

Abstract
Background
Stromal progenitor cells (SPC) exhibit immunosuppressive effects in vitro that have led to speculation regarding their capacity to evade host immune recognition and to treat autoimmune diseases and GVHD. However, there is little in vivo experimental data to support these immunologic claims. To assess immune recognition of SPC in vivo, we evaluated the immune response of animals transplanted with SPC.
Methods
C57BL/6 (B6) or Balb/c adult, murine, bone marrow derived SPC (AmSPC) were administered by intraperitoneal injection into B6 recipients. T cell proliferation and alloantibody response was assessed from spleens and peripheral blood harvested from transplanted animals and analyzed by cell proliferation assay and flow cytometry. To assess tolerance induction, transplanted animals also received allogeneic skin grafts.
Results
Animals injected with allogeneic AmSPC mounted an accelerated CD4 response to alloantigen compared to syngeneic AmSPC injected and uninjected controls. Allogeneic AmSPC injected animals also demonstrated high titers (≥1:1000) of antibody directed against allogeneic AmSPC targets. Animals primed with donor or host matched AmSPC also failed to induce tolerance and all animals exhibited rejection of allogeneic skin grafts (n = 7, p<0.0001).
Conclusions
In contrast to their in vitro behavior, our data demonstrate that AmSPC are recognized by the host immune system in vivo, elicit a cellular and humoral immune response, and fail to induce tolerance. These findings have significant implications for all allogeneic SPC based therapeutic strategies.
Introduction
Mesenchymal “Stem” Cells (MSC) are multipotent cells derived from bone marrow and a variety of other tissues.(1, 2) Although the term MSC is widely used, all populations described thus far are heterogeneous and contain cells with a hierarchy of potentiality. They are therefore more appropriately referred to as bone marrow (or other tissue derived) stromal progenitor cells (SPC) which is the terminology that we will use throughout this report. They are of potential therapeutic value because of their extensive capacity for in vitro expansion, their capacity for induced differentiation into at least some mesenchymal lineages, and their relative ease of genetic manipulation.(3) In addition, SPC have interesting immunologic properties in vitro which have led to speculation and controversy regarding their in vivo immunologic behavior. Although the mechanisms are not fully defined, SPC have been demonstrated in vitro by multiple investigators to suppress stimulated T cells in co-culture experiments.(4–19) Because of these in vitro observations, it has been suggested that SPC may evade alloimmune surveillance, induce specific immunologic tolerance, and suppress graft versus host disease (20–28), but there is little clinical or experimental data to support these claims. In fact, a recent study demonstrated the in vivo elimination of subcutaneously implanted, gene-modified, allogeneic SPC in a murine system.(29) The immune rejection of allogeneic SPC would complicate their use in therapeutic applications requiring the long term persistence of allogeneic cells for sustained clinical benefit.
In this study we utilize a well characterized population of murine adult bone marrow derived stromal progenitor cells (AmSPC), that are devoid of hematopoietic cells, and satisfy defining criteria that are widely used for so called MSC, to characterize the alloresponse to SPC after intraperitoneal administration. In contrast to their in vitro behavior, our data demonstrate that SPC are recognized by the host immune system in vivo, and elicit both host cellular and antibody immune responses. To test the immunomodulatory capacity of AmSPC in vivo, we also performed allogeneic skin grafting and demonstrate skin graft rejection in animals instead of tolerance induction. These findings have significant implications for all allogeneic SPC based therapeutic strategies.
Materials and Methods
AmSPC Isolation and Expansion
Femurs and tibias were removed from euthanized 4—12 week old C57BL/6 (B6) and Balb/c male mice (obtained from Jackson Laboratories, Bar Harbor, ME). The bone marrow was flushed via a 23 gauge needle with complete MesenCult Basal Medium for Murine Mesenchymal Stem Cells (StemCell Technologies, Vancouver, Canada) containing mesenchymal stem cell stimulatory supplements, 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies/Invitrogen, Carlsbad, CA). Bone marrow from each mouse was filtered through a 70 μm nylon filter. Red cell lysis was performed by the addition of Ammonium Chloride (StemCell Technologies, Vancouver, Canada). Cells were plated into 6-well tissue culture plates (Falcon, Franklin Lakes, NJ), and kept in a humidified incubator at 37°C and 5% CO2. Adherent cells were lifted once 90% confluent using 0.25% Trypsin 0.05mM EDTA (Life Technologies/Invitrogen, Carlsbad, CA) and expanded by replating at 5000 cells/cm2. Complete MesenCult media was used to feed expanding cells every 3—4 days.
Flow cytometric analysis of Immune Phenotype
Both untreated AmSPCs and AmSPCs treated with interferon gamma (IFN-γ-200 ng/ml for 72 hours) were suspended in 100 μl of PBS and incubated in FC blocking antibody (CD16/CD32) (BD Biosciences Pharmingen, San Diego, CA) for 10 min at room temperature. AmSPCs were incubated with primary antibody directly conjugated to phycoerythrin (PE) (1 μg/l × 106 cells) for 30 min at 4°C, washed twice with PBS, and stained with 1μg propidium iodine prior to analysis. Primary antibodies with specificity for the surface antigens CD3, CD11b, CD13, CD31, CD44, CD45, CD90, C-kit (CD117), MHC I, MHC II, and Sca-1 were used (BD Pharmingen, San Diego, CA).
Primary Mixed Lymphocyte Cultures in the presence of AmSPC
B6 responder matched AmSPC were trypsinized at passage 13. Cells were counted by trypan blue exclusion (greater than 95% viable). AmSPC were plated at 5,000 cells/cm2 into 12 well tissue culture treated plates (Falcon, Franklin Lakes, NJ). AmSPC were incubated alone overnight in complete Mesencult media to allow adherence to the dish. Spleens were harvested from 12 week old female B6 mice as responder cells, and from 12 week old Balb/c mice as stimulator cells. Spleens were processed using 70 μm cell strainers and 5 ml syringes (BD Biosciences, San Jose, CA) to achieve a single cell suspension. Once a single cell suspension was prepared, red blood cells were lysed using Ammonium Chloride (StemCell Technologies, Vancouver, Canada). Responder cells were stained with CFDA SE tracking dye (CFSE - Molecular Probes, Eugene, OR) using the manufacturers instructions. Stimulator splenocytes were given 2500 cGy irradiation in a Cs135 gamma irradiator. After staining was complete, cells were washed three times in complete assay medium, Iscove’s Modified Dulbecco Media with non-essential amino acids, sodium pyruvate, 100U penicillin, 100μg streptomycin, 55μM Beta-Mercaptoethanol (all from Life Technologies/Invitrogen, Carlsbad, CA) and 10% Fetal 6Bovine Serum (HyClone, Logan, UT). Media was removed from the 24hr adherent AmSPC cultures and responder and stimulator splenocytes were added to appropriate wells at the concentration of 1.2 × 106 cells/cm2 and 6 × 105 cells/cm2 (responder:stimulator ratio of 2:1). Alternatively, the polyclonal lymphocyte stimulant phytohemagluttin in (PHA) (Sigma-Aldrich, St. Louis, MO) was added to stimulate cultures at a final concentration of 10 μg/ml. All experiments were performed a minimum of three times and all samples prepared in triplicate. Optimal culture conditions and time periods required for peak proliferation in PHA and alloantigen stimulated cultures were determined from pilot studies. Proliferation was observed at 3 days in mitogen stimulated cultures with no proliferation observed in alloantigen stimulated cultures until 7 days in culture. As a result, PHA stimulated cultures were incubated at 37° C, 5% CO2 for 3 days and alloantigen stimulated cultures were incubated for 7 days under the same conditions. The above experiments were also performed with stimulator matched Balb/c AmSPC and repeated in a transwell assay system. For transwell assays, either B6 or Balb/c AmSPC were plated in the lower and separated from splenocytes in the upper chamber by a 4μm pore size semi-permeable membrane insert.
AmSPC Preparation for Injections
Animals were housed in the Laboratory Animal Facility of the Abramson Research Center at the Children’s Hospital of Philadelphia. The experimental protocols were approved by the Institutional Animal Care and Use Committee followed guidelines set forth in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. AmSPC were trypsinized at passage 13 and a single cell suspension created. Cells were washed 3 times with 1X dPBS (Life Technologies/Invitrogen, Carlsbad, CA). Cells were counted by trypan blue exclusion (greater than 95% viable). Cells were resuspended at 5 × 106 cells/ml in 1X dPBS with 1% normal mouse serum. A volume of 200 μl of the cell suspension was immediately injected into the peritoneal cavity of 6–8 week old female B6 mice. This injection was considered the zero-week time point. A second injection prepared exactly as described above was administered at the four-week time point.
Analysis of T cell proliferation in response to alloantigen
Spleens were removed from treated B6 mice 2 weeks after the second immunization (6 week time point) and Balb/c spleens were harvested as stimulator cells. In order to increase the sensitivity of detection for discrete proliferation peaks, the CD4+ lymphocyte fraction of responder splenocytes cell populations was selected using a CD4+ T cell isolation kit and an autoMACS cell separator according to the manufacturer’s instructions (Miltenyi BioTech, Auburn, CA). CD4+ cells were stained with CFSE and stimulator cells treated with irradiation as previously described. Control CD4+ cells were obtained from age-matched animals for comparison. All samples were prepared in triplicate. Responder and stimulator cells were added to appropriate wells at the concentration of 1.2 × 106 cells/cm2 and 6 × 105 cells/cm2 (responder:stimulator ratio of 2:1) in complete assay medium. Purified anti CD3 antibody (BD Pharmingen, San Jose, CA) was added to the control wells at a final concentration of 1 μg/μl. Cultures were incubated at 37° C, 5% CO2 and analyzed at 3.5 and 5.5 day time points. Three samples per group were analyzed by flow cytometry with a FACS Calibur system and Cell Quest Pro software.
Alloantibody analysis
Mice were bled from the retro-orbital vein at 0, 2, 4, 5, and 6 weeks. Blood was placed in a clot activating tube and centrifuged for 10 minutes at 500g. Serum was aspirated and frozen at −80° C. Target B6 (H2b), Balb/c (H2d) p13 AmSPC were treated with IFN-γ (200 ng/ml) for 72 hours prior to coincubations. Cells were counted by trypan blue exclusion (greater than 95% viable for both IFN-γ treated and untreated target AmSPC). Treated and untreated target AmSPC were coincubated for 1 hour with dilutions of serum from injected animals (1:10, 1:100, 1:1000) in FACS staining buffer, 1X dPBS (Life Technologies/Invitrogen, Carlsbad, CA), 0.5% BSA (Sigma-Aldrich, St. Louis, MO), and 0.1% Sodium Azide (Sigma-Aldrich, St. Louis, MO). Cells were washed with FACS staining buffer and then incubated with anti-Mouse IgG (FITC conjugated) and IgM (PE conjugated) antibodies (Jackson ImmunoResearch Inc., West Grove, PA) for 30 minutes. Additional experiments using p13 CBA (H2k) AmSPC targets with allogeneic primed animal sera was also performed. Cells were washed using FACS staining buffer. Three cell samples for each condition were then analyzed by dual color flow cytometry.
Skin grafting
Six-8 week old, female B6 mice received intraperitoneal injections with either syngeneic (B6) AmSPC, allogeneic (Balb/c) AmSPC, syngeneic (B6) splenocytes, or allogeneic (Balb/c) splenocytes at the zero and four week timepoints as described above. At six weeks, two weeks after the second injection, skin grafting was performed by a modification of the technique described by Billingham and Medawar.(30) Briefly, full thickness donor skin grafts (1.5 × 0.5 cm2) were prepared from the ventral skin of Balb/c mice and transferred to recipient sites on B6 mice. Recipient sites were created on the lateral thorax of both AmSPC or splenocyte injected and non-injected control mice while carefully preserving the panniculus carnosus. Autografting was performed using skin removed from the allograft site to serve as a technical control. Each mouse received an autograft and allograft. The grafts were covered with petroleum gauze and held in place with a Band-Aid to create a pressure dressing. Dressings were removed after 5 days. Nonadherent grafts were considered technical failures and were excluded. Adherent grafts were monitored for signs of rejection (hardening of the graft, and necrosis) and photographs were taken daily. Grafts were considered rejected when >90% of the surface area was necrotic and the graft hardened. Photographs of the grafts were also reviewed in series at the end of the experiment by an investigator blinded to the group and treatment.
