

Abstract
The Raf kinase inhibitory protein (RKIP) binds to Raf-1 interfering with binding of the MEK substrate and potentially also Raf-1 activation. In response to mitogen stimulation RKIP dissociates from Raf-1 and later re-associates. Here, using a combination of mutational approaches, biochemical studies, peptide arrays and plasmon surface resonance (BIAcore), we fine map and characterize a minimal 24 amino acid long RKIP binding domain in the Raf-1 N-region, which consists of constitutive elements at both flanks and a center element that is regulated by phosphorylation and enhances the re-binding of RKIP to Raf-1 in the later phase of mitogen stimulation.
Keywords: RKIP, Raf-1, kinetic binding analysis, surface plasmon resonance
INTRODUCTION
The mitogen-activated protein (MAP) kinases are important components of conserved signaling pathways controlling embryogenesis, cell differentiation, proliferation, and death. The MAPK signaling cascades are organized in a hierarchical three-tiered module: MAP kinases are activated by MAP kinase kinase (MAPKK), which is activated by a MAP kinase kinase kinase (MAPKKK) [1]. ERKs (extracellular signal-regulated kinases) are the first characterized subfamily of MAP kinases activated by growth factors and other stimuli via a cascade involving Ras, Raf-1 (MAPKKK), and MEK1/2 (MAPKK). Mammals have three Raf protein isoforms, A-Raf, B-Raf, and Raf-1, which despite sharing many common features of regulation can differentially modulate cellular responses [2,3]. Raf-1 regulation is very complex, and involves amongst other processes the phosphorylation of the N-region at Ser338 and Tyr341. PAK1 and 2 have been shown to phosphorylate Ser338 and SRC kinases have been shown to induce phosphorylation of Tyr341 [2].
One protein that binds to and negatively regulates Raf-1 signaling is Raf kinase inhibitor protein (RKIP) [4,5]. A member of the highly conserved phosphatidylethanolamine binding protein family, RKIP is widely expressed in mammals [6,7]. RKIP inhibits phosphorylation and activation of MEK by Raf-1 by dissociating the physical interaction between Raf-1 and MEK, thus acting as a competitive inhibitor of substrate binding [4,5]. The overexpression of RKIP leads to decreased activation of MEK by Raf-1 and the reduction of transformation mediated by Raf-1. In contrast, the down-regulation of RKIP increases MEK and ERK phosphorylation and AP-1 dependent transcription [4]. In addition to Raf-1, B-Raf and two other members of the MAPKKK family, TAK1 and NIK, are also targets of RKIP [8,9]. Recently the role of RKIP in signal transduction was found to extend to feedback inhibition for G-protein-coupled receptors [10]. Although the mechanism of suppression of the signaling pathways initiated by some of these kinase targets by RKIP has been partially delineated, little is known about the molecular events regulating the inhibition. We have previously shown that RKIP is released from Raf-1 upon mitogen stimulation, and that this release coincides with the activation of the MEK-ERK pathway [4,5]. Thus, it is important to understand the mechanisms which regulate RKIP binding to and dissociation from Raf-1. Recently Trakul et al. [11] reported that RKIP interferes with Raf-1 activation by inhibiting the phosphorylation of the Raf-1 N-region, Ser338 and Tyr340/341, by PAK and SRC family kinases, respectively. As a consequence Raf-1 activation was inhibited, and in turn the phosphorylation of these residues curbed RKIP binding to Raf-1 [11]. Thus, the current state of knowledge suggests that RKIP inhibits both Raf-1 activation [11] as well as the ability of Raf-1 to phosphorylate its substrate MEK [4,5]. These mechanisms are not mutually exclusive and rather indicate that RKIP may regulate Raf-1 at multiple levels. However, the mechanistic details and how these inhibitory events are coordinated are unresolved. Here we start to address these questions by mapping the minimal region of Raf-1 required for RKIP binding, and then using specific point mutants and quantitative surface plasmon resonance (Biacore) to analyze the role of Raf-1 phosphorylation in the RKIP interaction with Raf-1. Our results indicate that N-region plays a coordinating role for both types of inhibition and that the affinity of RKIP to Raf-1 is decisively modulated by the phosphorylation of the N-region at Ser338/339 and Tyr341.
