supplemented with 10% fetal bovine serum). To induce differentiation, cells were switched to differentiation medium, DM (DMEM supplemented with 2% horse serum and 1X insulin, transferrin and selenium) 24 hours after transfection and cultured for additional 48 hours. C3H10T1/2 mouse fibroblasts (ATCC) were cultured in growth medium, GM (DMEM supplemented with 10% newborn calf serum). Twenty-four hours after transfection, cells were switched to differentiation medium, DM (DMEM supplemented with 2% horse serum and 1X insulin, transferrin and selenium) and allowed to differentiate for 48 hours. The pcDNA-mETF expression vector encoding mouse Tead2 was kindly provided by Dr. Hiroaki Ohkubo (Kumamoto University, Japan) (30). Transfections were performed with FuGENE 6 transfection reagent (Roche) and luciferase activity assayed with Luciferase Reporter Assay kit (Promega) on the microtiter luminescence detection system (MLX, Dynex, and Centro LB 9600, Berthold). Luciferase assays were done in triplicate points and repeated three times. Transfection efficiency was determined by co-transfection of green fluorescent protein.
Gel shift assay—Twenty base pair double-stranded oligonucleotide probes containing potential MyoD binding sites in the first intron of Tead2 gene were used for MyoD gel shift assay. The sequences on the plus strand are Tead2 E1: 5′ TCTCCAGCAGCTGCTGCCCT 3′; Tead2 E2: 5′ CTCTGAGCACCTGTTCTTTC 3′. A 21 bp MyoD binding probe from muscle creatine kinase (MCK) promoter (Geneka Biotech) was used as a control. The oligonucleotide probes were labeled with [gamma-32P] ATP and purified with MicroSpin™ G25 columns (Amersham Pharmacia Biotech). Gel shift assay was done using a MyoD gel shift kit (Geneka Biotech). Nuclear extracts (10μg) from C2C12 cells were incubated with binding buffer for 20 min at 4°C, and then incubated with 5ng labeled oligonucleotide probes for additional 20min at 4°C. For competition test, 100 times more of unlabeled wild-type or mutant MCK probes (500ng) were added to each reaction. The reaction mixture was subjected to 6% polyacrylamide gel electrophoresis at 90 volts for 2.5 hours. Gels were dried and exposed to X-ray film.
Chromatin immunoprecipitation assay (ChIP)—Murine myoblasts (C2C12) were cultured in 15 cm plates in 10% fetal bovine serum to 75% confluency. To induce differentiation, serum was withdrawn and cells were allowed to differentiate for 2 days. Cells were fixed with 1% of formaldehyde to cross-link protein and DNA. Cells were then harvested and chromatin was extracted. Chromatin was sonicated to 200bp to 600bp fragments. Chromatin fragments were precleared with protein A (Invitrogen) and then incubated with MyoD antibodies (M318X, Santa Cruz Biotech) or rabbit IgG and rotated overnight at 4°C. Chromatin bound by antibodies was then precipitated with protein A, washed as described previously (25), and eluted with 50 mM NaHCO3 and 1% SDS. Protein and DNA cross-linking was reversed by incubating at 67°C in 0.3M NaCl for 5 hours. DNA was then ethanol precipitated, digested with proteinase K and extracted with phenol/chloroform.
Primers were designed to amplify candidate promoter sequences. Tead2 forward: 5′ TTGTCCCTGGATCTCTCTGTCCC 3′, reverse: 5′ ATGGGGTTGAAGCCACCTGACC 3′. Gapdh forward: 5′ CAGCATAGAGCAGGTGGACCATG 3′, reverse: 5′ TCAGCCCACTCTCCAGAAGC 3′. Rapsn (Receptor-associated protein of the synapse) forward: 5′ TCAGAAGTGTCAAAGGGGACACC 3′, reverse: 5′ CACCCTGGGAACAAGGCTGGTTC 3′. Real time PCR was performed using Stratagene Mx3000P system and ABI Prism 7900HT Sequence Detection System.
RESULTS
Fgfr4-/- mice show abnormal muscle regeneration—To investigate the role of Fgfr4 in muscle regeneration, we induced staged degeneration/regeneration in gastrocnemii muscles in Fgfr4-/- mice and controls using CTX, as we have previously described (17,25,26). Six muscles at each time point of 0, 3, 4, 7, and 14 days post-injection were analyzed by H+E for both Fgfr4 null and normal controls, with three muscles in each group selected for more detailed study. The Fgfr4 null mice have an insertion of the NEO gene in exon 6, and have been reported to lack mRNA and protein (21). RT-PCR was done at day 3 of regeneration in both wild-type and Fgfr4 null muscles, with primer sets to amplify both exons 5-7, and 5-8. Wild-type muscle showed the expected RT-PCR products, while the Fgfr4 null muscles showed an absence of these products (data not shown). This confirmed both the loss-of-function mutation of the Fgfr4 strains used, and the expression of the Fgfr4 gene at the 3 day time point, as we have previously reported (17).
H+E histopathology showed similar staining patterns in both Fgfr4-/- and controls at days 0, 3, and 4 (data not shown). However, by day 7 there were clear distinctions between Fgfr4 null and wild type mice by immunostaining with embryonic myosin heavy chain antibodies (Fig. 1). Wild-type muscle showed maturing myofibers of relatively homogeneous size, central nuclei, and moderate staining with embryonic myosin heavy chain. Fgfr4 null mice showed considerable variability in myofiber size, with many small and intensely staining myofibers, suggesting a lag in the timing of regeneration. There were also very small fibers that stained intensely positive for embryonic myosin heavy chain, yet appeared to lack internal nuclei (Fig. 1).
Fig. 1.

Fgfr4-/- mouse muscle shows impaired regeneration with embryonic myosin heavy chain staining. Shown is immunostaining of day 7 regenerating muscle frozen sections from normal (A, B, C) and Fgfr4-/- (D, E, F) mice. Positive regenerating muscle fibers in normal mice appear to be larger and more homogeneous than these in Fgfr4-/- mice, suggesting they are in a more advanced regeneration stage. Muscle sections are from two independent normal mouse muscles and three independent Fgfr4-/- mouse muscles.
By 14 days, wild-type muscle showed characteristic large, closely packed regenerated myofibers with central nuclei (Fig. 2A). Fgfr4 null mice showed a highly variable histopathology, with some areas showing regenerated myofibers of variable size, with larger nuclei (Fig. 2B). Other areas showed extensive calcifications by the von Kossa technique (Fig. 2C), and others adipose tissue (Fig. 2D). Calcifications and adipocytes were not seen in any area of the wild-type regenerated mice. Inflammation, calcification and fat infiltration were quantified. Inflammation was characterized by staining of muscle sections with phycoerythrin-conjugated antibodies against CD16/CD32, which are expressed on a variety of inflammatory cells including natural killer cells, monocytes, macrophages, dendritic cells, granulocytes, mast cells, B lymphocytes, immature thymocytes, and some activated mature T lymphocytes. Fgfr4 null mice showed 2-3 times more CD16/CD32+ cells relative to the total muscle area than wild type mice (Fig. 2E). Fgfr4 null muscle showed large numbers of calcifications (three representative fields of three Fgfr4 null mouse muscles), with an average of 60 calcified foci per 10-X field (Fig. 2F). Fgfr4 null mice also showed 8-30% of the muscle area replaced by adipocytes, while no adipocytes were seen in wild-type muscle (Fig. 2G)
Fig. 2.

. Fgfr4-/- mouse muscle shows impaired regeneration at late stage. Shown is H&E staining of day 14 regenerating muscle frozen sections from normal (A) and Fgfr4-/- (B, D), and von Kossa staining of calcifications in Fgfr4-/- mice (C). Wild-type mice show effective muscle regeneration (A), whereas Fgfr4-/- mice show poorly staged regeneration (B), extensive inflammation (C, E), calcified regions (C, F), and fatty replacement (D, G). *, p<0.05.
The calcifications were particularly striking in size and number. Chronic degeneration/regeneration in muscle can lead to foci of calcification, presumably due to poor clearance of macrophages. However, the large number and size of the calcifications suggested that these may be bone-like. To test this, we analyzed the mineral composition of the muscle using Fourier Transform infrared microspectroscopy (31). We compared the infrared spectra from normal murine muscle (Fig 3A), Fgfr4 null muscle 14 days after injection with cardiotoxin (Fig. 3B), and normal murine bone (Fig. 3C). The mineral peaks at ∼900-1200 cm-1 in the injected Fgfr4-/- muscle are comparable to normal bone, indicating that hydroxyapatite is present. The increased intensity of the mineral relative to the amide I peak in the injected specimen (Fig. 3B) in contrast to the bone demonstrates that the calcifications are not truly bone-like because the ratio of mineral to collagen differs (either less collagen or more mineral), but have similar components.
Fig. 3.
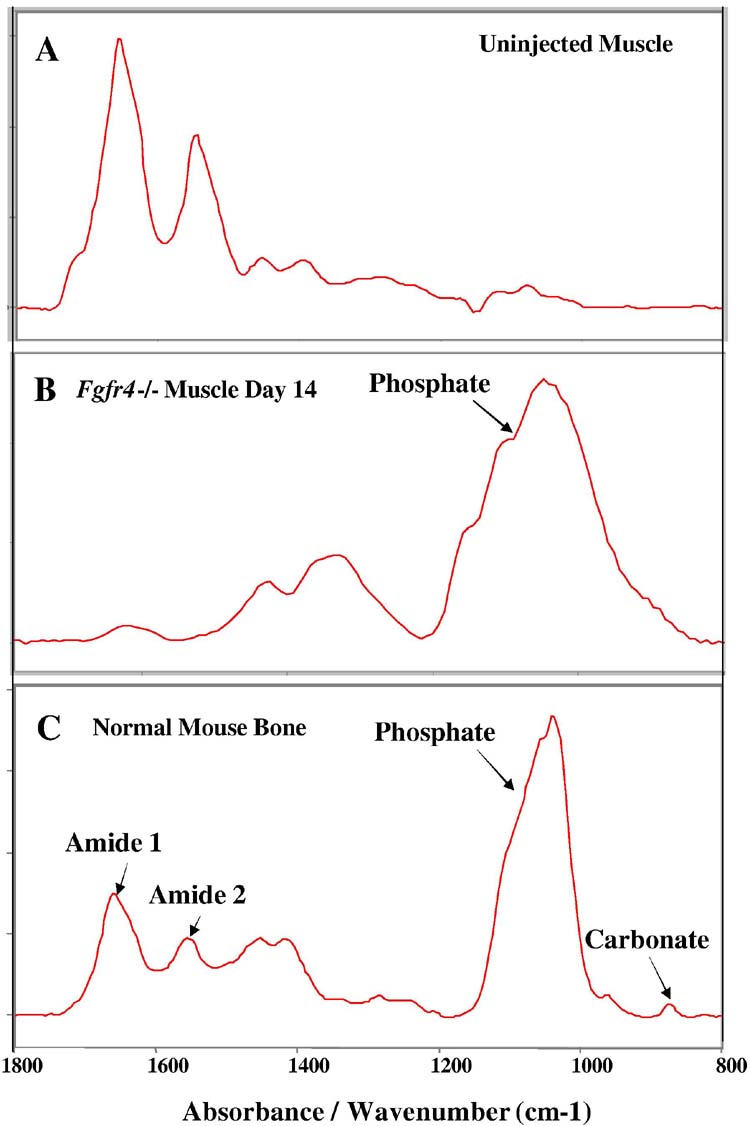
Fourier Transform infrared microspectroscopy of injected Fgfr4-/- muscle indicates the mineral deposit is hydroxyapatite. A, spectrum from a control, uninjected mouse muscle compared with panel B, the spectrum from a Fgfr4-/- muscle containing calcifications 14 days after injection with 10 uM cardiotoxin. Spectrum of B is comparable with panel C, the spectrum for normal bone. The mineral peaks at ∼900-1200 cm-1 in the injected muscle are comparable to normal bone, indicating that hydroxyapatite is present. The increased intensity of the mineral relative to the amide I peak in the injected specimen (B) in contrast to the bone demonstrates that the calcifications are not truly cortical bone-like because the ratio of mineral to collagen differs (either less collagen or more mineral).
Taken together, this data shows that Fgfr4 deficient muscle regenerates abnormally, with poorly timed regeneration after day 4 (myotube formation).
Activators of Fgfr4: Identification of Tead as a candidate—To determine transcriptional activators for Fgfr4 relevant to myogenesis, we obtained mouse Fgfr4 (NM_008011) promoter sequence from Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway), and queried the promoter region (600 bp) and first intron of the Fgfr4 genomic sequence for potential transcriptional factor binding sites using Transcription Element Search System (TESS, http://www.cbil.upenn.edu/tess/). We identified an M-CAT motif (CATTCCT) 49 bp from the transcription start site of mouse Fgfr4. This M-CAT motif is conserved in human FGFR4. The M-CAT motif has been found in promoters of muscle specific genes, and has been shown to be bound by TEA domain proteins (Tead) proteins. To determine which Tead isoform (gene) was a possible regulator of Fgfr4, we queried our previously reported 27-time point muscle regeneration expression profiling series for all Tead transcripts (17,25,26). The transcripts corresponding to Tead1 (TEF-1) and Tead3 (TEF-5) were not detectable at any time points by Affymetrix GeneChips (absent calls). There are two different probe sets for Tead4 (TEF-3). One showed expression at all time points without differential regulation and the other showed upregulation at day 3.5. Tead2 (TEF-4) showed a characteristic expression pattern similar to MyoD and Fgfr4, with ∼10-fold upregulation at peak time point (day 3) (Fig. 4A). Upregulation of Tead2 at this critical time point in muscle regeneration was confirmed by both quantitative multiplex fluorescent (QMF) RT-PCR using LI-COR DNA Analyzer and real time RT-PCR using ABI Prism 7900HT Sequence Detection System (Fig. 4B, C, D). The fold change difference observed by QMF RT-PCR and real time RT-PCR is probably caused by the different signal quantitation methods each system employed. Because Tead2 expression is commensurate with Fgfr4, whereas Tead4 is half day later, we focused on Tead2 in this study.
Fig. 4.
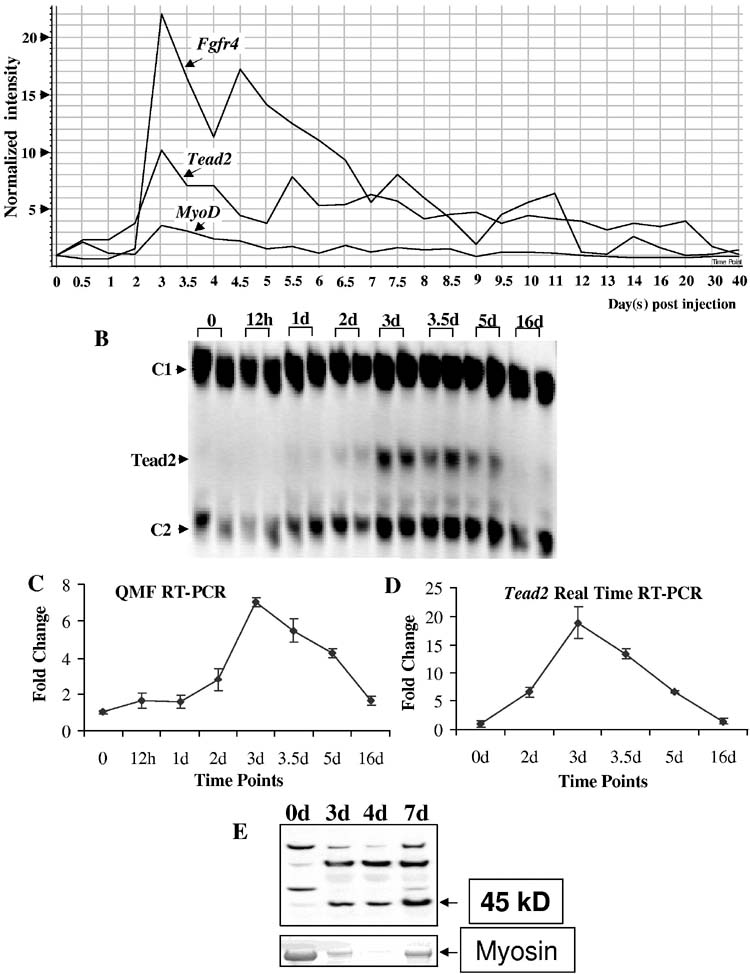
Expression of Tead2 in muscle regeneration. A, shown is the temporal expression of Tead2, Fgfr4 and MyoD over the entire 27 time point series. Tead2 was transiently upregulated at day 3 post-injection, corresponding to a stage of proliferating myoblast differentiation. B, shown is the expression of Tead2 confirmed by QMF-RT-PCR at selected time points (0, 12h, 1d, 2d, 3d, 3.5d, 5d, 16d). Two independent muscle samples studied at each of the time points are shown. The intensity of Tead2 bands was normalized to each control transcript (C1- control 1, NIPI-like protein; C2- control 2, Cmas). The two different relative measurements of Tead2 were then plotted relative to the time 0 values, with two replicate values shown with standard error (C). D, shown is the expression of Tead2 confirmed by real time RT-PCR at selected time points (0, 2d, 3d, 3.5d, 5d, 16d). Tead2 expression was normalized to the control gene, Cmas. The error bar shows the range of two replicates. The fold change difference between C and D is possibly caused by the two different systems used. E, shown is expression of Tead2 confirmed by Western blot at selected time points (0, 3d, 4d, 7d). 10μg of total proteins were loaded for each time point. Tead2 protein (45kD) also showed increased expression during muscle regeneration. Myosin was stained with Coomassie blue and used as loading control. Because of the extensive myofiber necrosis at early time points of muscle degeneration/regeneration, the amount of myosin was not equal across all the time points, as expected.
To further study Tead2 expression, we produced affinity purified polyclonal antibodies to a 15 amino acid sequence unique to Tead2 N-terminal region before the TEA DNA-binding domain. Immunoblotting showed a band of the predicted molecular weight (45 kD), as well as additional bands (Fig. 4E). Immunoprecipitation of muscle cell nuclear extracts with the same Tead2 antibodies pulled down only the expected 45 kD protein, suggesting that the additional bands seen by immunoblot were non-specific (data not shown). Tead2 was not detectable in normal non-regenerating muscle, but was induced by degeneration/regeneration by day 3, consistent with the mRNA data (microarray and RT-PCR).
Tead2 antibodies were then used for immunolocalization of the protein in regenerating muscle (Fig. 5). Double immunostaining with antibodies to Tead2 and a basal membrane protein Laminin a2 showed Tead2 protein to be localized to regenerating myofiber nuclei, with some cells showing staining patterns suggestive of concentration of Tead2 at the nuclear envelope. Tead2 was not detectable in non-injected control muscle nuclei.
Fig. 5.

Tead2 is expressed in regenerating muscle fibers. Shown are normal murine muscle (A, B, C, D), and regenerating muscle at 7 days (E, F, G, H). Cryosections were immunostained with Tead2 (A and E), Laminin a2 (B and F), or stained with H&E (D and H), and visualized using fluorescent microscopy. Superimposed immunostaining images are also shown (C and G). Tead2 is seen strongly expressed in the central nuclei of regenerating muscle fibers.
Tead2 activates the Fgfr4 promoter—The data presented above was consistent with Tead2 as a candidate for regulating Fgfr4. To test this hypothesis, a 226 bp Fgfr4 genomic region containing the M-CAT motif was subcloned into two different reporter constructs. One of these constructs harbored the viral SV40 promoter directing expression of the luciferase gene (SV40P-luc), the second contained the luciferase gene only (Fgfr4-SV40P-luc and Fgfr4-luc reporters) (Fig. 6A). These Fgfr4 constructs were transiently transfected into mouse C2C12 skeletal muscle cells with or without the co-transfection of Tead2 expression constructs. C2C12 cells were grown in growth medium (GM) for 24 hours after transfection. C2C12 cell differentiation was then induced by switching to differentiation medium (DM) and continuing culture for 2 days. Cells were harvested and luciferase activity measured. In myoblasts and myotubes transfected with Fgfr4-SV40P-luc, luciferase activity significantly increased in the presence of Tead2 constructs compared to that in the absence of Tead2 constructs (p<0.0048 and 0.0009, respectively; data not shown). In myotubes transfected with Fgfr4-luc, luciferase activity showed a 3-fold increase in the presence of Tead2 constructs relative to that in the absence of Tead2 constructs (p<0.0002) (Fig. 6B). Luciferase activity did not show significant difference between myoblasts transfected with Fgfr4-luc alone and myoblasts transfected with Fgfr4-luc and Tead2 (Fig. 6B).
Fig. 6.
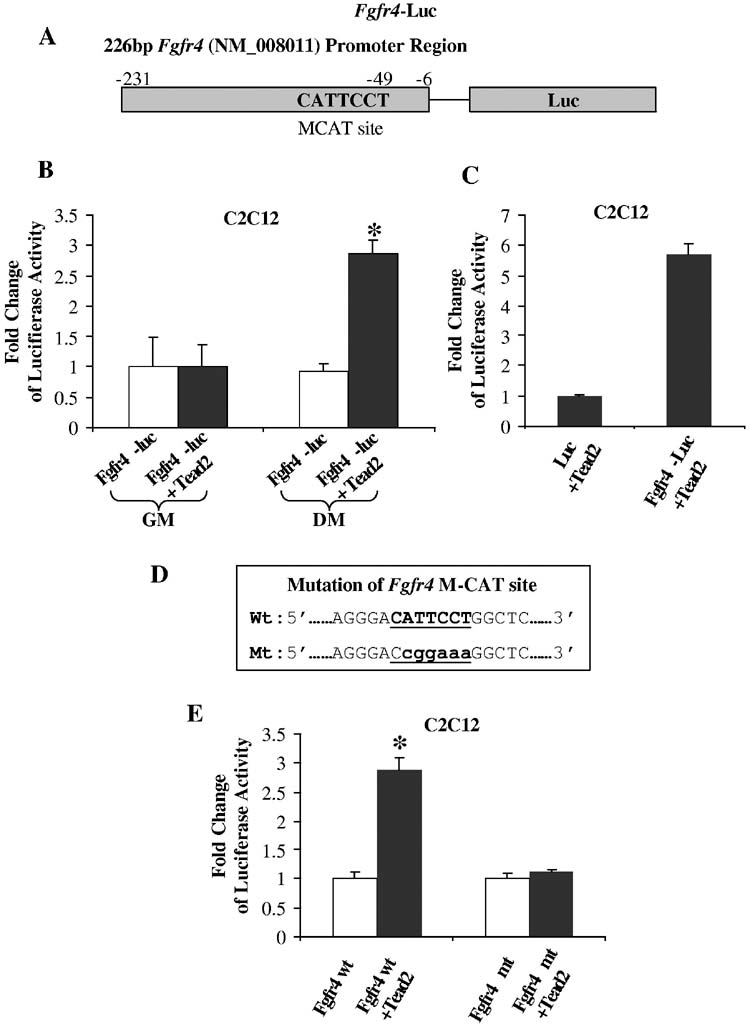
Fgfr4 promoter contains Tead2 sensitive positive regulatory elements. A, shown is a schematic of the constructs containing the 226 bp of Fgfr4 promoter and the luciferase reporter. B, shown is the luciferase activity in proliferating myoblast C2C12 cells or differentiated myotubes after transfection of Fgfr4-luciferase constructs in the presence or absence of Tead2 constructs. Luciferase activity was normalized to transfection without Tead2 in each condition. The Fgfr4 genomic element acted as a positive promoter in response to Tead2 in differentiated myogenic cells, but not in proliferating myoblasts. GM, growth medium; DM, differentiation medium. Standard deviations are shown (n=3). *, p<0.05 relative to vector controls. C, shown is luciferase activity in differentiated myotubes after co-transfection of Tead2 constructs and luc constructs/Fgfr4-Luc constructs. Luciferase activity was normalized to transfection of luc constructs. Luciferase activity significantly increased in the presence of Fgfr4 promoter element. D, shown is Fgfr4 promoter sequence containing wild type (Wt) or mutant (Mt) M-CAT binding sites. E, shown are luciferase activity changes in differentiated myotubes after transfection of wild type Fgfr4-luc constructs or mutant Fgfr4-luc constructs in the presence or absence of Tead2 constructs. Luciferase activity was normalized to transfection without Tead2 constructs. Tead2 increased luciferase activity in the presence of wild type Fgfr4 promoter. This effect was abolished when the M-CAT binding site was mutated. *, p<0.05 relative to vector controls. Fold induction difference between C and E is due to normalization to luc only constructs and Fgfr4-luc constructs respectively.
These results indicate that Tead2 can enhance the transcriptional activation of the SV40 promoter linked to the Fgfr4 genomic region in both myoblasts and myotubes. Tead2 alone is sufficient to activate transcription of the Fgfr4-luc construct in myotubes, but not in myoblasts. This suggests that co-activators of Tead2 are present in differentiated myotubes, but not myoblasts.
To test whether the Fgfr4-luc construct responds to Tead2 in a dose-dependent manner, the Fgfr4-luc construct was co-transfected with 0, 50ng, 100ng, 150ng and 200ng of the Tead2 construct respectively. Luciferase activity showed a slight increase with increasing amount of Tead2 from 50ng to 200ng, although the dose-response was not dramatic (data not shown).
Transcriptional activation of Fgfr4 is dependent on the presence of the M-CAT motif—We further investigated whether the Fgfr4 promoter region was necessary for this activation. Fgfr4-luc constructs or constructs containing only luciferase (luc) were transfected into C2C12 cells in the presence of Tead2 constructs. Transfected cells were then allowed to differentiate for 48 hours. Cells transfected with the Fgfr4-luc construct showed a significant upregulation of luciferase activity compared to cells transfected with the luc construct (Fig. 6C). These results suggest that the Fgfr4 genomic region is necessary for the transcriptional activation in response to Tead2.
Finally, we tested whether regulation of Fgfr4 promoter region by Tead2 was dependent on the M-CAT motif. We interrupted the integrity of the Fgfr4 M-CAT motif by introducing six single point mutations (Fig. 6D) and co-transfected the resulting mutated Fgfr4-luc construct in C2C12 cells with a Tead2 expression vector. While the Fgfr4-luc wild-type construct could be efficiently transactivated by Tead2, the Fgfr4-M-CAT mutant-luc did not respond to Tead2 indicating that Tead2 activates the Fgfr4 promoter through the M-CAT site (Fig. 6E). These results suggest that Tead2 is a direct regulator of the Fgfr4 promoter.
Co-activators of Tead2 are specific to differentiated myogenic cells—Our finding of activation of Fgfr4-luc by co-transfected Tead2 in myotubes, but not in myoblasts suggested that Tead2 co-activators may be specific to differentiated myogenic cells. To test this, we transfected Fgfr4-luc constructs into non-myogenic 10T1/2 cells with or without co-transfection of Tead2 constructs, with increasing amounts of the myogenic differentiation factor, MyoD. Co-transfection of Fgfr4 promoter reporter with Tead2 and MyoD expression constructs did not result in significant reporter expression in growth medium (GM) (Fig. 7A). When cells were switched into differentiation medium (DM) for 48 hours, Tead2 was still unable to activate Fgfr4-luc, suggesting the co-activator of Tead2 is not present in 10T1/2 cells (Fig. 7B). However, co-transfection of MyoD with Tead2 constructs activated Fgfr4-luc in a dose dependent manner (Fig. 7B).
Fig. 7.
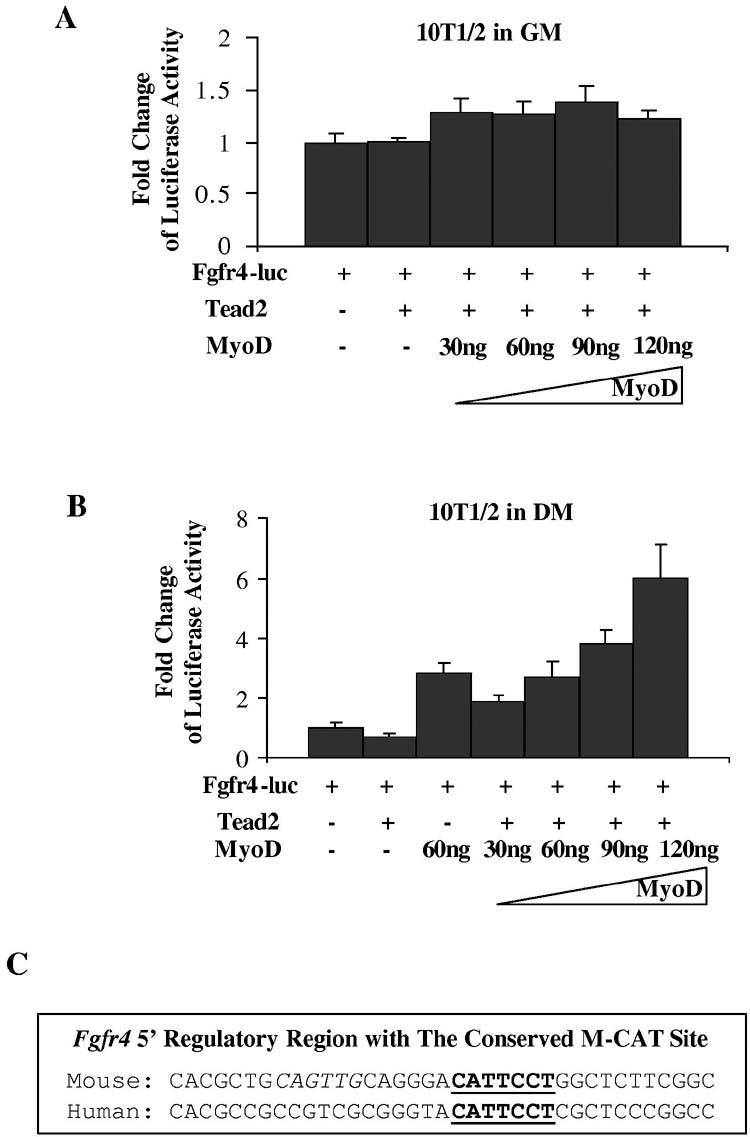
Tead2 activation of the Fgfr4 promoter requires co-factors in differentiated myogenic cells. A, shown is transfection into non-myogenic 10T1/2 cells of Fgfr4-luc reporter construct, with or without co-transfection of Tead2 expression construct, and with increasing amount of MyoD expression construct. The Fgfr4 promoter failed to be activated by Tead2 in proliferating 10T1/2 cells in growth media (GM). Co-transfection of increasing amount of MyoD constructs did not significantly increase luciferase expression. B, shown is a similar transfection series to that conducted in A, but with cells cultured in myogenic differentiation media (DM). Additionally, 60ng of MyoD constructs were co-transfected with Fgfr4-luc without Tead2. Tead2 alone was unable to activate Fgfr4-luc. Co-transfection of MyoD with Tead2 constructs activated Fgfr4-luc in a dose dependent manner. However, 60ng of MyoD constructs alone activated Fgfr4-luc to a similar extent compared to transfection of the same amount of MyoD plus Tead2 constructs. This indicates that MyoD activates the Fgfr4 promoter directly, rather than through the induction of Tead2 co-activators. C, shown is the mouse and human Fgfr4 5′ regulatory region. The M-CAT motif (bold and underlined) is conserved, whereas the E-box (bold and italic) is not.
This data suggested that either MyoD could activate Fgfr4-luc directly, or co-activators of Tead2 were induced by MyoD during 10T1/2 cell differentiation. To distinguish the two possibilities, we transfected 10T1/2 cells with MyoD and Fgfr4-luc without co-transfection of Tead2 constructs. MyoD alone activated Fgfr4-luc to a similar extent compared to transfection of the same amount of MyoD plus Tead2 constructs (Fig. 7B). This indicates that MyoD activated Fgfr4-luc constructs directly rather than through the induction of Tead2 co-activators. This is not surprising because the 226bp Fgfr4 promoter region contains an E-box 6bp before the M-CAT motif (Fig. 7C). Although the M-CAT motif is conserved between mouse and human FGFR4, the E-box is not conserved in human. This suggests that the E-box may play a less important role in Fgfr4 regulation across species boundaries.
Expression of Vestigial like 2 in muscle regeneration—Vestigial like 2 (Vgl2) is a co-activator of Tead transcription factors (32,33). Vgl2 appears to be the only known Tead interacting protein expressed specifically in both embryonic and adult muscles, although a potential role of Vgl2 has not been investigated in muscle regeneration. The U74Av2 GeneChips do not contain a probe set for Vgl2. The probe set for Vgl2 on MOE 430 2.0 array showed that Vgl2 is present in day 0 normal muscle, down-regulated at early muscle regeneration, and then expression increased towards normal level. We further examined the expression of Vgl2 in muscle regeneration using real time RT-PCR. Vgl2 mRNA was detected in uninjected control muscle. Following CTX injection, the expression of Vgl2 was down-regulated with the onset of necrosis (day 2). Although the level of Vgl2 at day 3 was still lower than that of uninjected muscle, its expression increased when compared to day 2 and gradually regained commensurate with satellite cell proliferation and differentiation (day 3 and after; Fig. 8). This observation suggests Vgl2 is a relevant co-activator for Tead2 mediated activation of transcription in muscle regeneration.
Fig. 8.

Expression of Vgl2 is regulated during muscle regeneration. Shown is expression of Vgl2 detected by real time RT-PCR at selected muscle regeneration time points (0, 2d, 3d, 3.5d, 5d, 16d). Two independent muscle samples were studied at each of the time points. The expression of Vgl2 bands was normalized to a control transcript (Cmas). The two different relative measurements of Tead2 were then plotted relative to the time 0 values, with two replicate values shown on the error bar. Expression of Vgl2 showed down-regulation at day 2, and then gradually regained towards time 0 expression levels.
MyoD directly binds to the first intron of Tead2—The transient upregulation of Tead2 at day 3 is the characteristic expression pattern of MyoD and some MyoD downstream targets (26). We therefore investigated whether the Tead2 gene is a potential direct target of MyoD transcriptional activation. MyoD binds to a consensus sequence (CANNTG) called the E-box (34). By searching Tead2 genomic sequence, we identified multiple E-boxes within 1 kb of the upstream promoter sequence and first intron. However, it has been shown that MyoD does not bind to all E-boxes equally. The affinity of MyoD to E-boxes is affected by the bases in the middle of E-boxes, as well as in the flanking region (25,35,36). By carefully examining the sequences of these E-boxes, we found two closely located (15 bp interval) E-boxes in the first intron region have high similarity to an expanded 9 bp consensus high affinity MyoD binding site. We have previously reported this expanded 9 bp consensus site on both the muscle creatine kinase (MCK) promoter and Slug promoter where we had previously shown functionality by both gel shift and chromatin immunoprecipitation assays (25; Fig. 9A). The two Tead2 intron E-boxes are conserved between mouse and human (Fig. 9B).
Fig. 9.
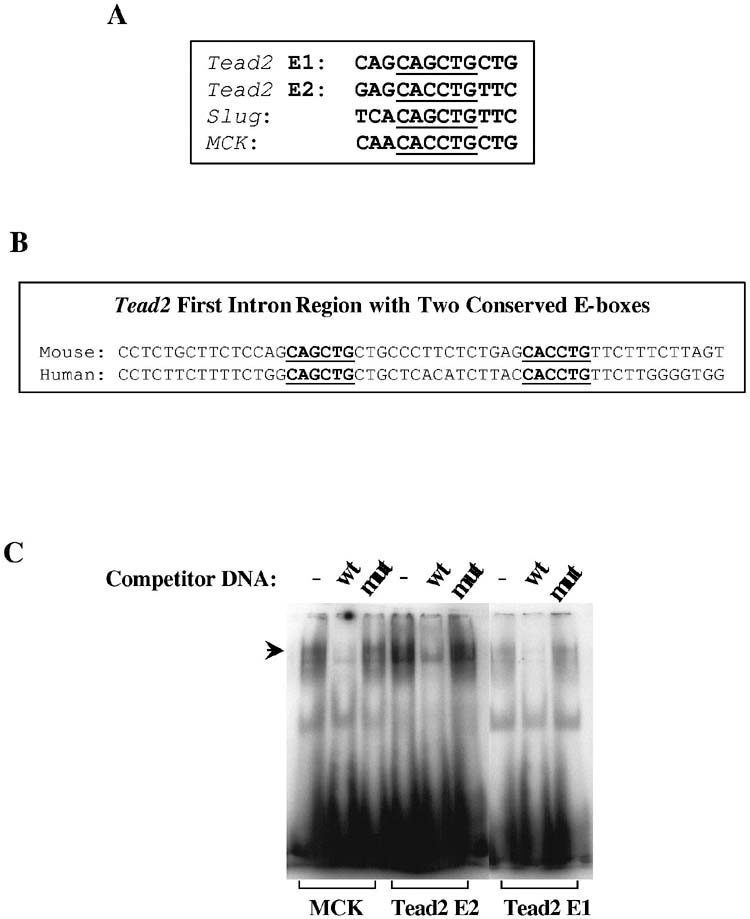
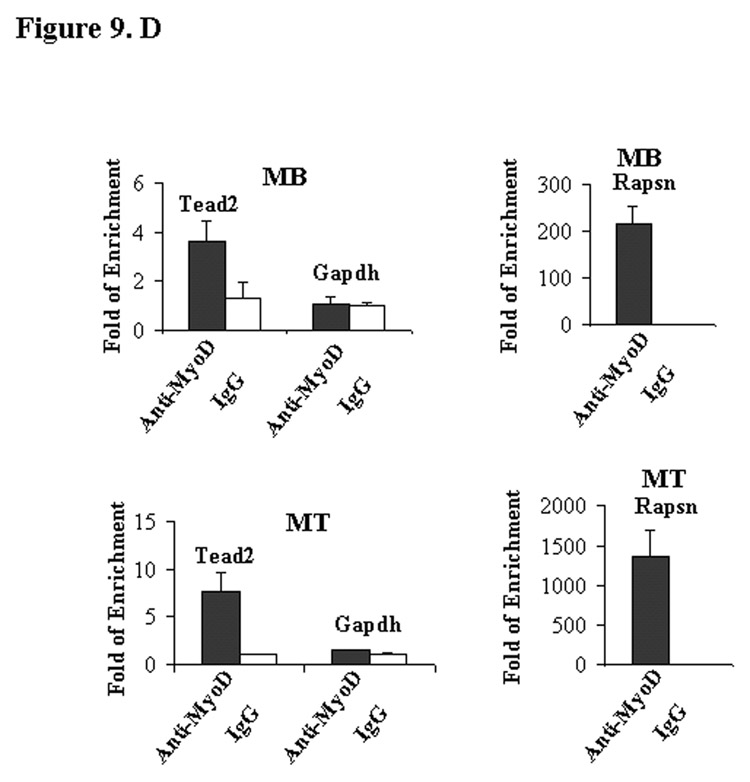
MyoD directly binds to the first intron of Tead2 gene. A, shown are E-boxes and flanking sequences in the mouse Tead2 first intron, Slug promoter and muscle creatine kinase (MCK) promoter. Tead2 E-boxes (Tead2 E1 and E2) show high similarity to Slug and MCK E-boxes. B, shown are gel shift assays with oligonucleotides corresponding to putative E box MyoD consensus binding site sequences from the mouse Tead2 first intron. Incubation of labeled oligonucleotides with C2C12 myonuclear extracts leads to band shift patterns with that are indistinguishable from a known downstream target of MyoD, MCK. The presence of 100 times more of unlabeled wild type (wt) competitor DNA displaces the band shift, whereas mutant (mut) competitor DNA does not affect band shifting. This in vitro assay suggests Tead2 is a likely MyoD downstream target. C, shown is chromatin immunoprecipitation assay with MyoD antibodies and control IgG anibodies in myoblast (MB) and myotubes (MT). In both cases, Tead2 first intron region was enriched in MyoD precipitated DNA, suggesting MyoD directly binds to Tead2 first intron. Enrichment was higher in myotubes than in myoblasts. Gapdh was used as a negative control. Rapsn was used as a positive control.
To test whether MyoD was able to bind to the two Tead2 E-boxes, an in vitro gel shift assay was performed using myoblast nuclear extracts and synthesized oligonucleotides containing each E-box. A band shift similar to that of the positive control E-box from MCK promoter was seen with both Tead2 E-boxes (Fig. 9C). The DNA/protein complexes were competed by excess unlabeled wild-type MCK probes, but not excess unlabeled mutant MCK probes. We further tested whether MyoD binds to the Tead2 first intron region by chromatin immunoprecipitation assays in both myoblasts and myotubes. In myoblasts, Tead2 showed a 3.6-fold enrichment in MyoD precipitates compared to IgG precipitates. In myotubes, Tead2 was 7.4-fold enriched in MyoD precipitates compared to IgG precipitates (Fig. 9D). This suggests that Tead2 is a direct downstream target of MyoD, and that MyoD binding to Tead2 intron increases with myoblast differentiation. As a positive control, we used Rapsn promoter; this is one of the genes we previously reported as a strong MyoD target (26). The Rapsn promoter showed very high affinit y to MyoD in both myoblasts (∼220 fold enrichment) and myotubes (∼1300 fold enrichment). Gapdh was used as a negative control.
MyoD activates the first intron of Tead2—To investigate whether MyoD can bind and activate the Tead2 gene via sequences in the first intron, a 213bp Tead2 intron region containing the two E-boxes were subcloned into pGL3-Promoter luciferase reporter vector contain ing a luciferase gene driven by a SV40 promoter (Fig. 10A). The constructs were transiently transfected into 10T1/2 cells without MyoD or with co-transfection of increasing amount of MyoD constructs. Cells were switched from growth medium to differentiation medium 24 hours after transfection, and allowed to differentiate for 48 hours before being harvested. Luciferase activity increased when MyoD constructs were added from a lower amount to a higher amount (Fig. 10B). When the two E-boxes were mutated (Fig. 10C), activation the constructs by MyoD was greatly reduced (Fig. 10D). These results suggest that the Tead2 first intron region can serve as an enhancer and be activated by MyoD.
Fig. 10.
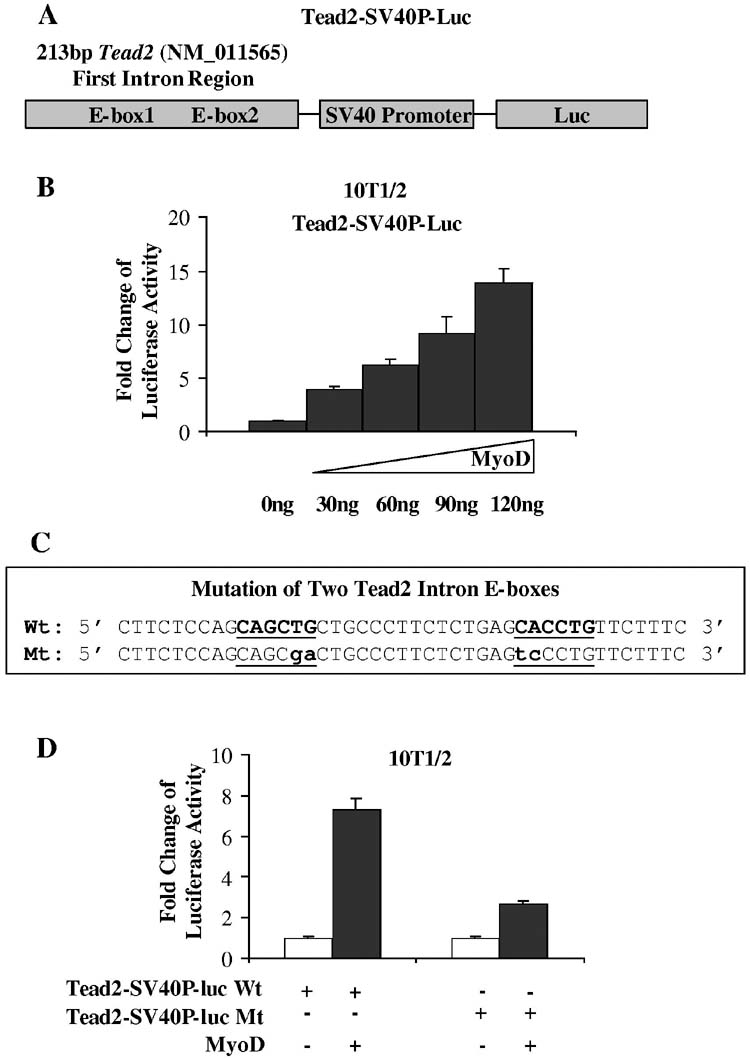
MyoD activates Tead2 intron region through the E-boxes. A, shown is a schematic of the constructs containing the 213 bp of Tead2 first intron region, a SV40 promoter and the luciferase reporter. B, shown is the dose response of Tead2-SV40P-luc constructs to MyoD. Tead2-SV40P-luc constructs were transfected into 10T1/2 cells with co-transfection of increasing amounts of MyoD expression construct. Luciferase activity increased with the increasing amount of MyoD. C, shown is Tead2 intron region containing wild type (Wt) or mutant (Mt) E-boxes. D, shown is transfection of wild type or mutant Tead2-SV40P-luc with or without co-transfection of MyoD. Activation of luciferase by MyoD was greatly reduced when the E-boxes were mutated.
DISCUSSION
Fgfr4 null muscle shows defective regeneration. While Fgfr4-/- mice have been reported to show no overt phenotype, our previous studies of Fgfr4 gene expression suggested that this protein could be critical for muscle regeneration (17). Induction of staged degeneration/regeneration in Fgfr4-/- and wild-type controls showed poorly coordinated and inefficient regeneration (Fig. 1,2). Particularly intriguing was the striking calcifications and extensive fat replacement in the Fgfr4-/- muscle. Fibrofatty replacement is a common feature of “end-stage” human dystrophic muscle, where chronic repeated bouts of degeneration/regeneration occur over many years. However, such fatty replacement is not typically seen after a single bout of damage in either human or mouse muscle. Likewise, calcifications can be seen in chronic inflammatory states in muscle, as in other tissues, but the dramatic number and size of calcifications seen in the Fgfr4-/- muscle after a single bout of degeneration/regeneration is highly unusual. We tested the chemical structure of the calcifications using infrared spectroscopy, and found spectra similar to immature murine bone, but with more hydroxyapatite mineral and less amount of protein. Future studies are needed to determine if the early and abundant calcifications reflect poor clearance of macrophages, or transdifferentiation of myogenic cells towards the bone lineages. The poor regeneration of Fgfr4 null muscle was consistent with importance of Fgfr4 expression during regeneration, and led us to define the molecular pathways involved in induction of Fgfr4 gene expression during muscle regeneration.
MyoD-Tead2 pathway in muscle regeneration—In this study, we present evidence supporting a MyoD-Tead2/Vgl2-Fgfr4 regulatory cascade in muscle regeneration in vivo. Tead proteins, also called transcription enhancer factors (TEF), are a family of transcriptional factors all sharing a highly conserved TEA DNA binding domain. In vertebrates, at least four family members have been identified, including Tead1 (TEF-1), Tead2 (TEF-4), Tead3 (TEF-5) and Tead4 (TEF-3). Homologues are also identified in Drosophila (scalloped), fungus (AbbA), and yeast (TEC1) (37-39). Tead1 was the first isolated in this family, as a binding protein of the GT-IIC and Sph sites in the simian virus 40 (SV40) enhancer (40). Tead2, 3, and 4 were cloned subsequently (30,41,42). Tead2, originally named as embryonic TEA domain-containing factor, was initially identified in neural precursor cells (30). Tead2 was recently shown to activate Pax3 transcription in neural crest cells (43). Tead2 mRNA is the earliest (as early as 2-cell stage) expressed Tead gene in mouse embryos (44). Tead1, Tead3 and Tead4 are expressed soon after (30,41,42,44,45). Tead1 is expressed in many embryonic tissues during mouse development (41). It is particularly important for cardiogenesis as Tead1 mutant mice show malformed hearts and embryonic lethality (46). Tead4 has been shown enriched in embryonic skeletal muscles and increased in expression in differentiated skeletal muscle C2C12 cells, suggesting it may have a role in myogenesis (41,42). Most recently, Tead4 was shown as a novel MyoD target by ChIP-on-chip assays (47). In this study, we found strong induction of another TEA domain protein, Tead2 in regenerating muscle nuclei. We further show that MyoD directly binds to and activates the first intron of Tead2, suggesting Tead2 is a downstream target of MyoD. It is worth mentioning that the positive control we used in ChIP assay, Rapsn, functions to cluster the nicotinic acetylchoine receptor on neuromuscular junctions (48). Interestingly, E-box mutations in the RAPSN promoter region are associated with some cases of congenital myasthenic syndrome (49). This E-box region is highly conserved between human and mouse. We identified Rapsn as a high potential MyoD target in our previous study (26), and the new ChIP data shown here validates the binding of MyoD to this expanded E box, and the importance of the patient mutations.