Statistical Analysis
Data are presented as the mean ±SD. Means were compared by student’s t-test.. Kaplan-Meier survival curves were created to compare skin graft rejection. Differences between groups were determined by log rank analysis. Probability values of p<0.05 were interpreted as statistically significant.
Results
Characteristics of AmSPC
AmSPC isolated by the described methodology are morphologically similar to human and rat “MSC”, contain a high percentage of CFU-f after passaging (up to 80%), and demonstrate linear expansion for more than 100 population doublings. Osteogenic, adipogenic, and chondrogenic differentiation can be induced by defined culture conditions. Phenotypically, AmSPC are Sca-1+, VCAM-1+, CD44+, CD31−, CD45−, and CD90−. The cells display the following immune phenotype, MHC Class I+, MHC II−, CD40−, CD80+/−, CD86−. Upon treatment with interferon gamma, upregulation of MHC I is observed, MHC II expression is induced, and co-stimulatory expression remains unchanged (Figure 1).
Figure 1. Immune phenotype of AmSPC +/− IFN-γ.
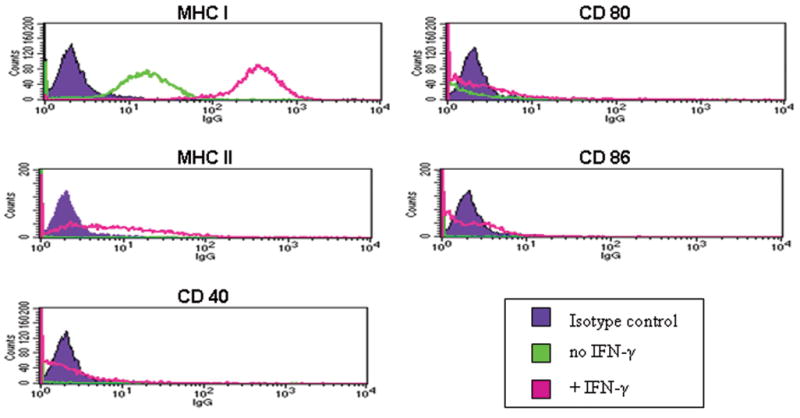
AmSPC express MHC I constitutively but do not express MHC II or the costimulatory molecules CD 40, CD80, and CD86. With IFN-γ pre-treatment (200 ng/ml for 72 hours), MHC I expression is upregulated and MHC II expression is induced, but costimulatory marker expression remains unchanged.
In Vitro Suppression of T-cell Proliferation
In the presence of AmSPC, B6 responder splenocyte proliferation was inhibited in MLR cultures. This suppression was observed for both CD4+ and CD8+ T cells in response to both alloantigen (Balb/c splenocytes) and polyclonal mitogen stimulation in vitro (Figure 2, panel A). This suppression occurred in the presence of both responder matched B6 AmSPC and stimulator matched Balb/c AmSPC and was therefore independent of MHC matching between responder and AmSPC cell populations. Furthermore, the suppression was contact dependent with no suppression observed in a transwell system (Figure 2, panel B).
Figure 2. AmSPC inhibition of T cell proliferation.
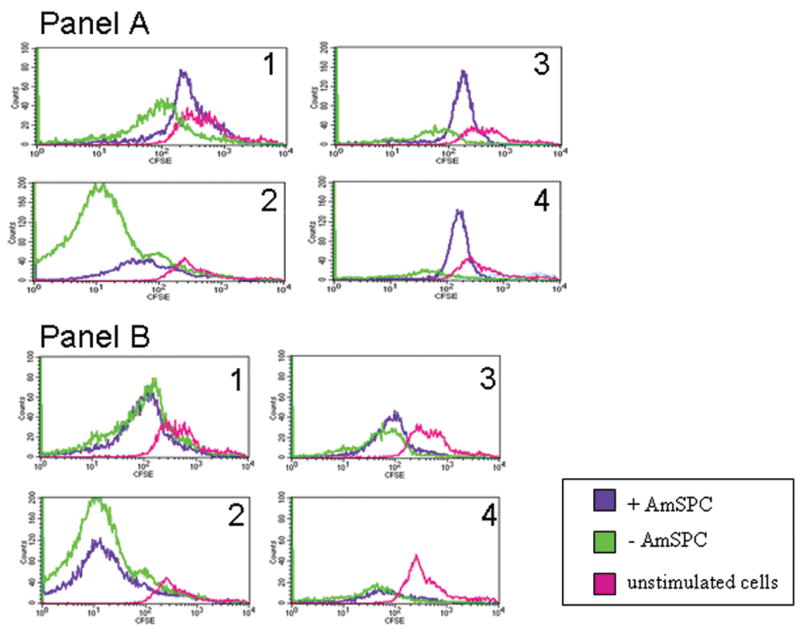
Representative histogram demonstrating direct co-culture of AmSPC with mixed lymphocyte cultures. The presence of B6 responder-matched or Balb/c stimulator-matched AmSPC results in inhibition of B6 responder CD4+ and CD8+ proliferation in both mitogen and alloantigen (Balb/c splenocyte) stimulated cultures (Panel A). Transwell separation of AmSPC (lower chamber) from splenocytes (upper chamber) does not result in T cell inhibition (Panel B). Response is measured by CFDA-SE divisions – each division halves the fluorescence intensity moving the population to the left on the histogram. (1 = CD4+ fraction of PHA stimulated cells, 2 = CD8+ fraction of PHA stimulated culture, 3 = CD4+ fraction of alloantigen stimulated culture, 4 = CD8+ fraction alloantigen stimulated culture.)
In Vivo Cellular and Humoral Response to AmSPC Alloantigen
In order to examine the in vivo host cellular response to injected AmSPC, spleens from B6 animals injected with either B6 or Balb/c AmSPC were harvested for T cell proliferation assays in response to alloantigen (Balb/c splenocytes). Nine of 10 animals injected with Balb/c (allogeneic) AmSPC mounted an accelerated CD4+ T-cell proliferation response (as indicated by decreases in the fluorescence intensity shifting histogram peaks to the left) compared to animals injected with syngeneic AmSPC or uninjected controls at 3.5 days after re-stimulation with alloantigen. (Figure 3A) At 5.5 days after alloantigen stimulation, all animals injected with B6 (syngeneic) AmSPC demonstrated initiation of T cell proliferation. Allogeneic primed cells exhibited continued proliferation with the majority of cells having shifted to the left and represented as a single heterogenous peak of cell divisions. (Figure 3B) No such increase in T cell proliferation was observed in uninjected animals. At the 7 day time point, all uninjected control CD4+ cell populations exhibited proliferation compared to unstimulated control cultures (data not shown).
Figure 3. 6 week post treatment T cell response after 7 day incubation with alloantigen.
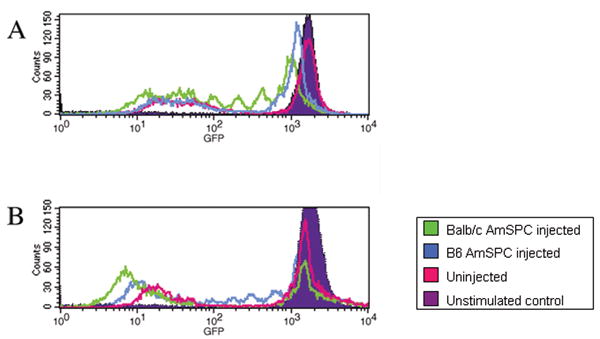
CD4+ T cells were isolated from spleens of B6 animals injected with either Balb/c (allogeneic) AmSPC, B6 (syngeneic) AmSPC, or uninjected animals. B6 CD4+ responder cells were stained with CSFE and T cell proliferation in response to stimulation with alloantigen (Balb/c splenocytes) is indicated by decreases in the fluorescence intensity and histogram peak shifts to the left. (A) After 3.5 days of alloantigen stimulation, alloantigen primed cells (green line) initiate T-cell proliferation while syngeneic primed (blue line) and uninjected cells (pink line) have not begun to divide. (B) After 5.5 days in coculture, more than half of allogeneic primed cells (green line) have proliferated and are represented as a single left peak comprised of a heterogenous number of cell divisions. In comparison syngeneic primed cells (blue line) have just begun to proliferate. At the end of 7 days in co-culture, all responder populations demonstrate a proliferative response compared to unstimulated controls (purple filled curve).
Mice primed with allogeneic AmSPC demonstrated both primary and secondary alloantibody responses as indicated by the presence of both IgM and IgG antibody isotypes. Extremely high IgG alloantibody titers (≥1:1000) were found in all 5 of the animals injected with allogeneic AmSPC (Figure 4a). Alloantibody binding was increased in samples where AmSPC were treated with interferon γ. Low IgG titers (≤ 1:100) were generated in animals injected with syngeneic AmSPC (Figure 4b). Sera from allogeneic AmSPC primed animals was also incubated with third party CBA (H2k) AmSPC. Compared to IgG titers detected with Balb/c targets, lower IgG titers were seen against CBA targets (n=5, Figure 5).
Figure 4. Alloantibody Response of animals transplanted with AmSPC.
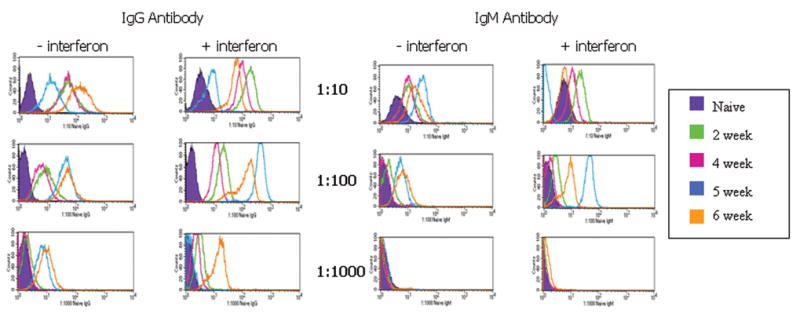
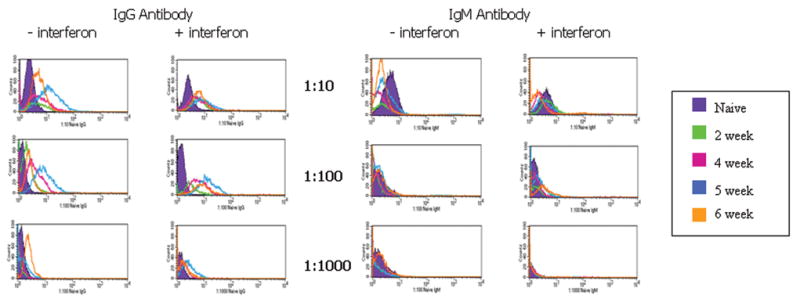
Untreated target Balb/c AmSPC and IFN-γ treated Balb/c target AmSPC were incubated with varying dilutions of mouse serum from 0,2,4,5, and 6 weeks post priming with Balb/c (allogeneic) AmSPC (Figure 4a) and B6 (syngeneic) AmSPC (Figure 4b). IgG and IgM antibody binding was measured by flow cytometric analysis. High alloantibody titers were found in animals injected with Balb/c (allogeneic) AmSPC compared to low titers exhibited in animals treated with B6 (syngeneic) AmSPC.
Figure 5. Antibody binding to Third-Party AmSPC Targets.
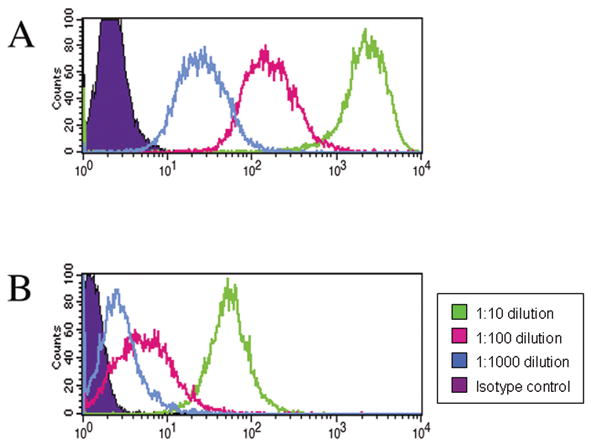
(A) IgG titers detected from serum of animals primed with Balb/c (allogeneic) AmSPC at 6 weeks post treatment and incubated with Balb/c AmSPC targets. (B) IgG titers detected using serum from the same animals incubated with CBA (third-party) AmSPC targets. Green line = 1:10 dilution, pink line = 1:100 dilution, blue line = 1:1000 dilution.
Skin Graft Rejection with Alloantigen Priming
B6 mice primed with intraperitoneal injection of Balb/c, allogeneic AmSPC (n=7) rejected Balb/c allogeneic skin grafts with a mean time to rejection of 6.71 ± 0.39 days compared to 8.375 ± 0.07 days for non-injected controls (n=12, p < 0.0001). B6 animals injected with B6, syngeneic AmSPC rejected Balb/c, allogeneic skin grafts at a mean of 7.5±0.2 days (n=10; p < 0.0001 compared individually to both allogeneic AmSPC injected and non-injected controls). Skin graft rejection of AmSPC primed B6 animals was also compared to B6 animals primed with Balb/c, allogeneic or B6, syngeneic splenocytes. Kaplan-Meier analysis of graft survival did not demonstrate a statistically significant difference between AmSPC primed animals and their splenocyte counterparts. Balb/c allogeneic primed animals exhibited statistically significant acceleration in graft rejection compared to B6 syngeneic injected and uninjected control animals (p<0.05, Figure 6). Autografts were not rejected. The technical success rate for all skin grafting was 97%.
Figure 6. Kaplan-Meier analysis of skin graft survival.
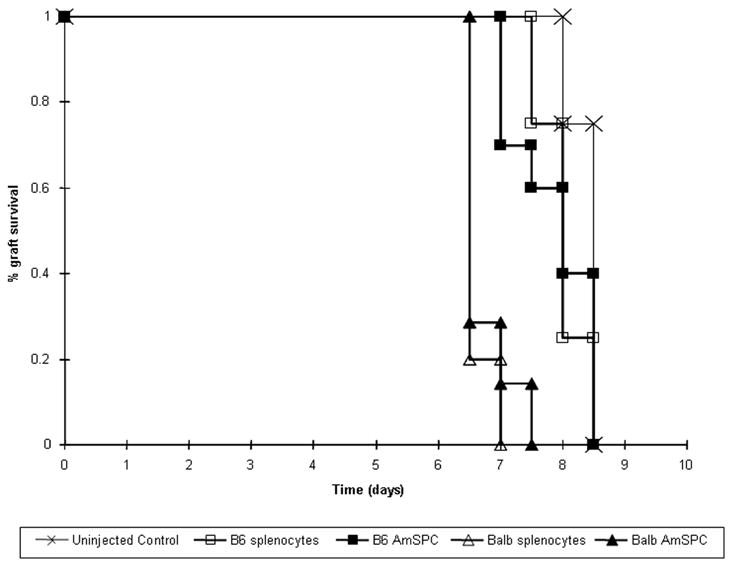
B6 mice treated with Balb/c (allogeneic) AmSPC or allogeneic splenocytes exhibited earlier rejection of Babl/c skin grafts compared to animals treated with B6 (syngeneic) AmSPC, syngeneic splenocytes, or untreated controls. (p<0.05).
Discussion
In agreement with previous studies of “MSC” immune function in vitro, AmSPC clearly inhibit T-cell proliferative responses to allogeneic stimulation in co-culture experiments. The multitude of studies demonstrating such in vitro suppression has formed the basis of claims that MSC are similarly immunosuppressive and poorly immunogenic in vivo. Our data, however, demonstrates that the introduction of allogeneic AmSPC into an immunologically competent animal elicits both a cellular and humoral host immune response. Sensitization of host CD4+ lymphocytes is evidenced both by the increased proliferation of T-cells observed upon restimulation with alloantigen and the detection of high titer alloantibody after immunization with allogeneic AmSPC. Furthermore, injection of donor matched AmSPC fail to induce host tolerance to allogeneic skin grafting.
The mechanism of SPC mediated in vitro suppression of T-cell proliferation remains to be fully defined. There is agreement that suppression of T cell proliferation after stimulation by polyclonal mitogens, anti CD3 antibody, or alloantigen appears to be independent of MHC matching between the SPC and the T cells(8, 10, 12). There have been conflicting reports on whether suppression is mediated by a soluble factor in supernatants or whether cell-cell contact is required(5, 7–10, 17). In our own study, the inhibition of T-cell proliferation was contact dependent. This implies that these effects on lymphocyte function in vitro are mediated by different mechanisms. A variety of molecular mechanisms have been proposed to explain MSC mediated in vitro suppression including IDO type reactions (16), veto-like activity (31), induction of T-cell anergy (19), inhibition of cytotoxic lymphocytes (4, 5, 17) and NK cells (13) in mixed lymphocyte culture, alteration of antigen presenting cell function (4, 7, 9), and production of T-cells with regulatory or suppressive phenotypes (4, 13). Failure to arrive at a consensus regarding the precise mechanisms responsible for these in vitro observations may be due in part to study differences in the cell population or type of stimulation used in co-culture experiments. (18)
Suppressive culture conditions are associated with extremely high cell densities and local concentrations of suppressive cytokines relative to physiologic conditions. In contrast, systemic administration of SPC would generally result in exposure of host immune cells to SPC at a much lower frequency. Thus, the likelihood of formation of the cell-cell networks and local cytokine milieus that occur in vitro would be minimal. In this context, it is likely that in vivo presentation of SPC may have entirely different immune consequences than those anticipated from in vitro studies such as the alloimmunization observed in this study.
One other group has shown the in vivo immune recognition of allogeneic SPC in a well designed and controlled study(29). Using murine SPC engineered to release erythropoietin, they demonstrate high recipient plasma levels of antibody to erythropoietin and decreasing host hematocrit with repeat challenges of subcutaneously implanted allogeneic SPC. Histological analysis of the subcutaneous implants for CD4+ and CD8+ cells yields indirect evidence of in vivo host cellular immune response. In our study, we utilized a non-transduced cell population of similar immune phenotype and passage and demonstrate evidence of a humoral response directed at SPC antigens rather than at a transgene product. Furthermore, host cellular recognition of AmSPC antigens is indicated by the accelerated proliferative response of CD4+ lymphocytes from 6 week posttreatment B6 animals upon re-stimulation with Balb/c, allogeneic stimulator splenocytes and suggests the induction of a memory T-cell response following initial antigen presentation in vivo.. In comparison to animals injected with AmSPC, animals initially injected with allogeneic splenocytes showed both increased magnitude and kinetics in their response (data not shown). This is most likely due to a potent response against MHC Class II which is expressed on the majority of splenocytes and is only inducible on AmSPC.