MATERIALS AND METHODS
Plasmids and cell culture
cDNA for GST-Raf-1 was cloned as described in pEBG as described [12]. The Raf-1 mutants pEF-mRaf-1Y340F and Y341F were a gift from Richard Marais. The mammalian expression vector for myc-tagged Rab24 was a gift from William Maltese. The other plasmids have been described before [4,5]. Cells were cultured in Dulbeccos’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Transfection, GST-pulldowns and immunoprecipitation
For in vivo RKIP - Raf-1 binding assays 293FT cells (Invitrogen) were co-transfected with pEBG Raf-1 (GST-Raf-1) and pCMV HA-RKIP constructs using the calcium phosphate precipitation method. Forty-eight hours after transfection, cells were scraped from plates and washed twice in phosphate buffered saline (PBS). Pellets were resuspended in PBS containing 1mM DTT, mammalian protease inhibitor cocktail (1:100 dilution; Sigma) and phosphatase inhibitors and lysed by sonication. 200μg total cell lysates were tumbled with 25μl Glutathione agarose beads (Sigma) for one hour, washed three times with PBS, resolved by 10% SDS-PAGE, and transferred onto PVDF membranes (Millipore) for Western blotting.
Binding studies with Raf-1 N-region point mutants were performed using COS-1 cells. Varying amounts of plasmid expression vectors for myc-tagged Rab24, B-Raf, Raf-1, Raf-1 Y340F, Raf-1 Y341, Raf-1 YY340/341FF, Raf-1 S338A, Raf-1 S339A, and Raf-1 SS338/339AA were cotransfected with HA-RKIP cDNA such that the expressed proteins were of equal levels. The transfection was done using FuGene6 (Roche) according to the manufacturer’s instructions. Twenty-four hours post transfection cells were made quiescent by serum starvation for 24 hours. Then, i.e. 48 hours post transfection, cells were stimulated with 30nM epidermal growth factor (EGF; Sigma) and 100nM 12,13-tetradecanoyl phorbol (TPA; Sigma) for 10 minutes. Cells were lysed in TBST buffer (20mM Tris pH7.4, 150 mM NaCl, 2mM EDTA, 1% Triton X-100,) containing protease and phosphatase inhibitors. For immunoprecipitation 200μg of cell extracts were incubated with 2μg 9E10 anti-myc monoclonal antibody for one hour at 4°C, then Protein G agarose beads (Upstate USA, Inc.) were added for a further 1.5 hours at 4°C. The beads were washed twice in TBST, once in TBS, resolved on 12% SDS-PAGE and transferred onto PVDF membranes. The blots were blocked in PBS plus 0.2% Tween-20 (PBST) with 5% non-fat milk solution for one hour. HA-MEK immunoprecipitates were prepared as above except that the 12CA5 antibody was used for immunoprecipitation. Anti-phospho MEK antibody (Cell Signaling Technology) was used to monitor activating MEK phosphorylation.
MCF-7 cells were transfected with 1 μg pcDNA3.1-FLAG-RKIP and 2 μg pEGFP-Raf-1 per 10 cm dish using an Amaxa Nucleofector according to the manufacturer’s protocol. Forty-eight hours post transfection, cells were starved overnight in DMEM containing 0.1% FBS. Cells were stimulated with 100 ng/ml EGF for 5, 10, 20, 40, 80 min and lysed in HKMEN buffer (10 mM HEPES pH 7.2, 140 mM KCl, 5 mM MgCl2, 2 mM EGTA, 0.1% NP-40) containing protease and phosphatase inhibitors. Afterwards, the lysates were sonicated three times for 5 sec each, and centrifuged at 13.000 rpm for 10 minutes. Each supernatant was immunoprecipitated with 20 μl anti-M2-FLAG agarose beads (Sigma) for 3 hours at 4°C. Beads were washed three times with HKMEN buffer, and separated on a 4-12% SDS-PAGE gel (Invitrogen).
Western blotting
Western blots of GST-Raf-1 and mutants and HA-RKIP were incubated for one hour at room temperature with the indicated antibodies (anti-GST antibody; anti-phospho-Raf-1 (Ser338), both from Upstate; mouse monoclonal anti-HA antibody, 12CA5; mouse monoclonal anti-myc antibody, 9E10, both from Roche Diagnostics). All primary antibodies were diluted in primary antibody solution (PBST, 5% bovine serum albumin, 0.002% sodium azide). Following three washes in PBST, blots were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) in PBST 5% milk for one hour at room temperature. HA-tagged proteins were detected by incubating blots with anti-HA antibody conjugated to horseradish peroxidase overnight at 4°C. Proteins were visualized with enhanced chemiluminescence using the BioRad ChemiDoc EQ system.
Raf-1 kinase assays
were done as described previously [4]. Briefly, Raf-1 was expressed in COS-1 cells together with HA-RKIP. Raf-1 was immunoprecipitated using an antiserum made against the 12 C-terminal amino acids or Raf-1 [13]. Cells were serum starved overnight, treated with 30ng/ml EGF for the timepoints indicated. Cells were lysed in TBST substituted with phosphatase and protease inhibitors [13]. Raf-1 immunoprecipitates were washed three times in TBST and once in Raf kinase buffer (TBS plus 5mM MgCl2). