

Abstract
Prostaglandins are key regulators of ion transport in the kidney. In MDCK cells, which model distal tubule cells, the transcription of the Na,K-ATPase β1 subunit is regulated by PGE1 and PGE2. To identify the EP receptors that mediate transcriptional regulation, transient transfection studies are conducted using the human β1 promoter/luciferase construct, pHβ1-1141 Luc. The involvement of EP1 and EP2 receptors is indicated by studies with the EP1 selective agonist 17-phenyl trinor PGE2, and the EP2 selective agonist butaprost (which stimulate), as well as by studies with the antagonists SC-51089 (EP1 specific) and AH 6809 (EP1 and EP2 specific). Consistent with the involvement of Gs coupled EP2 receptors, is that the PGE1 stimulation is inhibited by the PKAI expression vector (encoding the protein kinase A (PKA) inhibitory protein), as well as by the myristolated PKA inhibitory peptide PKI. In addition to this evidence (for the involvement of EP2 receptors), evidence for the involvement of EP1 receptors in the PGE1 mediated stimulation of Na,K-ATPase β subunit gene transcription includes the stimulatory effect of 17-phenyl trinor PGE2, as well as the inhibitory effects of SC-51089. Also consistent with the involvement of Gq coupled EP1 receptors, the PGE1 stimulation is inhibited by the PKCI vector (encoding the PKC inhibitory domain), the PKC inhibitor Go 6976, thapsigargin, as well as the calmodulin antagonists W7 and W13.
Keywords: Prostaglandins; Kidney; MDCK; Na,K-ATPase; Transcription; EP receptors
1. Introduction
Prostaglandins (PGs) are biologically active lipid mediators, which are products of the metabolism of arachidonic acid by cyclooxygenase. Prostaglandins play important roles systemic blood pressure regulation and volume control [1]. In the kidney, endogenously produced prostaglandins regulate renal blood flow as well as ion transport, so as to maintain homeostasis under conditions of physiologic stress [2]. Prostaglandin E2 (PGE2) [2] the major renal prostaglandin, is produced by all of the nephron segments, the macula densa, and the renal interstitium [3]. The renal prostaglandins produced in these various locales have a variety of affects on different types of renal cells, acting in both an autocrine and paracrine manner.
PGE2 in particular has profound effects on a number of the transport systems of the renal tubular epithelium [2,4], including the Na+/H+ antiport system [5], the Na,K-ATPase [4] and the epithelial Na+ channel [6,7]. The mechanisms by which prostaglandins affect transport systems are poorly understood, and the signaling pathways are to a large extent undefined. These issues have been further complicated by the results of microperfusion and micropuncture studies, that indicate that prostaglandin effects vary considerably between nephron segments. Some of the differences can be explained by the presence of different types of prostanoid receptors, and transport systems in the cell types that are in the different nephron segments.
One manner through which prostaglandin-mediated affects on transport can be more precisely defined is by means of studies with cultured renal cells. The Madin Darby Canine Kidney (MDCK) cell line is a well-defined cell culture system, which is amenable for studies concerning prostaglandin action [8]. MDCK cells retain the morphological polarity distinctive of renal tubule epithelial cells, including an apical surface which faces the culture medium, and a basolateral surface which faces the plastic dish [9]. Adjacent cells are interconnected by tight junctions. At confluence, MDCK monolayers possess the capacity for transepithelial ion transport [10–13]. The Ussing chamber studies of Cereijido et al. [14] have shown that (1) the entry of sodium into MDCK cells is dependent upon an amiloride-sensitive Na+/H+ antiport system, and (2) the active extrusion of Na+ in the apical to the basolateral direction is dependent upon a ouabain-sensitive Na,K-ATPase, which is localized on the basolateral surface [13].
Typical of renal distal tubule cells, MDCK cells have been shown to retain appropriate hormone responses, as well as transport systems [9]. Indeed, arginine vasopressin, norepinephrine, glucagon, PGE1 and PGE2 all cause the activation of adenylate cyclase in MDCK cells [10,15,16]. In vitro studies concerning the actions of such effector molecules are facilitated by a hormonally defined culture conditions. When MDCK cells are cultured in serum free Medium K-1, that contains insulin, transferrin, triiodothyronine, hydrocortisone and PGE1 [17], the cells grow over the long term, at the same rate obtained in serum-supplemented medium [18].
Previously, we reported that PGE1, and 8-Bromocyclic AMP (8Br-cAMP) cause an increase in the Na,K-ATPase activity of MDCK cells [19], which was associated with an increase in the level of the Na,K-ATPase in this cell type [4]. The Na,K-ATPase is a protein which is composed of an α subunit with catalytic activity, as well as a β subunit, which facilitates the integration of the protein into the plasma membrane [20]. We observed that the level of both the α and the β subunit increased in PGE1 treated MDCK cells [4,19]. In addition, the level of the mRNA for the α and β subunit of the Na,K-ATPase was increased [4,19]. Notably, the level of β subunit mRNA increased to a greater extent than α subunit mRNA [4,19]. As the β subunit level is known to limit the assembly of α/β heterodimers [21], the regulation of the β1 subunit was examined in greater detail.
Transient transfection studies were conducted with MDCK cells, using a human Na+,K+-ATPase β1 subunit promoter/luciferase construct (pHβ1-1141Luc) [22]. The results of these studies indicated that both PGE1 and PGE2 stimulated transcription [4,22]. Subsequently, we identified a Prostaglandin Response Element (PGRE) on the human β1 subunit promoter that was responsible for the observed regulation by PGE1 and PGE2 [23,24]. A number of different classes of prostanoid receptors are involved in mediating signaling events initiated by prostaglandins. For this reason, in this report we further define the prostanoid receptors that are present in MDCK cells which are responsible for mediating signaling events initiated by PGE1 and PGE2, which result in increased the transcription of the Na,K-ATPase β1 subunit gene.
2. Experimental methods
2.1. Materials
Hormones, transferrin, prostaglandins, and other chemicals were from Sigma-Aldrich (St. Louis, MO). Dulbecco’s modified Eagle’s medium (DMEM), Ham’s nutrient mixture F-12, soybean trypsin inhibitor, molecular weight markers, and lipofectamine reagent were from Invitrogen (Carlsbad, CA). Nitrocellulose membranes, acrylamide, molecular weight markers, and other reagents for electrophoresis were from Bio-Rad (Hercules CA). The Galacto-Star™ system was obtained from Applied Biosystems (Bedford, MA). The pSVβ gal plasmid, and reporter lysis buffer were from Promega. The Prism 4 program was obtained from GraphPad Software, Inc. (San Diego, CA). The myristolated PKA inhibitory peptide (14–22) was from EMD Biosciences (La Jolla, CA). Prostaglandin agonists and antagonists were from Cayman Chemicals (Ann Arbor, MI).
2.2. Antibodies
Rabbit polyclonal antibodies against a synthetic peptide homologous to the C-terminal of the human EP1 receptor (amino acids 380–402; GLTPSAWESSLRSSRHSGLSHF), the C-terminal of the human EP2 receptor (amino acids 335–358; SLRTQDATQTSCSTQSDASKQADL), amino acids 308–327 of the human EP3 receptor (NQTSVE-HCVKTHTEKQKECNF), and the C-terminal of the human EP4 receptor (amino acids 459–488; GSGRAGPAPKGSS-LVTFPSETLNLSEKCI) were from Cayman Chemicals (Ann Arbor, MI). Previously, the anti EP1 antibody does not cross-react with human EP2, EP3 and EP4, but is cross-reactive with the rat and murine EP1 receptor (which has 70% homology with amino acids 380–402 in human EP1). Canine EP1 (gi 50978908) has 90% homology with amino acids 380–402 in human EP1 (gi 38505193). The EP2, EP3 and EP4 antibodies have been shown to be cross-reactive with human EP2, EP3 and EP4 receptors, respectively, without cross-reacting with each of the remaining three classes of human EP receptors. The EP2, EP3 and EP4 antibodies have also been shown to be cross-reactive with mouse and rat EP2 (homology 83%(mouse), 72% (rat)), mouse and rat EP3 (homology of 65% for both mouse and rat) as well as mouse and rat EP4 (homology of 79% (mouse) and 76% (rat)), respectively. Canine EP2 (gi 50978908), EP3 (gi 50950179) and EP4 (gi 50978708) receptors are 80% homologous to amino acids 335–358 in human EP2 (gi 31881630), 95% homologous to amino acids 308–327 in human EP3 (gi 38505174), and 78% homologous to amino acids 459–488 in human EP4 (gi 4506259), respectively.
2.3. Expression vectors
The human Na,K-ATPase beta 1 promoter/Luciferase construct pHβ1-1141 Luc [22] was a gift from Dr. Jerry Lingrel (U. Cincinnati), and the constitutive β-galactosidase expression vector pSVβ gal was purchased from Promega (Madison, WI). The pKAI and the pKCI expression vectors were gifts from Dr. Te-Chung Lee, State University of New York at Buffalo. The pKAI vector was generated from the pTC21 expression vector [25] by inserting a cDNA to the PKA inhibitor [26], TCGACCACCATGACTTATGCCGATTTCATTGCCTCTGGCCGCACTGGCCGC-CGCAACGCCATTCATTAG, into the Sal I and Bam HI sites of PTC21 [27], while pKCI was generated by similarly inserting cDNA to the auto-inhibitory domain of rat PKC (AATTCCACCATGCGCTTCGCCCGCAAAGGCGCC-CTCCGGCAGAAGAACGTGTG, encoding for amino acid residues 19–31 of PKC as well as EwRI sequences) into the Eco RI site of PTC21.
2.4. Cell culture
The basal medium is a 50:50 mixture of DME and Ham’s nutrient mixture F-12 supplemented with 15 mM HEPES (pH 7.4), 20 mM sodium bicarbonate, 92 units/ml penicillin, and 200 μg/ml streptomycin (DME/F12) [17]. The growth medium for stock cultures of MDCK cells (Medium K-1) consists of basal medium further supplemented with 5 μg/ml bovine insulin, 5 μg/ml human transferrin, 5 × 10−12 M triiodothyronine (T3), 5 × 10−8 M hydrocortisone, 25 ng/ml PGE1, and 5 × 10−8 M selenium. MDCK stock cultures were routinely subcultured by detachment of the cells using 0.53 mM EDTA and 0.05% trypsin in phosphate-buffered saline (PBS), followed by inhibition of trypsin action using 0.1% soybean trypsin inhibitor in PBS [17].
2.5. Transient transfection studies
MDCK cells were plated at 105 cells/35-mm dish into basal medium supplemented with 5 μg/ml bovine insulin and 5 μg/ml human transferrin. The day after plating, the cultures were cotransfected utilizing lipofectamine, with a reporter plasmid (1 μg), as well as pSVβ gal (0.2 μg), to adjust for transfection efficiency. During the transfection the cultures were maintained in antibiotic free basal medium supplemented with 5 μg/ml insulin and 5 μg/ml transferrin. The following day, the medium was changed to basal medium supplemented with 5 μg/ml insulin and 5 μg/ml transferrin. After a 2-h incubation, appropriate effector molecules were added, followed by a 4-h incubation (or, as otherwise indicated). At the end of the incubation, the cells were solubilized in reporter lysis buffer, and centrifuged for 1 min. Cell lysates were then utilized for luciferase and β-galactosidase assays.
To determine the luciferase activity, aliquots of the cell lysates were placed in 100 μl of luciferase assay buffer (20 mM Tricine, 1.07 mM MgCO3·4 Mg(OH)2, 2.67 mM MgSO4, 0.1 mM EDTA, 33.3 mM dithiothreitol, 270 μM Coenzyme A, 470 μM luciferin, and 530 μM adenosine triphosphate), and the emitted light measured in a Packard Tri-Carb 4530 scintillation counter (coincidence off). The luciferase activity of the cell lysates was normalized with respect to β-galactosidase activity. The β-galactosidase activity was measured by incubating aliquots of the cell lysates in a reaction buffer containing Galacton-Star® substrate (provided in the Galacto-Star™ system) for 90 min at 23 °C, and followed by measurements of the emitted light, as described above.
The values obtained from the luciferase assays were the mean (±S.E.) of quadruplicate determinations. The level of stimulation (or inhibition) of luciferase activity, the values were divided by the indicated control value from the same experimental culture set. The significance of observed stimulation (or inhibition) was determined by a one-way analysis of variance (ANOVA), and the Newman–Keuls multiple comparison test, using Prism 4 software. Differences were significant when p < 0.05.
2.6. Western analysis
MDCK monolayers were washed with PBS, and lysed in 50 mM Tris, pH 7.4, 150 mM NaCl, Na+ deoxycholate, 1% NP-40, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM sodium orthovanadate and 1 mM NaF. The cell lysates were removed from the culture dishes with rubber policemen, and transferred to microfuge tubes. Samples, were equalized for protein content by the Bradford method [28], separated on 10% SDS-polyacrylamide gels, and transferred to nitrocellulose membranes, using a Trans-Blot apparatus (Bio-Rad). The blots were blocked for 1 h in Tris-buffered saline (TBS) containing 0.1% (v/v) Tween 20 (TTBS), followed by a 2-h incubation with primary antibody, also in TTBS. After five washes with TTBS (5 min/wash), the blots were incubated 1 h in TTBS containing a goat anti-rabbit IgG-biotin conjugate, followed by five washes with TTBS. Finally, the blots were then incubated 1 h in TTBS containing a mouse anti-biotin-alkaline phosphatase conjugate. Signals were visualized using 0.33 mg/ml nitroblue tetrazolium (NBT) and 0.17 mg/ml 5-bromo-4-chloro-3- indolyl phosphate (BCIP) in 0.1 M NaCl and 0.05 M MgCl2. Blots were scanned with a Bio-Rad scanning densitometer, and band intensities quantified using the Quantity One program.
3. Results
3.1. PGE1 stimulation of the β1 promoter: signaling through EP1 and EP2 receptors
To determine the effects of prostaglandins on Na,K-ATPase β1-subunit gene expression in MDCK cells, transient transfection studies were conducted with pHβ1-1141Luc, a human β1promoter/luciferase construct [22]. Initially, the effects of PGE1, carbaprostacyclin and fluprostanol were examined. Fig. 1 shows a 6.4 ± 0.6-fold stimulation by 70 nM PGE1, an EP receptor agonist, and a 2.8 ± 0.4-fold stimulation by carbaprostacyclin (0.5 μM), an IP receptor agonist, whereas fluprostanol (7.5 nM), an FP receptor agonist, had no significant effect.
Fig. 1.
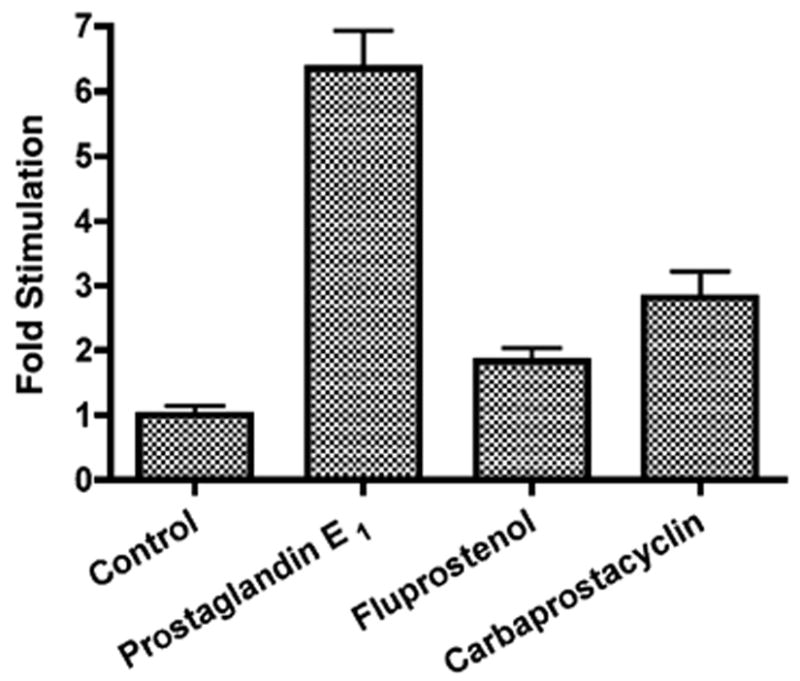
Prostaglandin effects on Na,K-ATPase β1 subunit gene expression. MDCK cells were transiently transfected with pHβ1-1141Luc. Twenty-four hours later, the effect of either 70 nM PGE1, 7.5 nM fluprostanol or 0.5 μM carbaprostacylin was examined. Luciferase activity was compared to untreated controls. Values are mean ± S.E.M.