We also show that a co-activator of Tead protein, Vgl2 expression was commensurate with Tead2 in muscle regeneration, suggesting it is a co-activator of Tead2. Vgl2 has been shown expressed in C2C12 myoblasts and upregulated upon differentiation. Transfected Vgl2 protein translocated from cytoplasm to nucleus during C2C12 differentiation (32). This agrees with our observation that Fgfr4 promoter was not activated in Tead2 transfected myoblasts, but activated in Tead2 transfected myotubes.
Fgfr4, transcriptional regulation and effects in muscle regeneration—All TEF proteins can bind to the same DNA sequence (M-CAT motif 5′-CATTCCT-3′). Recent studie s have also suggested that Tead1 binds to and activates A/T-rich and MEF2 elements on some muscle genes (50). The M-CAT motif was initially identified in the cardiac troponin T promoter, and the interaction of M-CAT motif and an M-CAT binding factor is required for the activation of cardiac troponin T promoter in muscle cells (51). The M-CAT binding factor was later found closely related and possibly identical to TEF-1 (52). Additionally, the GT-IIC binding site for TEF-1 in the SV40 enhancer is highly similar to the M-CAT motif. Subsequently, M-CAT has also been found present in the promoter of other muscle specific genes such as a and β myosin heavy chain, and skeletal a actin (53-57). In this study, we identified an M-CAT motif in the Fgfr4 promoter region, and showed that Tead2 could induce transcription through this region. Furthermore, activation of transcription was dependent on the integrity of the M-CAT site. These results suggest that Fgfr4 is a novel downstream target of Tead2 in muscle regeneration. Both genes are induced at day 3 post-injection, suggesting an association with the transition from myoblasts to differentiated myotubes in muscle regeneration. Fgfr4-/- mouse muscle appeared to regenerate less efficiently than normal mouse muscle, with lagged expression of muscle cytoskeletal genes, persistent inflammation, calcification and fat infiltration in late stage. This finding agrees with the role of Fgfr4 in embryonic muscle development (18).
Our data suggests that MyoD acts upstream of Tead2 and Fgfr4. Ectopic expression of MyoD rapidly induces Fgfr4 in chick embryos in vivo, which had previously suggested a direct regulation of Fgfr4 by MyoD (58). The data presented here suggests a MyoD-Tead2-Fgfr4 pathway in regeneration, including an indirect regulatory loop between MyoD and Fgfr4. We also showed that MyoD can activate mouse Fgfr4 promoter region through an E-box close to the M-CAT motif, although this E-box is not conserved in human FGFR4 whereas the M-CAT motif is highly conserved. This suggests that the MyoD-Tead2-Fgfr4 pathway may coexist with a direct MyoD-Fgfr4 pathway in mouse muscle .
In conclusion, our data suggests a MyoD-Tead2-Fgfr4 regulatory pathway in muscle regeneration. This pathway is critical for regeneration, as Fgfr4-/- mice show impaired muscle regeneration with slowed maturation of regenerating fibers, and development of intramuscular adipose and calcifications. It will be of interest to investigate signaling pathways downstream of Fgfr4 in muscle regeneration in future studies.
Footnotes
Supported by grants from the Muscular Dystrophy Association USA to EPH, Department of Defense W81XWH-04-01-0081 to EPH, and National Institutes of Diabetes, Digestive and Kidney Disease grant DK35310 to WLM. The authors thank Dr. Hiroaki Ohkubo (Kumamoto University, Japan) for providing the pcDNA-mETF expression vector. The embryonic myosin heavy chain antibody developed by Dr. Helen M. Blau was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA 52242.
- FGF
- fibroblast growth factor
- Fgfr
- fibroblast growth factor receptor
- M-CAT
- muscle-CAT
- MCK
- muscle creatine kinase
- Tead
- TEA domain
- TEF
- transcription enhancer factor
- Vgl
- vestigial-like
- Cmas
- cytidine monophospho-N-acetylneuraminic acid synthetase
- Rapsn
- receptor-associated protein of the synapse
- SV40
- simian virus 40
- GM
- growth medium
- DM
- differentiation medium
- CTX
- cardiotoxin
- ChIP
- chromatin immunoprecipitation
- QMF-PCR
- quantitative multiplex fluorescent-polymerase chain reaction
REFERENCES
- 1.Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Cytokine Growth Factor Rev. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Eswarakumar VP, Lax I, Schlessinger J. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Muenke M, Schell U, Hehr A, Robin NH, Losken HW, Schinzel A, Pulleyn LJ, Rutland P, Reardon W, Malcolm S, Winter RM. Nature Genet. 1994;8:269–274. doi: 10.1038/ng1194-269. [DOI] [PubMed] [Google Scholar]
- 4.Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Nat. Genet. 1994;8:98–103. doi: 10.1038/ng0994-98. [DOI] [PubMed] [Google Scholar]
- 5.Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, Le Merrer M, Munnich A. Nature. 1994;371:252–254. doi: 10.1038/371252a0. [DOI] [PubMed] [Google Scholar]
- 6.Shiang R, Thompson LM, Zhu YZ, Church DM, Fielder TJ, Bocian M, Winokur ST, Wasmuth JJ. Cell. 1994;78:335–342. doi: 10.1016/0092-8674(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 7.Rutland P, Pulleyn LJ, Reardon W, Baraitser M, Hayward R, Jones B, Malcolm S, Winter RM, Oldridge M, Slaney SF, Poole MD, Wilkie AOM. Nat. Genet. 1995;9:173–176. doi: 10.1038/ng0295-173. [DOI] [PubMed] [Google Scholar]
- 8.deLapeyriere O, Ollendorff V, Planche J, Ott MO, Pizette S, Coulier F, Birnbaum D. Development. 1993;118:601–611. doi: 10.1242/dev.118.2.601. [DOI] [PubMed] [Google Scholar]
- 9.Coulier F, Pizette S, Ollendorff V, deLapeyriere O, Birnbaum D. Prog. Growth Factor Res. 1994;5:1–14. doi: 10.1016/0955-2235(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 10.Shaoul E, Reich-Slotky R, Berman B, Ron D. Oncogene. 1995;10:1553–1561. [PubMed] [Google Scholar]
- 11.Pizette S, Coulier F, Birnbaum D, DeLapeyriere O. Exp. Cell Res. 1996;224:143–151. doi: 10.1006/excr.1996.0122. [DOI] [PubMed] [Google Scholar]
- 12.Sheehan SM, Allen RE. J. Cell Physiol. 1999;181:499–506. doi: 10.1002/(SICI)1097-4652(199912)181:3<499::AID-JCP14>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 13.Dusterhoft S, Pette D. Differentiation. 1999;65:161–169. doi: 10.1046/j.1432-0436.1999.6530161.x. [DOI] [PubMed] [Google Scholar]
- 14.Kastner S, Elias MC, Rivera AJ, Yablonka-Reuveni Z. J. Histochem. Cytochem. 2000;48:1079–1096. doi: 10.1177/002215540004800805. [DOI] [PubMed] [Google Scholar]
- 15.Cool SM, Sayer RE, van Heumen WR, Pickles JO, Nurcombe V. Histochem. J. 2002;34:291–297. doi: 10.1023/a:1023326524562. [DOI] [PubMed] [Google Scholar]
- 16.Armand AS, Launay T, Pariset C, Della Gaspera B, Charbonnier F, Chanoine C. Biochim. Biophys. Acta. 2003;1642:97–105. doi: 10.1016/s0167-4889(03)00103-4. [DOI] [PubMed] [Google Scholar]
- 17.Zhao P, Hoffman EP. Dev. Dyn. 2004;229:380–392. doi: 10.1002/dvdy.10457. [DOI] [PubMed] [Google Scholar]
- 18.Marics I, Padilla F, Guillemot JF, Scaal M, Marcelle C. Development. 2002;129:4559–4569. doi: 10.1242/dev.129.19.4559. [DOI] [PubMed] [Google Scholar]
- 19.Yu SJ, Zheng L, Ladanyi M, Asa SL, Ezzat S. Clin. Cancer Res. 2004;10:6750–6758. doi: 10.1158/1078-0432.CCR-04-0223. [DOI] [PubMed] [Google Scholar]
- 20.Yu S, Zheng L, Trinh DK, Asa SL, Ezzat S. Lab. Invest. 2004;84:1571–1580. doi: 10.1038/labinvest.3700187. [DOI] [PubMed] [Google Scholar]
- 21.Weinstein M, Xu X, Ohyama K, Deng CX. Development. 1998;125:3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- 22.Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, Deng CX, McKeehan WL. J. Biol. Chem. 2000;275:15482–15489. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 23.Yu C, Wang F, Jin C, Wu X, Chan WK, McKeehan WL. Am. J. Pathol. 2002;161:2003–2010. doi: 10.1016/S0002-9440(10)64478-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu C, Wang F, Jin C, Huang X, McKeehan WL. J. Biol. Chem. 2005;280:17707–17714. doi: 10.1074/jbc.M411771200. [DOI] [PubMed] [Google Scholar]
- 25.Zhao P, Iezzi S, Carver E, Dressman D, Gridley T, Sartorelli V, Hoffman EP. J. Biol. Chem. 2002;277:30091–30101. doi: 10.1074/jbc.M202668200. [DOI] [PubMed] [Google Scholar]
- 26.Zhao P, Seo J, Wang Z, Wang Y, Shneiderman B, Hoffman EP. C. R. Biol. 2003;326:1049–1065. doi: 10.1016/j.crvi.2003.09.035. [DOI] [PubMed] [Google Scholar]
- 27.Zhou JH, Hoffman EP. J. Biol. Chem. 1994;269:18563–18571. [PubMed] [Google Scholar]
- 28.Chen YW, Zhao P, Borup R, Hoffman EP. J. Cell. Biol. 2000;151:1321–1336. doi: 10.1083/jcb.151.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mendelsohn R, Paschalis EP, Boskey AL. J Biomed. Opt. 1999;4:14–21. doi: 10.1117/1.429916. [DOI] [PubMed] [Google Scholar]
- 30.Yasunami M, Suzuki K, Houtani T, Sugimoto T, Ohkubo H. J. Biol. Chem. 1995;270:18649–18654. doi: 10.1074/jbc.270.31.18649. [DOI] [PubMed] [Google Scholar]
- 31.Boskey A, Mendelsohn R. J Biomed. Opt. 2005;10:031102. doi: 10.1117/1.1922927. [DOI] [PubMed] [Google Scholar]
- 32.Maeda T, Chapman DL, Stewart AFR. J. Biol. Chem. 2002;277:48889–48898. doi: 10.1074/jbc.M206858200. [DOI] [PubMed] [Google Scholar]
- 33.Mielcarek M, Gunther S, Kruger M, Braun T. Mech. Dev. 2002;119:S269–S274. doi: 10.1016/s0925-4773(03)00127-8. [DOI] [PubMed] [Google Scholar]
- 34.Buskin JN, Hauschka SD. Mol. Cell. Biol. 1989;9:2627–2640. doi: 10.1128/mcb.9.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blackwell TK, Weintraub H. Science. 1990;250:1104–1110. doi: 10.1126/science.2174572. [DOI] [PubMed] [Google Scholar]
- 36.Huang J, Blackwell TK, Kedes L, Weintraub H. Mol. Cell Biol. 1996;16:3893–3900. doi: 10.1128/mcb.16.7.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laloux I, Dubois E, Dewerchin M, Jacobs E. Mol. Cell Biol. 1990;10:3541–3550. doi: 10.1128/mcb.10.7.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell S, Inamdar M, Rodrigues V, Raghavan V, Palazzolo M, Chovnick A. Genes Dev. 1992;6:367–379. doi: 10.1101/gad.6.3.367. [DOI] [PubMed] [Google Scholar]
- 39.Aramayo R, Timberlake WE. EMBO J. 1993;12:2039–2048. doi: 10.1002/j.1460-2075.1993.tb05853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davidson I, Xiao JH, Rosales R, Staub A, Chambon P. Cell. 1988;54:931–942. doi: 10.1016/0092-8674(88)90108-0. [DOI] [PubMed] [Google Scholar]
- 41.Jacquemin P, Hwang JJ, Martial JA, Dolle P, Davidson I. J. Biol. Chem. 1996;271:21775–21785. doi: 10.1074/jbc.271.36.21775. [DOI] [PubMed] [Google Scholar]
- 42.Yockey CE, Smith G, Izumo S, Shimizu N. J. Biol. Chem. 1996;271:3727–3736. doi: 10.1074/jbc.271.7.3727. [DOI] [PubMed] [Google Scholar]
- 43.Milewski RC, Chi NC, Li J, Brown C, Lu MM, Epstein JA. Development. 2004;131:829–837. doi: 10.1242/dev.00975. [DOI] [PubMed] [Google Scholar]
- 44.Kaneko KJ, Cullinan EB, Latham KE, DePamphilis ML. Development. 1997;124:1963–1973. doi: 10.1242/dev.124.10.1963. [DOI] [PubMed] [Google Scholar]
- 45.Yasunami M, Suzuki K, Ohkubo H. Biochem. Biophy. Res. Comm. 1996;228:365–370. doi: 10.1006/bbrc.1996.1667. [DOI] [PubMed] [Google Scholar]
- 46.Chen Z, Friedrich GA, Soriano P. Genes Dev. 1994;8:2293–2301. doi: 10.1101/gad.8.19.2293. [DOI] [PubMed] [Google Scholar]
- 47.Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD. Genes Dev. 2005;19:553–569. doi: 10.1101/gad.1281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Froehner SC, Luetje CW, Scotland PB, Patrick J. Neuron. 1990;5:403–410. doi: 10.1016/0896-6273(90)90079-u. [DOI] [PubMed] [Google Scholar]
- 49.Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG. Hu. Mol. Genet. 2003;12:739–748. doi: 10.1093/hmg/ddg089. [DOI] [PubMed] [Google Scholar]
- 50.Karasseva N, Tsika G, Ji J, Zhang A, Mao X, Tsika R. Mol. Cell Biol. 2003;23:5143–5164. doi: 10.1128/MCB.23.15.5143-5164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mar JH, Ordahl P. Mol. Cell Biol. 1990;10:4271–4283. doi: 10.1128/mcb.10.8.4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farrance IK, Mar JH, Ordahl CP. J. Biol. Chem. 1992;267:17234–17240. [PubMed] [Google Scholar]
- 53.Flink IL, Edwards JG, Bahl JJ, Liew CC, Sole M, Morkin E. J. Biol. Chem. 1992;267:9917–9924. [PubMed] [Google Scholar]
- 54.Shimizu N, Prior G, Umeda PK, Zak R. Nucleic Acids Res. 1992;20:93–1799. doi: 10.1093/nar/20.7.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knotts S, Rindt H, Neumann J, Robbins J. J. Biol. Chem. 1994;269:31275–31282. [PubMed] [Google Scholar]
- 56.MacLellan WR, Lee T-C, Schwartz R, Schneider MD. J. Biol. Chem. 1994;269:16754–16760. [PubMed] [Google Scholar]
- 57.Molkentin JD, Markham BE. Mol. Cell Biol. 1994;14:5056–5065. doi: 10.1128/mcb.14.8.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delfini MC, Duprez D. Development. 2004;131:713–723. doi: 10.1242/dev.00967. [DOI] [PubMed] [Google Scholar]