In addition to eliciting host immune responses, AmSPC also fail to prevent skin graft rejection. In contrast to other studies demonstrating skin graft prolongation (6, 32), our results demonstrate that animals injected with allogeneic AmSPC reject allogeneic skin grafts with the same kinetics as animals injected with allogeneic splenocytes. In the above mentioned studies skin graft prolongation is between 1 and 4 days and while statistically significant, such prologation is unlikely to be of clinical or therapeutic significance. In our study, animals primed with allogeneic AmSPC exhibited a statistically significant acceleration in the time to skin graft rejection compared to syngeneic AmSPC controls. Although the window of skin graft rejection between the groups in this study is narrow, we believe that at the very least, the data suggests that allogeneic AmSPC are not tolerogenic.
The rejection of allogeneic skin grafts seen in primed animals in our study also provides indirect evidence of the generation of T cell responses to allogeneic AmSPC. Acute graft rejection is a T cell mediated immune response. In all likelihood, host T cells, sensitized to donor alloantigen by intraperitoneal SPC injections, recognize alloantigen within the graft and generate cytotoxic responses resulting in acute graft rejection. Animals primed with syngeneic SPC exhibited a mean graft rejection time intermediate to that of allogeneic injection recipients and uninjected controls but no difference in graft survival curve compared to uninjected controls. We attribute the faster mean graft rejection time of syngeneic injection recipients to an enhanced nonspecific inflammatory state produced by intraperitoneal injections and the presence of foreign material within the peritoneal cavity. Peritoneal macrophages and resident natural killer cells phagocytose foreign material as part of the innate immune response. Once activated, macrophages produce a variety of cytokines which influence lymphocyte effector functions and the adaptive immune response. (33) In this fashion, the introduction of cellular material can serve as an adjuvant thereby enhancing the rejection of allogeneic skin grafts in animals primed with syngeneic AmSPC. Accelerated skin graft rejection argues against the capacity of SPC to escape immune surveillance or to be tolerogenic in immune competent animals.
Additionally, alloantibody is produced in high titers against allogeneic AmSPC. Binding of alloantibody is increased with interferon gamma treatment, suggesting that upregulation of class I contributes to this phenomena. The higher antibody titers detected when allogeneic primed animal serum was incubated with allogeneic AmSPC compared to third-party AmSPC targets also suggests that the alloantibody response is MHC mediated. An interesting point to further investigate would be to determine if there is antibody generated against MHC Class II. A recent study examining host immune responses to multipotent adult progenitor cells (MAPC), a cell type with similar MHC class I and II antigen expression profile to SPC, showed indirect evidence of upregulation of MHC class II antigen expression in vivo contributing to MAPC immune clearance(34). Given that an area of injection would be prone to inflammation, and interferon gamma would be produced, upregulation of Class I and Class II expression is very likely.
Lower levels of antibody activity were observed in animals injected with syngeneic AmSPC. We interpret this as a low level immune response to fetal bovine serum (FBS) antigens presented by the AmSPC. AmSPC are subjected to long term culture in FBS. During this time the cells incorporate many xenoproteins that can be expressed on their MHC. It has been well documented that FBS sensitization occurs when cultured cells are transplanted into immunocompetent animals (35–37). As FBS derived proteins should be presented equally by allogeneic, syngeneic, or third-party cells, the greater magnitude of humoral and cellular immune response observed with allogeneic cells can only be explained by response to alloantigen.
Since species related differences in SPC have been well documented, it is important to note that we have confirmed that the murine SPC utilized in this study have similar in vitro suppressive properties to those described for the human and rat cells used in previous immunologic studies of so called MSCs. Specifically, AmSPC suppress T-cell proliferation in vitro to polyclonal mitogen and allogeneic stimulation in a non-MHC restricted manner. In agreement with many studies, our cells require cell-cell contact for suppression to occur. Thus, the results of this study would seem relevant to the clinical application of human SPC, and would suggest that clinical systemic administration of allogeneic SPC in the absence of immunosuppression would elicit an alloimmune response.
Perhaps the strongest evidence for the immunogenicity of SPC is provided by the disconnect between in vitro studies and supporting in vivo data. Despite the large number of studies supporting the immune suppressive capacity of SPC in vitro, there is a paucity of in vivo data in the immunologically competent host that supports the capacity of allogeneic or xenogeneic SPC to evade the immune response or induce tolerance to donor antigen. The vast majority of studies documenting in vivo persistence of allogeneic or xenogeneic SPC were performed in either immunodeficient or irradiated hosts, or were site directed into immune privileged sites such as the brain.(38–43) The few studies demonstrating long term engraftment of allogeneic SPC have been methodologically flawed, either by utilization of a hematopoietically contaminated cell population(44, 45) or by the use of non-rigorous detection methodology such as membrane associated dyes (28, 46, 47) as the sole evidence of donor cell persistence. The issue is also complicated by the attribution of beneficial physiologic effect to assumed engraftment and differentiation of SPC. There are now many documented instances of beneficial effect via paracrine mechanisms(48–50) that can transiently be provided by allogeneic cells. If in fact SPC were immune privileged cells that could escape immune surveillance, one would anticipate that after years of effort by many laboratories, a large number of studies clearly documenting engraftment and persistence of allogeneic SPC in immunocompetent hosts would be available.