The PGE1 stimulation of transcription may be the consequence of the activation of a number of different subtypes of EP receptors, including EP1 and EP2. In order to evaluate the possibility of EP2 involvement, the effect of butaprost was examined. Butaprost is a structural analogue of PGE1, which is a selective EP2 receptor agonist [29]. The effect of butaprost on transcription was examined as a function of the butaprost concentration from 0 to 4 × 10−6 M. Fig. 2A shows a stimulatory effect of butaprost on transcription, which increased as a function of concentration. The maximal stimulation (22 ± 5-fold) was observed at a butaprost concentration of 4 × 10−7 M, which is close to the previously reported Ki value obtained for butaprost (1.1 × 10−7 M) for displacement of radiolabeled PGE2 binding to the murine EP2 receptor [29].
Fig. 2.
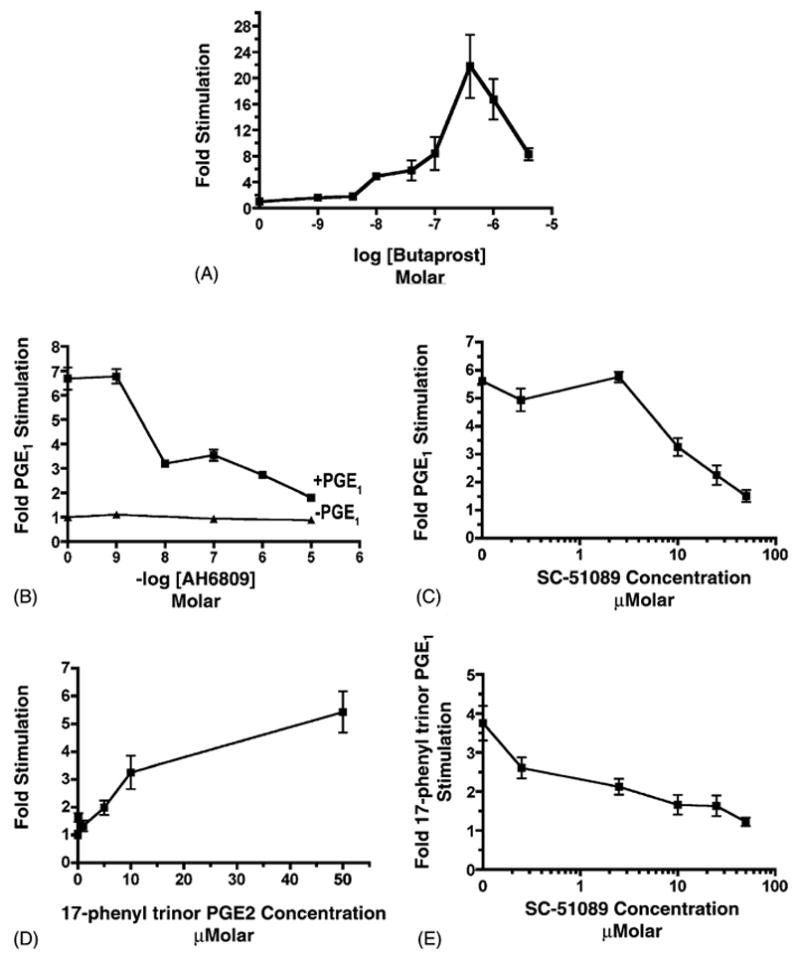
Effects of EP agonists and antagonists on Na+,K+-ATPase β1 subunit gene expression. MDCK cells were transiently transfected with pHβ1-1141Luc. Twenty-four hours later, cultures were treated for 4 h with either: (A) butaprost at concentrations ranging from 0–4 × 10−6 M; (B) AH6809 at concentrations ranging from 0–10−5 M in either the presence or in the absence of 28 nM PGE1; (C) SC-51089 at concentrations ranging from 0–50 μM in the presence of 28 nM PGE1 or in the absence of PGE1; (D) 17-phenyl trinor PGE2 at concentrations ranging from 0–50 μM; and (E) 10 μM 17-phenyl trinor PGE2 in the presence of SC-51089 at concentrations ranging from 0 to 50 μM. In the experiments with AH6809 and with SC-51089 cultures were preincubated with these antagonists for 30 min prior to the initiation of the 4-h incubation with PGE1. Control experiments indicated that SC-51089 (0–50 μM) had no significant effect in the absence of PGE1. The luciferase activity of cell lysates was determined in quadruplicate after the 4-h incubation period, and compared with untreated controls. At least three experiments were performed. Values are mean ± S.E.M.
In order to examine the possibility of EP2 receptor involvement further, the effect of AH6809 on the PGE1 stimulation was examined. AH6809 is a competitive EP2 antagonist, which has been shown to be specific for the murine EP2 receptor subtype [29]. Because AH6809 is a competitive antagonist, the effect of increasing concentrations of AH6809 on the PGE1 stimulation of transcription were examined when PGE1 at a concentration of 0.028 μM, which, as we previously showed, causes 67% of the maximal PGE1 stimulation [4]. Fig. 2B shows that AH6809 did indeed inhibit the PGE1 stimulation, and caused 50% inhibition at an AH6809 concentration of 6.2 μM. When the AH6809 concentration was increased to 10 μM, a 73 ± 3% inhibition of the PGE1 stimulation was obtained. Similarly, 10 μM AH6809 caused a 70% inhibition of the binding of 0.005 μM 3H-PGE2 to the recombinant human EP2 receptor [30].
In order to determine whether EP1 receptors are also involved, the EP1 receptor antagonist SC-51089 [32–34] was employed. Initially, the effect of increasing concentrations of SC-51089 on the stimulatory effect of 0.028 μM PGE1 was examined. Fig. 2C indicates that the PGE1 stimulation was inhibited by SC-51089 at concentrations ≥ 10 μM, unlike the case with the control level (data not included). An IC50 value of 15 μM was derived from the curve. Fig. 2C also shows that when the SC-51089 concentration was raised to 50 μM, the PGE1 stimulation was inhibited 91 ± 4%. When utilizing a 30 μM SC-19220 [29,35,36], an SC-51089 analogue, similarly a 40 ± 5% inhibition of the stimulatory effect of 0.028 μM PGE1 was obtained (unpublished observation).
In order to assess the involvement of EP1 receptors further, the effect of the EP1 agonist 17-phenyl trinor PGE2 [29] was examined, as illustrated in Fig. 2D. A 1.6 ± 0.2-fold stimulation was observed at 0.01 μM, and increased to as high as 5.4 ± 0.7 when the 17-phenyl trinor PGE2 concentration was raised to 50 μM. In order to evaluate whether the stimulatory effect of 17-phenyl trinor PGE2 is indeed mediated by EP1 receptors, the effect of SC-51089 on the 17-phenyl trinor PGE2 stimulation was examined. Fig. 2E shows that the stimulatory effect of 10 μM 17-phenyl trinor PGE2 was inhibited by SC-51089 by up to 94 ± 4% at 50 μM, and that the IC50 value was 4 μM.
Four major classes of EP receptors have been identified, including EP1, EP2, EP3 as well as EP4. In order to determine whether MDCK cells do indeed possess EP1, EP2, EP3 and/or EP4 receptors, a Western analysis was conducted. In addition, the effected of a 4-h incubation with PGE1 (70.5 nM) on the levels of these receptors was examined. Fig. 3A shows that all four classes of EP receptors were present in MDCK cells, and that the molecular weights of the EP receptors were between 52 and 53 kDa. The results (shown in Fig. 3A) also indicate that the level of EP1, EP2, EP3 and EP4 receptors increased by 6.5-, 3.1-, 7.9- and 3.0-fold, respectively, following a 4-h incubation with PGE1. In contrast (shown in Fig. 3B) the level of GAPDH was unchanged in PGE1 treated MDCK cells.
Fig. 3.
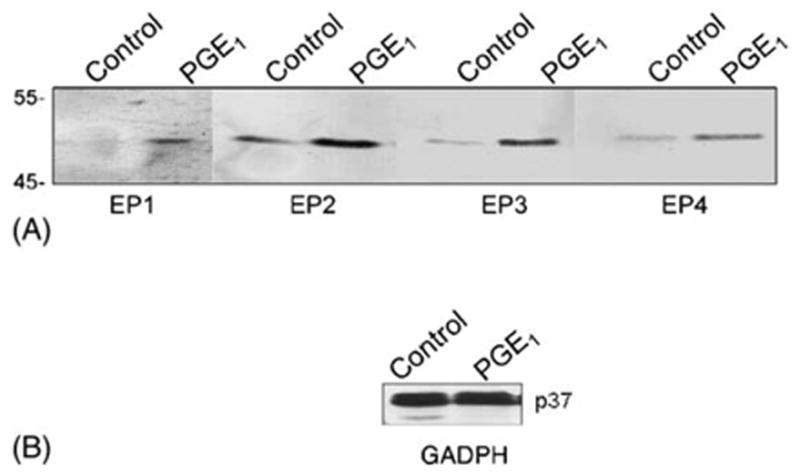
EP receptors in MDCK Cells. Confluent MDCK monolayers were either treated with 70 nM PGE1 for 4 h, or untreated. Cell lysates were analyzed by Western analysis using an antibody against either (A) EP1,EP2, EP3 or EP4, or (B) GAPDH.
3.2. PGE1 and 8Br-cAMP stimulates the Na+,K+-ATPase β1 promoter activity
All 4 classes of EP receptors are G-protein coupled, including EP2 and EP4 receptors (coupled to Gs, which activates adenylate cyclase (AC)), EP3 receptors (coupled to Gi, which inhibits AC), and EP1 receptors (coupled to Gq which activates Phospholipase C (PLC)) [29,31]. Consistent with the involvement of Gs coupled receptors was the observation (Fig. 4) that both 1.4 μM PGE1 (Fig. 4A) and 1 mM 8BrcAMP (Fig. 4B) were stimulatory. Also consistent with the involvement of Gs was the partial inhibition of the PGE1 and 8 BrcAMP stimulation by PKI, a cell-permeable myristo-lated PKA inhibitory peptide (36 nM) [4,38]. The PGE1 stimulation was reduced from 6.4 ± 0.4-fold to to 4.6 ± 0.2-fold by PKI (Fig. 4A), while the 8Br-cAMP stimulation was reduced from 8.4 ± 0.4-fold to 5.1 ± 0.4-fold (Fig. 4B).
Fig. 4.
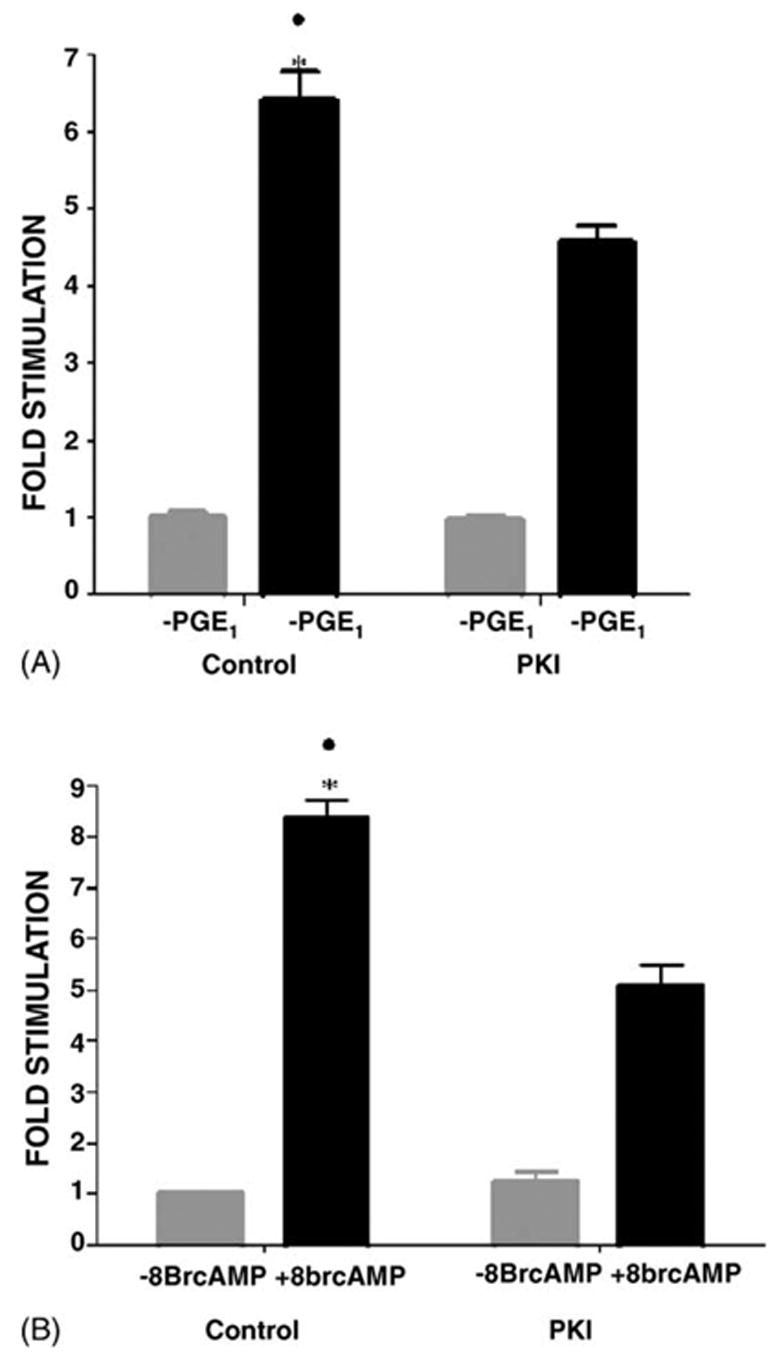
Role of PKA in the PGE1 stimulation. MDCK cells were transfected with the pHβ1-1141Luc. Twenty-four hours later, the cells were treated for 4 h with either (A) 1.4 μM PGE1or 1 mM 8Br-cAMP. (B) 1.4 μM PGE1 in the presence or the absence of 36 nM PKI. (C) 1 mM 8Br-cAMP in the presence or absence of 36 nM PKI. After the 4-h incubation the luciferase activity was determined in quadruplicate, and compared with untreated controls. At least three experiments were performed. Values are mean ± S.E.M. *, ●, p < 0.001 compared to control without PGE1 and PGE1 + PKI, or PGE1 + 8Br-cAMP.
In order to examine the role of PKA further, the effect of the PKAI vector (encoding for the Protein Kinase A inhibitory peptide) on the PGE1 and the 8Br-cAMP stimulation was examined. Fig. 5A shows that in the presence of PKAI both the PGE1 stimulation, and the 8Br-cAMP stimulation were reduced (45 ± 10%, and 52% ± 10%, respectively), when compared with control cultures cotransfected with the empty vector. However, the PGE1 stimulation was also reduced by 65 ± 5% by the vector PKCI (which expresses the inhibitory domain of PKC) [27] (as shown in Fig. 5B). In the same experiment (Fig. 5B), PKCI reduced the TPA stimulation by 82 ± 2%, as compared with cultures cotransfected with the empty vector. Thus, the results in Fig. 5B suggest that a component of the PGE1 stimulation is dependent upon PKC as well as PKA.
Fig. 5.
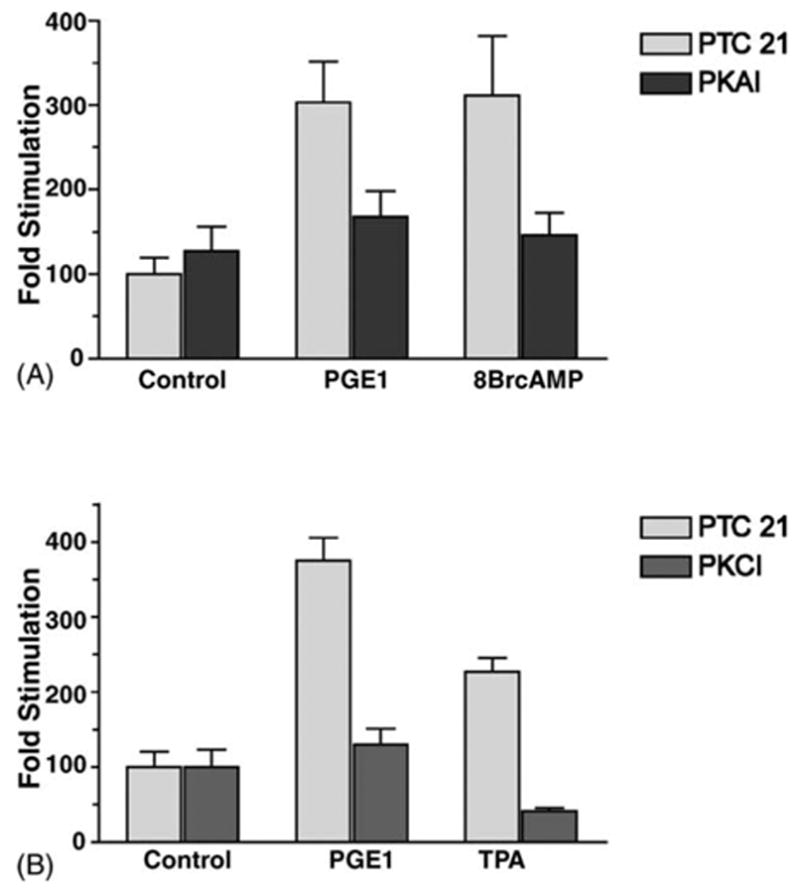
Role of PKA and PKC in the PGE1 stimulation. MDCK cells were cotransfected with pβ1-1141 Luc and either PKAI, PKCI or the empty vector PTC 21. (A) A portion of the PKAI and PTC 21 transfected cultures were then treated 3 h with either 1.4 μM PGE1 or 1 mM 8 BrcAMP. (B) A portion of the PKCI and PTC 21 transfected cultures were treated 3 h with either 1.4 μM PGE1 or 1 μM TPA. Luciferase determinations (in quadruplicate) were compared with untreated controls. Values are mean ± S.E.M.
3.3. Other signaling pathways involved in mediating PGE1 effects
Because PGE1 was previously causes an increase in intracellular Ca2+, as well as cAMP in MDCK cells [2,16], the involvement of Ca2+ and Ca2+/calmodulin-dependent protein kinase (CaM kinase) was also examined. CaM Kinase II activity is affected by phosphorylation by CaM Kinase Kinase, and reduced following dephosphorylation by Protein Phosphatase I (PP-1) and Protein Phosphatase 2A (PP-2A). Okadaic acid, a PP-1 and PP-2A inhibitor, caused a 3.2-fold stimulation of β1 subunit gene transcription (Fig. 6A), consistent with the involvement of CaM Kinase in the PGE1 stimulation.
Fig. 6.
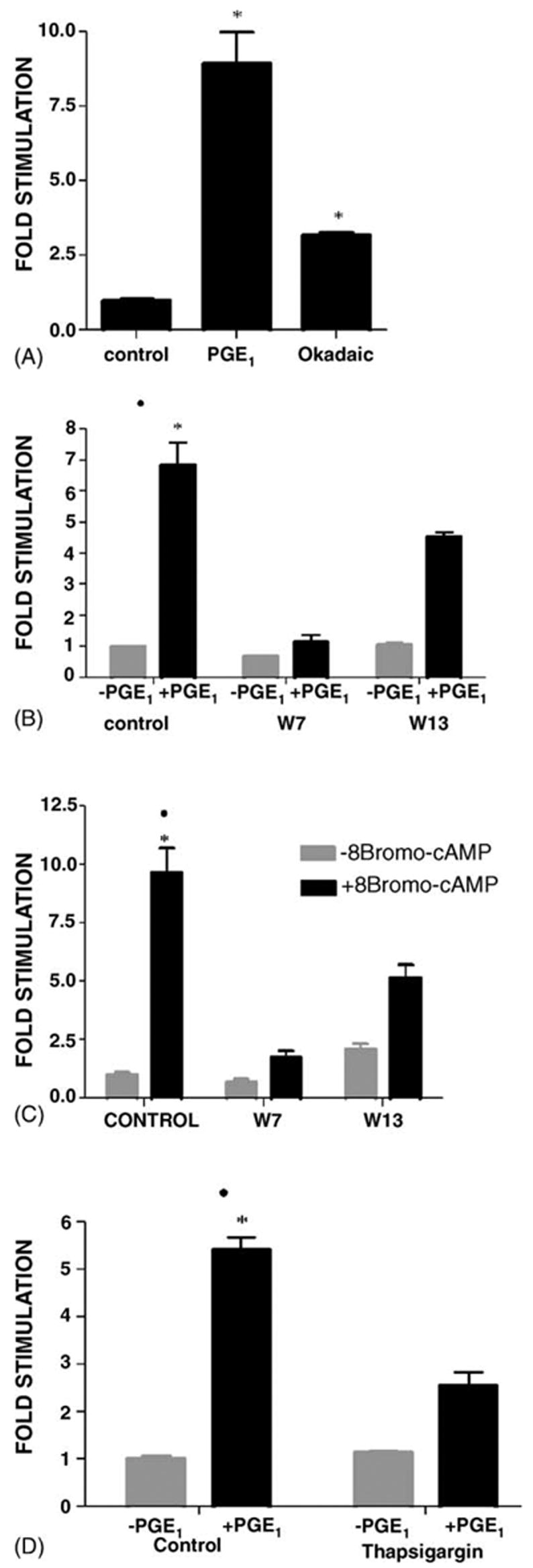
Role of calcium. MDCK cells were transfected with the pHβ1-1141Luc. Cultures were treated for 4 h with either (A) 1.4 μM PGE1 or 1 μM okadaic acid (B) 1.4 μM PGE1 or (C) 1 mM 8Br-cAMP. (D) 1.4 μM PGE1. In B and C a portion of the cultures were pretreated with either 56 μM W7 or, 1 μM W13 for 30 min prior to the 4-h incubation, as well as during the incubation period. In (D) a portion of the cultures were treated with 0.1 μM thapsigargin 30 min prior to, and during the 4-h incubation. Untreated controls were controls. Determinations were in quadruplicate. At least three experiments were performed. Values are mean ± S.E.M. * or ●, p < 0.001 relative to control, PGE1 + W7/W13, and for PGE1 + Thapsigargin.
The effects of the calmodulin antagonist N-(6 aminohexyl)-5-chloro-1-naphthalenesulfonamide (W7) and W13 were also examined (Fig. 6B). The observation that W7 (56 μM) reduced the PGE1 stimulation by 6.2 ± 0.7-fold (Fig 6B) was also consistent with the involvement of CaMK in mediating the PGE1 stimulation. Even W13 (added at only 1 μM) caused a 1.5 ± 0.7-fold reduction in the PGE1 stimulation. Fig. 6C shows that W7 and W13 also inhibited the 8Br-cAMP stimulation of transcription. These results may be explained if the PKA and CaMK pathways converge, and CaMK acts downstream of PKA.
Ultimately, CaMK activation is Ca2+ dependent. Consistent with the involvement of intracellular Ca2+, Fig. 6D shows that the PGE1 stimulation (5.4 ± 0.3-fold) was reduced to 3.3 ± 0.3-fold in the presence of 0.1 μM thapsigargin. Thapsigargin releases sequestered Ca2+ by inhibiting endoplasmic reticulum Ca2+ ATPases, without affecting PKC [39–41].
4. Discussion
Previously, we reported that PGE1 and PGE2 stimulate transcription of the Na,K-ATPase β subunit gene in MDCK cells [24]. In this report we present evidence consistent with the involvement of at least two different classes of G protein coupled prostaglandin receptors in mediating the PGE1 stimulation of β1 subunit gene transcription, including the EP1 and the EP2 receptor subtypes. The hypothesis that EP2 receptors are involved in mediating the prostaglandin stimulation of Na,K-ATPase β1 subunit gene transcription is supported by (1) the observed stimulatory effect of the EP2 agonist butaprost, and (2) the observed inhibitory effect of AH6809 on the PGE1 stimulation. However, the EP1 selective agonist 17-phenyl trinor PGE2 stimulated transcription by pHβ1-1141 Luc. In addition, the EP1 selective antagonist SC-51089 was also found to be capable of inhibiting the PGE1 stimulation by up to 91 ± 4%. Thus, EP1 receptors (coupled to Gq) as well as EP2 receptors (coupled to Gs) are very likely involved in mediating the stimulatory effect of PGE1 on Na,K-ATPase β1 subunit gene transcription. Consistent with this hypothesis are the results of our Western analysis which indicate that all four subtypes of PGE receptors (EP receptors) are expressed in MDCK cells, including EP1, and EP2, as well as EP3 and EP4.
While the binding of PGE1 and PGE2 to Gs coupled EP1 receptors results in the activation of adenylate cyclase, the binding of PGE1 and PGE2 to Gq coupled EP1 receptors results in the activation of PLC, an increase in the levels of diacylglycerol and inositol 1,4,5-trisphosphate (IP3), and the activation of PKC [38]. In this report the involvement of EP1 as well as EP2 receptors in mediating the effects of PGE1 and PGE2 was also supported by our observations concerning the cellular signaling pathways involved in mediating the PGE1 stimulation. Evidence for the involvement of both cAMP and PKC as mediators of the PGE1 effect on β1 subunit transcription includes the observed inhibitory effects the PKAI and the PKCI expression vectors on the PGE1 stimulation. The PKAI and PKCI expression vectors encode for the PKA inhibitory peptide, and the inhibitory domain of PKC, respectively. Also consistent with the involvement of PKA, was the observation that a myristolated PKA inhibitory peptide (PKI) (like the PKAI vector) caused a partial inhibition of the PGE1 stimulation. In contrast, the PGE1 stimulation was completely inhibited by the PKCI vector. Similarly, we previously observed that the PKC inhibitor Go 6976 partially inhibited the PGE1 stimulation of β1 transcription in MDCK cells [4]. Because the PGE1 stimulation was completely inhibited by PKCI, PKA may possibly act by augmenting the stimulation caused by PKC, rather than being responsible for a separate component of the PGE1 stimulation. However, 8 BrcAMP was indivually observed to stimulate transcription by pHβ1-1141Luc, as was observed in the case of TPA.
Previous observations that PGE1 and PGE2 cause an increase in intracellular Ca2+ and cAMP in MDCK cells are consistent with the involvement of EP1 and EP2 receptors in mediating the effects of these two prostaglandins in MDCK cells [10,16,19,30,39–42]. The increase in intracellular Ca2+ that occurs in PGE1 and PGE2 treated MDCK cells may cause a number of Ca2+ mediated pathways to be activated (in addition to PKC), which may all contribute to the PGE1 stimulation. Consistent with this hypothesis, Thapsigargin, which increases cytoplasmic Ca2+ (without affecting PKC), and okadaic acid caused a stimulation of β1 subunit gene transcription. The calmodulin antagonists W7 and W13 inhibited the stimulatory effects PGE1 on β1 subunit gene transcription. Such calmodulin mediated effects on transcription can be explained by the activation of CaMK, and the phosphorylation of the cAMP regulatory element binding protein (CREB) by CAMK. However CaMK may also regulate β1 subunit gene transcription indirectly, by modulating the effects of PKA and/or PKC. Consistent with this latter hypothesis is the observation that W7 and W13 inhibited the stimulatory effects of 8Br-cAMP on β1 subunit gene transcription.
The results of our transient transfection studies have indicated the involvement of EP1 as well as EP2 receptors in regulating Na,K-ATPase β1 subunit gene expression in our MDCK cell line, which is a distal tubule model. Previously, the results of in situ hybridization have indicated that EP1 receptor mRNA is found primarily in the collecting duct [43,44], while EP2 receptors are present predominantly in distal tubule cells as well as glomeruli [7,30]. Of particular interest in these regards, targeted disruption of EP2 was observed to cause salt-sensitive hypertension, supporting a role for EP2 in renal salt handling [44,45]. Our Western analysis also indicates that EP3 and EP4 receptors are present in MDCK cells. Previously studies indicate that in the kidney, EP3 is localized primarily in the collecting duct and thick ascending limb, while EP4 is primarily in the glomerulus [46].
There is an extensive literature regarding the effects of prostaglandins on renal transport, which consists primarily of in vivo microperfusion and micropuncture studies [47]. The results of these studies have indicated that the effects of prostaglandins on renal ion transport differ considerably as a function of nephron segment [47]. There are many conflicting reports, which may be explained by differences in responses between species, as well as other methodological details [47].
For this reason, in our studies utilizing EP receptor agonists and antagonists with MDCK cells, we cannot exclude the possibility of that the observed effects of prostaglandin agonists and antagonists on MDCK cells may differ from previously reported effects on human EP receptors. Indeed species differences in antagonist binding have been reported [29]. In addition is the possibility of different binding affinities of antagonists for the different isoforms of each receptor subtype. Two isoforms of the EP1 receptor, and eight isoforms of the EP3 receptor have been identified [3,30,48]. Altered responses to agonists and antagonists may also result from the cell culture situation. However, all of the EP receptors have been expressed in cultured cells as recombinant receptors [30], and the results obtained from these studies are generally in agreement with previous pharmacological and biochemical data [29].
Our investigations regarding PGE1 and PGE2 have been concerned with the effects of chronic treatments on MDCK cells [4]. PGE1 and PGE2 also have acute effects, exemplified by the reported inhibitory effects of PGE1 and PGE2 on the activity of the Na,K-ATPase in a clonal isolate of MDCK cells with collecting duct properties [49]. In this report the reduction in Na,K-ATPase activity following a 10 min incubation, was presumably due to a decrease in the level of the Na,K-ATPase in the plasma membrane, rather than to changes in overall gene expression. Such acute effects of PGE1 and PGE2 may possibly be mediated by EP3 receptors (which are localized in the collecting duct) [43,44,50,51]. However, the inhibitory effect of PGE2 on Na+ absorption in the collecting duct has been shown to occur via a Ca2+ dependent mechanism, and was not prevented by a potent EP3 agonist [44,51]. Nonsteroidal anti-inflamatory drugs (NSAIDs) enhance the urinary concentrating ability of the kidney by preventing such inhibitory effects of PGE2 in the collecting duct. However, even in this case EP3 receptors are not necessarily involved, because the targeted disruption of EP3 did not prevent such effects of NSAIDs [44,52,53].
Although PGE2 inhibits Na+ absorption in the collecting duct [52], PGE2 infusion studies with conscious dogs suggest that prostaglandins can also enhance renal Na+ absorption [54]. Consistent with this hypothesis are the results of studies with another subclone of MDCK cells (MDCK C7), which models renal collecting duct principal cells [7]. In these studies, PGE2 was observed to stimulate sodium reabsorption via an amiloride sensitive epithelial Na+ channel (ENaC) [7]. The investigators proposed that the stimulatory effect of PGE2 on ENaC was mediated by the EP2 receptors that they detected by RT-PCR.
Another proposed site for stimulatory effects of renal prostalandins on Na+ absorption is the distal tubule [46]. Indeed, enhanced Na,K-ATPase activity has been reported in isolated microdisected rat distal tubules following PGE2 treatment [55]. In our own studies we observed that PGE1 and PGE2 increase the Na,K-ATPase activity of MDCK cells which model cells in the distal tubule. In addition our results indicate that the PGE1 and PGE2 stimulation are mediated by increases in cAMP and Ca2+. Ultimately, the transactivation CREB occurs as a consequence of PGE1 treatment, presumably affecting the CREB which binds to the prostaglandin response element on the human β1 subunit promoter [56]. The precise role played by specific EP receptors in modulating the effects of transcription factors on the β1 subunit promoter will be the subject of future studies.
Acknowledgments
We thank Dr. Jerry Lingrel for pHβ1-1141 Luc, Dr. Te Chung Lee for the PKAI and PKCI vectors, and Dr. Janet Geisel for assistance. This work was supported by NHLBI 1R01HL69676-01 to M.T.
Footnotes
References
- 1.Herschman HR, Xie W, Reddy S. Inflammation, reproduction, cancer and all that. The regulation and role of the inducible prostaglandin synthase. Bioessays. 1995;17:1031–7. doi: 10.1002/bies.950171207. [DOI] [PubMed] [Google Scholar]
- 2.Bonventre JV, Nemenoff R. Renal tubular arachidonic acid metabolism. Kidney Int. 1991;39:438–49. doi: 10.1038/ki.1991.55. [DOI] [PubMed] [Google Scholar]
- 3.Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–53. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 4.Taub M, Borsick M, Geisel J, Matlhagela K, Rajkhowa T, Allen C. Regulation of the Na K-ATPase in MDCK cells by prostaglandin E1: a role for calcium as well as camp. Exp Cell Res. 2004;299:1–14. doi: 10.1016/j.yexcr.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez MG, Reyes JL. Induction of alkalinization in cultured renal cells (MDCK line) by prostaglandin E2. Prostaglandins. 1995;49:79–91. doi: 10.1016/0090-6980(95)00004-t. [DOI] [PubMed] [Google Scholar]
- 6.Worrell RT, Bao HF, Denson DD, Eaton DC. Contrasting effects of cPLA2 on epithelial Na+ transport. Am J Physiol Cell Physiol. 2001;281:C147–56. doi: 10.1152/ajpcell.2001.281.1.C147. [DOI] [PubMed] [Google Scholar]
- 7.Wegmann M, Nusing RM. Prostaglandin E2 stimulates sodium reabsorption in MDCK C7 cells, a renal collecting duct principal cell model. Prostaglandins Leukot Essent Fatty Acids. 2003;69:315–22. doi: 10.1016/j.plefa.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Madin SH, Darby NB., Jr Established kidney cell lines of normal adult bovine and ovine origin. Proc Soc Exp Biol Med. 1958;98:574–6. doi: 10.3181/00379727-98-24111. [DOI] [PubMed] [Google Scholar]
- 9.Saier MH, Jr, Boerner P, Grenier FC, et al. Sodium entry pathways in renal epithelial cell lines. Miner Electrolyte Metab. 1986;12:42–50. [PubMed] [Google Scholar]
- 10.Rindler MJ, Chuman LM, Shaffer L, Saier MH., Jr Retention of differentiated properties in an established dog kidney epithelial cell line (MDCK) J Cell Biol. 1979;81:635–48. doi: 10.1083/jcb.81.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coyne DW, Mordhorst M, Bertrand W, Morrison AR. Bradykinin stimulated PGE2 production is independent of changes in intracellular calcium in MDCK cells. Biochem Biophys Res Commun. 1989;161:1333–40. doi: 10.1016/0006-291x(89)91389-2. [DOI] [PubMed] [Google Scholar]
- 12.Lever JE. Inducers of mammalian cell differentiation stimulate dome formation in a differentiated kidney epithelial cell line (MDCK) Proc Natl Acad Sci USA. 1979;76:1323–7. doi: 10.1073/pnas.76.3.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cereijido M, Ehrenfeld J, Meza I, Martinez-Palomo A. Structural and functional membrane polarity in cultured monolayers of MDCK cells. J Membr Biol. 1980;52:147–59. doi: 10.1007/BF01869120. [DOI] [PubMed] [Google Scholar]
- 14.Cereijido M, Robbins ES, Dolan WJ, Rotunno CA, Sabatini DD. Polarized monolayers formed by epithelial cells on a permeable and translucent support. J Cell Biol. 1978;77:853–80. doi: 10.1083/jcb.77.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valentich JD, Tchao R, Leighton J. Hemicyst formation stimulated by cyclic AMP in dog kidney cell line MDCK. J Cell Physiol. 1979;100:291–304. doi: 10.1002/jcp.1041000210. [DOI] [PubMed] [Google Scholar]
- 16.Aboolian A, Vander Molen M, Nord EP. Differential effects of phorbol esters on PGE2 and bradykinin-induced elevation of [Cai2+] in MDCK cells. Am J Physiol. 1989;256:F1135–43. doi: 10.1152/ajprenal.1989.256.6.F1135. [DOI] [PubMed] [Google Scholar]
- 17.Taub M, Chuman L, Saier MH, Jr, Sato G. Growth of Madin-Darby canine kidney epithelial cell (MDCK) line in hormone-supplemented, serum-free medium. Proc Natl Acad Sci USA. 1979;76:3338–42. doi: 10.1073/pnas.76.7.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taub M, Sato GH. Growth of kidney epithelial cells in hormone-supplemented, serum-free medium. J Supramol Struct. 1979;11:207–16. doi: 10.1002/jss.400110210. [DOI] [PubMed] [Google Scholar]
- 19.Taub ML, Wang Y, Yang IS, Fiorella P, Lee SM. Regulation of the Na,K-ATPase activity of Madin-Darby canine kidney cells in defined medium by prostaglandin E1 and 8-bromocyclic AMP. J Cell Physiol. 1992;151:337–46. doi: 10.1002/jcp.1041510215. [DOI] [PubMed] [Google Scholar]
- 20.Jorgensen PL. Mechanism of the Na+, K+ pump. Protein structure and conformations of the pure (Na+ +K+)-ATPase. Biochimica et Biophysica Acta. 1982;694:27–68. doi: 10.1016/0304-4157(82)90013-2. [DOI] [PubMed] [Google Scholar]
- 21.Geering K. The functional role of the beta-subunit in the maturation and intracellular transport of Na,K-ATPase. FEBS Lett. 1991;285:189–93. doi: 10.1016/0014-5793(91)80801-9. [DOI] [PubMed] [Google Scholar]
- 22.Feng J, Orlowski J, Lingrel JB. Identification of a functional thyroid hormone response element in the upstream flanking region of the human Na,K-ATPase beta 1 gene. Nucleic Acids Res. 1993;21:2619–26. doi: 10.1093/nar/21.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matlhagela K, Borsick M, Rajkhowa T, Taub M. Identification of a prostaglandin-responsive element in the Na,K-ATPase {beta}1 promoter that is regulated by cAMP and Ca2+: evidence for an interactive role of cAMP regulatory element-binding protein and Sp1. J Biol Chem. 2005;280:334–46. doi: 10.1074/jbc.M411415200. [DOI] [PubMed] [Google Scholar]
- 24.Taub M, Borsick M, Geisel J, Rajkhowa T, Allen C. Regulation of the Na,K-ATPase in MDCK cells by prostaglandin E1: a role for calcium as well as cAMP. Exp Cell Res. 2004;299:1–14. doi: 10.1016/j.yexcr.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 25.Lee TC, Shi Y, Schwartz RJ. Displacement of BrdUrd-induced YY1 by serum response factor activates skeletal alpha-actin transcription in embryonic myoblasts. Proc Natl Acad Sci USA. 1992;89:9814–8. doi: 10.1073/pnas.89.20.9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grove JR, Price DJ, Goodman HM, Avruch J. Recombinant fragment of protein kinase inhibitor blocks cyclic AMP-dependent gene transcription. Science. 1987;238:530–3. doi: 10.1126/science.2821622. [DOI] [PubMed] [Google Scholar]
- 27.Knopf JL, Lee MH, Sultzman LA, et al. Cloning and expression of multiple protein kinase C cDNAs. Cell. 1986;46:491–502. doi: 10.1016/0092-8674(86)90874-3. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122:217–24. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodward DF, Pepperl DJ, Burkey TH, Regan JW. 6-Isopropoxy-9-oxoxanthene-2-carboxylic acid (AH 6809), a human EP2 receptor antagonist. Biochem Pharmacol. 1995;50:1731–3. doi: 10.1016/0006-2952(95)02035-7. [DOI] [PubMed] [Google Scholar]
- 31.Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–29. [PubMed] [Google Scholar]
- 32.Hallinan EA, Hagen TJ, Husa RK, et al. N-substituted dibenzoxazepines as analgesic PGE2 antagonists. J Med Chem. 1993;36:3293–9. doi: 10.1021/jm00074a010. [DOI] [PubMed] [Google Scholar]
- 33.Hallinan EA, Hagen TJ, Tsymbalov S, et al. Aminoacetyl moiety as a potential surrogate for diacylhydrazine group of SC-51089, a potent PGE2 antagonist, and its analogs. J Med Chem. 1996;39:609–13. doi: 10.1021/jm950454k. [DOI] [PubMed] [Google Scholar]
- 34.Hallinan EA, Hagen TJ, Tsymbalov S, Stapelfeld A, Savage MA. 2,4-Disubstituted oxazoles and thiazoles as latent pharmacophores for diacylhydrazine of SC-51089, a potent PGE2 antagonist. Bioorg Med Chem. 2001;9:1–6. doi: 10.1016/s0968-0896(00)00229-7. [DOI] [PubMed] [Google Scholar]
- 35.Coleman RA, Kennedy I, Sheldrick RL. New evidence with selective agonists and antagonists for the subclassification of PGE2-sensitive (EP) receptors. Adv Prostaglandin Thromboxane Leukot Res. 1987;17A:467–70. [PubMed] [Google Scholar]
- 36.Zeng L, An S, Goetzl EJ. Selective regulation of RNK-16 cell matrix metalloproteinases by the EP4 subtype of prostaglandin E2 receptor. Biochemistry. 1996;35:7159–64. doi: 10.1021/bi960036x. [DOI] [PubMed] [Google Scholar]
- 37.Abramovitz M, Adam M, Boie Y, et al. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483:285–93. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- 38.Glass DB, Cheng HC, Mende-Mueller L, Reed J, Walsh DA. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J Biol Chem. 1989;264:8802–10. [PubMed] [Google Scholar]
- 39.Jan CR, Ho CM, Wu SN, Tseng CJ. Bradykinin-evoked Ca2+ mobilization in Madin Darby canine kidney cells. Eur J Pharmacol. 1998;355:219–33. doi: 10.1016/s0014-2999(98)00481-6. [DOI] [PubMed] [Google Scholar]
- 40.Alonso GL, Gonzalez DA, Takara D, Ostuni MA, Sanchez GA. Calcium additional to that bound to the transport sites is required for full activation of the sarcoplasmic reticulum Ca-ATPase from skeletal muscle. Biochim Biophys Acta. 1998;1405:47–54. doi: 10.1016/s0167-4889(98)00101-3. [DOI] [PubMed] [Google Scholar]
- 41.Jean D, Harbison M, McConkey DJ, Ronai Z, Bar-Eli M. CREB and its associated proteins act as survival factors for human melanoma cells. J Biol Chem. 1998;273:24884–90. doi: 10.1074/jbc.273.38.24884. [DOI] [PubMed] [Google Scholar]
- 42.Parker D, Ferreri K, Nakajima T, et al. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devis PE, Grohol SH, Taub M. Dibutyryl cyclic AMP resistant MDCK cells in serum free medium have reduced cyclic AMP dependent protein kinase activity and a diminished effect of PGE1 on differentiated function. J Cell Physiol. 1985;125:23–35. doi: 10.1002/jcp.1041250105. [DOI] [PubMed] [Google Scholar]
- 44.Taub M, Saier MH, Jr, Chuman L, Hiller S. Loss of the PGE1 requirement for MDCK cell growth associated with a defect in cyclic AMP phosphodiesterase. J Cell Physiol. 1983;114:153–61. doi: 10.1002/jcp.1041140203. [DOI] [PubMed] [Google Scholar]
- 45.Taub M, Devis PE, Grohol SH. PGE1-independent MDCK cells have elevated intracellular cyclic AMP but retain the growth stimulatory effects of glucagon and epidermal growth factor in serum-free medium. J Cell Physiol. 1984;120:19–28. doi: 10.1002/jcp.1041200104. [DOI] [PubMed] [Google Scholar]
- 46.Bansal V, Syres KM, Makarenkova V, et al. Interactions between fatty acids and arginine metabolism: implications for the design of immune-enhancing diets. JPEN J Parenter Enteral Nutr. 2005;29:S75–80. doi: 10.1177/01486071050290S1S75. [DOI] [PubMed] [Google Scholar]
- 47.Sugimoto Y, Namba T, Shigemoto R, Negishi M, Ichikawa A, Narumiya S. Distinct cellular localization of mRNAs for three subtypes of prostaglandin E receptor in kidney. Am J Physiol. 1994;266:F823–8. doi: 10.1152/ajprenal.1994.266.5.F823. [DOI] [PubMed] [Google Scholar]
- 48.Breyer MD, Breyer RM. G protein-coupled prostanoid receptors and the kidney. Annu Rev Physiol. 2001;63:579–605. doi: 10.1146/annurev.physiol.63.1.579. [DOI] [PubMed] [Google Scholar]
- 49.Tilley SL, Audoly LP, Hicks EH, et al. Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest. 1999;103:1539–45. doi: 10.1172/JCI6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breyer MD, Breyer RM. Prostaglandin E receptors and the kidney. Am J Physiol Renal Physiol. 2000;279:F12–23. doi: 10.1152/ajprenal.2000.279.1.F12. [DOI] [PubMed] [Google Scholar]
- 51.Bonvalet JP, Pradelles P, Farman N. Segmental synthesis and actions of prostaglandins along the nephron. Am J Physiol. 1987;253:F377–87. doi: 10.1152/ajprenal.1987.253.3.F377. [DOI] [PubMed] [Google Scholar]
- 52.Slipetz D, Buchanan S, Mackereth C, et al. Sequestration and phosphorylation of the prostaglandin E2 EP4 receptor: dependence on the C-terminal tail. Biochem Pharmacol. 2001;62:997–1012. doi: 10.1016/s0006-2952(01)00742-0. [DOI] [PubMed] [Google Scholar]
- 53.Cohen-Luria R, Rimon G, Moran A. Department of Physiology, F. o. H. S. B.-G. U. o. t. N. B.-S. I. PGE2 inhibits Na-K-ATPase activity and ouabain binding in MDCK cells. Am J Physiol. 1993;264(1):F61–5. doi: 10.1152/ajprenal.1993.264.1.F61. Pt 2. [DOI] [PubMed] [Google Scholar]
- 54.Good DW, George T. Regulation of HCO3- absorption by prostaglandin E2 and G proteins in rat medullary thick ascending limb. Am J Physiol. 1996;270:F711–7. doi: 10.1152/ajprenal.1996.270.5.F711. [DOI] [PubMed] [Google Scholar]
- 55.Hebert RL, Jacobson HR, Breyer MD. Prostaglandin E2 inhibits sodium transport in rabbit cortical collecting duct by increasing intracellular calcium. J Clin Invest. 1991;87:1992–8. doi: 10.1172/JCI115227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fleming EF, Athirakul K, Oliverio MI, et al. Urinary concentrating function in mice lacking EP3 receptors for prostaglandin E2. Am J Physiol. 1998;275:F955–61. doi: 10.1152/ajprenal.1998.275.6.F955. [DOI] [PubMed] [Google Scholar]
- 57.Breyer MD, Jacobson HR, Davis LS, Breyer RM. In situ hybridization and localization of mRNA for the rabbit prostaglandin EP3 receptor. Kidney Int. 1993;44:1372–8. doi: 10.1038/ki.1993.391. [DOI] [PubMed] [Google Scholar]
- 58.Kirschenbaum MA, Stein JH. The effect of inhibition of prostaglandin synthesis on urinary sodium excretion in the conscious dog. J Clin Invest. 1976;57:517–21. doi: 10.1172/JCI108304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scherzer P, Wald H, Rubinger D, Popovtzer MM. Indomethacin and sodium retention in the rat: role of inhibition of prostaglandin E2 synthesis. Clin Sci. 1992;83:307–11. doi: 10.1042/cs0830307. [DOI] [PubMed] [Google Scholar]
- 60.Matlhagela K, Borsick M, Rajkhowa T, Taub M. Identification of a prostaglandin-responsive element in the Na,K-ATPase β1 promoter which is regulated by cAMP and Ca2+: evidence for a role of CREB and Sp1. J Biol Chem. :334–86. doi: 10.1074/jbc.M411415200. in press. [DOI] [PubMed] [Google Scholar]