Finally, the results of this study should be interpreted in appropriate context. Our results clearly demonstrate allogeneic AmSPC delivered by intraperitoneal injection elicit a primary and secondary alloimmune response. This argues against the capacity of SPC to avoid immune surveillance or, at least in this setting, to be tolerogenic. Furthermore, this immune response induces cytotoxic effector functions resulting in donor skin graft rejection. While the specific elimination of donor SPC has not been clearly demonstrated, prevailing evidence would suggest SPC to be cleared by these same mechanisms. It is possible however, that SPC are able to set up local immune suppressive environments after immune recognition allowing their persistence in host tissues. It is also possible that in studies in which microchimerism (pcr detectable chimerism) persists, the frequency of donor cells is below the threshold of immune activation. Clarification of these issues is important prior to clinical applications utilizing allogeneic SPC. At the very least, our data suggests that in vitro immune properties of SPC cannot be extrapolated into assertions that SPC are immunosuppressive, immunomodulatory, or tolerogenic in vivo. Such assertions should only be based on further experimental evidence from in vivo studies.
Footnotes
This work was supported by grants R01 HL73253-01 from the National Institutes of Health (AWF). AWF is also supported by funds from the Ruth and Tristram C. Colket, Jr. Chair of Pediatric Surgery.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 2.Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276:71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 3.Javazon EH, Beggs KJ, Flake AW. Mesenchymal stem cells: Paradoxes of passaging. Exp Hematol. 2004;32:414–425. doi: 10.1016/j.exphem.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 5.Angoulvant D, Clerc A, Benchalal S, et al. Human mesenchymal stem cells suppress induction of cytotoxic response to alloantigens. Biorheology. 2004;41:469–476. [PubMed] [Google Scholar]
- 6.Bartholomew A, Sturgeon C, Siatskas M, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30:42–48. doi: 10.1016/s0301-472x(01)00769-x. [DOI] [PubMed] [Google Scholar]
- 7.Beyth S, Borovsky Z, Mevorach D, et al. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce t-cell unresponsiveness. Blood. 2005;105:2214–2219. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- 8.Di Nicola M, Carlo-Stella C, Magni M, et al. Human bone marrow stromal cells suppress t-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 9.Jiang XX, Zhang Y, Liu B, et al. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105:4120–4126. doi: 10.1182/blood-2004-02-0586. [DOI] [PubMed] [Google Scholar]
- 10.Krampera M, Glennie S, Dyson J, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific t cells to their cognate peptide. Blood. 2003;101:3722–3729. doi: 10.1182/blood-2002-07-2104. [DOI] [PubMed] [Google Scholar]
- 11.Le Blanc K, Rasmusson I, Gotherstrom C, et al. Mesenchymal stem cells inhibit the expression of cd25 (interleukin-2 receptor) and cd38 on phytohaemagglutinin-activated lymphocytes. Scand J Immunol. 2004;60:307–315. doi: 10.1111/j.0300-9475.2004.01483.x. [DOI] [PubMed] [Google Scholar]
- 12.Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringden O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immunol. 2003;57:11–20. doi: 10.1046/j.1365-3083.2003.01176.x. [DOI] [PubMed] [Google Scholar]
- 13.Maccario R, Podesta M, Moretta A, et al. Interaction of human mesenchymal stem cells with cells involved in alloantigen-specific immune response favors the differentiation of cd4+ t-cell subsets expressing a regulatory/suppressive phenotype. Haematologica. 2005;90:516–525. [PubMed] [Google Scholar]
- 14.Maitra B, Szekely E, Gjini K, et al. Human mesenchymal stem cells support unrelated donor hematopoietic stem cells and suppress t-cell activation. Bone Marrow Transplant. 2004;33:597–604. doi: 10.1038/sj.bmt.1704400. [DOI] [PubMed] [Google Scholar]
- 15.Majumdar MK, Keane-Moore M, Buyaner D, et al. Characterization and functionality of cell surface molecules on human mesenchymal stem cells. J Biomed Sci. 2003;10:228–241. doi: 10.1007/BF02256058. [DOI] [PubMed] [Google Scholar]
- 16.Meisel R, Zibert A, Laryea M, et al. Human bone marrow stromal cells inhibit allogeneic t-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619–4621. doi: 10.1182/blood-2003-11-3909. [DOI] [PubMed] [Google Scholar]
- 17.Rasmusson I, Ringden O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit the formation of cytotoxic t lymphocytes, but not activated cytotoxic t lymphocytes or natural killer cells. Transplantation. 2003;76:1208–1213. doi: 10.1097/01.TP.0000082540.43730.80. [DOI] [PubMed] [Google Scholar]
- 18.Rasmusson I, Ringden O, Sundberg B, Le Blanc K. Mesenchymal stem cells inhibit lymphocyte proliferation by mitogens and alloantigens by different mechanisms. Exp Cell Res. 2005;305:33–41. doi: 10.1016/j.yexcr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 19.Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated t cells. Blood. 2005;105:2821–2827. doi: 10.1182/blood-2004-09-3696. [DOI] [PubMed] [Google Scholar]
- 20.Bacigalupo A. Mesenchymal stem cells and haematopoietic stem cell transplantation. Best Pract Res Clin Haematol. 2004;17:387–399. doi: 10.1016/j.beha.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Devine SM, Peter S, Martin BJ, Barry F, McIntosh KR. Mesenchymal stem cells: Stealth and suppression. Cancer J. 2001;7 (Suppl 2):S76–82. [PubMed] [Google Scholar]
- 22.El-Badri NS, Maheshwari A, Sanberg PR. Mesenchymal stem cells in autoimmune disease. Stem Cells Dev. 2004;13:463–472. doi: 10.1089/scd.2004.13.463. [DOI] [PubMed] [Google Scholar]
- 23.Kassem M. Mesenchymal stem cells: Biological characteristics and potential clinical applications. Cloning Stem Cells. 2004;6:369–374. doi: 10.1089/clo.2004.6.369. [DOI] [PubMed] [Google Scholar]
- 24.Le Blanc K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy. 2003;5:485–489. doi: 10.1080/14653240310003611. [DOI] [PubMed] [Google Scholar]
- 25.Le Blanc K, Rasmusson I, Sundberg B, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 26.Le Blanc K, Ringden O. Immunobiology of human mesenchymal stem cells and future use in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11:321–334. doi: 10.1016/j.bbmt.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Le Blanc K, Ringden O. Use of mesenchymal stem cells for the prevention of immune complications of hematopoietic stem cell transplantation. Haematologica. 2005;90:438a. [PubMed] [Google Scholar]
- 28.Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res. 2004;95:9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- 29.Eliopoulos N, Stagg J, Lejeune L, Pommey S, Galipeau J. Allogeneic marrow stromal cells are immune rejected by mhc class i- and class ii-mismatched recipient mice. Blood. 2005;106:4057–4065. doi: 10.1182/blood-2005-03-1004. [DOI] [PubMed] [Google Scholar]
- 30.Billingham RE, Medawar PB. The technique of free skin grafting in mammals. J Exp Biol. 1951;28:385–402. [Google Scholar]
- 31.Potian JA, Aviv H, Ponzio NM, Harrison JS, Rameshwar P. Veto-like activity of mesenchymal stem cells: Functional discrimination between cellular responses to alloantigens and recall antigens. J Immunol. 2003;171:3426–3434. doi: 10.4049/jimmunol.171.7.3426. [DOI] [PubMed] [Google Scholar]
- 32.Guo Z, Li H, Li X, et al. In vitro characteristics and in vivo immunosuppressive activity of compact bone-derived murine mesenchymal progenitor cells. Stem Cells. 2006;24:992–1000. doi: 10.1634/stemcells.2005-0224. [DOI] [PubMed] [Google Scholar]
- 33.Wu Q, Feng Y, Yang Y, et al. Kinetics of the phenotype and function of murine peritoneal macrophages following acute inflammation. Cell Mol Immunol. 2004;1:57–62. [PubMed] [Google Scholar]
- 34.Tolar J, O’Shaughnessy MJ, Panoskaltsis-Mortari A, et al. Host factors that impact the biodistribution and persistence of multipotent adult progenitor cells. Blood. 2006 doi: 10.1182/blood-2005-08-3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kievits F, Boerenkamp WJ, Ivanyi P. H-2-dependent binding of xenogeneic beta 2-microglobulin from culture media. J Immunol. 1988;140:4253–4255. [PubMed] [Google Scholar]
- 36.MacDermott RP, Bragdon MJ. Fetal calf serum augmentation during cell separation procedures accounts for the majority of human autologous mixed leukocyte reactivity. Behring Inst Mitt. 1983:122–128. [PubMed] [Google Scholar]
- 37.Spees JL, Gregory CA, Singh H, et al. Internalized antigens must be removed to prepare hypoimmunogenic mesenchymal stem cells for cell and gene therapy. Mol Ther. 2004;9:747–756. doi: 10.1016/j.ymthe.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 38.Bensidhoum M, Chapel A, Francois S, et al. Homing of in vitro expanded stro-1- or stro-1+ human mesenchymal stem cells into the nod/scid mouse and their role in supporting human cd34 cell engraftment. Blood. 2004;103:3313–3319. doi: 10.1182/blood-2003-04-1121. [DOI] [PubMed] [Google Scholar]
- 39.Francois S, Bensidhoum M, Mouiseddine M, et al. Local irradiation induces not only homing of human mesenchymal stem cells (hmsc) at exposed sites but promotes their widespread engraftment to multiple organs: A study of their quantitative distribution following irradiation damages. Stem Cells. 2005 doi: 10.1634/stemcells.2005-0260. [DOI] [PubMed] [Google Scholar]
- 40.Isakova IA, Baker K, Dufour J, Gaupp D, Phinney DG. Preclinical evaluation of adult stem cell engraftment and toxicity in the cns of rhesus macaques. Mol Ther. 2006 doi: 10.1016/j.ymthe.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 41.Lu D, Li Y, Wang L, et al. Intraarterial administration of marrow stromal cells in a rat model of traumatic brain injury. J Neurotrauma. 2001;18:813–819. doi: 10.1089/089771501316919175. [DOI] [PubMed] [Google Scholar]
- 42.Noort WA, Kruisselbrink AB, in’t Anker PS, et al. Mesenchymal stem cells promote engraftment of human umbilical cord blood-derived cd34(+) cells in nod/scid mice. Exp Hematol. 2002;30:870–878. doi: 10.1016/s0301-472x(02)00820-2. [DOI] [PubMed] [Google Scholar]
- 43.Liechty KW, MacKenzie TC, Shaaban AF, et al. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med. 2000;6:1282–1286. doi: 10.1038/81395. [DOI] [PubMed] [Google Scholar]
- 44.Saito T, Kuang JQ, Bittira B, Al-Khaldi A, Chiu RC. Xenotransplant cardiac chimera: Immune tolerance of adult stem cells. Ann Thorac Surg. 2002;74:19–24. doi: 10.1016/s0003-4975(02)03591-9. discussion 24. [DOI] [PubMed] [Google Scholar]
- 45.Niyibizi C, Wang S, Mi Z, Robbins PD. The fate of mesenchymal stem cells transplanted into immunocompetent neonatal mice: Implications for skeletal gene therapy via stem cells. Mol Ther. 2004;9:955–963. doi: 10.1016/j.ymthe.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 46.Shake JG, Gruber PJ, Baumgartner WA, et al. Mesenchymal stem cell implantation in a swine myocardial infarct model: Engraftment and functional effects. Ann Thorac Surg. 2002;73:1919–1925. doi: 10.1016/s0003-4975(02)03517-8. discussion 1926. [DOI] [PubMed] [Google Scholar]
- 47.Silva GV, Litovsky S, Assad JA, et al. Mesenchymal stem cells differentiate into an endothelial phenotype, enhance vascular density, and improve heart function in a canine chronic ischemia model. Circulation. 2005;111:150–156. doi: 10.1161/01.CIR.0000151812.86142.45. [DOI] [PubMed] [Google Scholar]
- 48.Gnecchi M, He H, Liang OD, et al. Paracrine action accounts for marked protection of ischemic heart by akt-modified mesenchymal stem cells. Nat Med. 2005;11:367–368. doi: 10.1038/nm0405-367. [DOI] [PubMed] [Google Scholar]
- 49.Mayer H, Bertram H, Lindenmaier W, et al. Vascular endothelial growth factor (vegf-a) expression in human mesenchymal stem cells: Autocrine and paracrine role on osteoblastic and endothelial differentiation. J Cell Biochem. 2005;95:827–839. doi: 10.1002/jcb.20462. [DOI] [PubMed] [Google Scholar]
- 50.Togel F, Hu Z, Weiss K, et al. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol. 2005;289:F31–42. doi: 10.1152/ajprenal.00007.2005. [DOI] [PubMed] [Google Scholar]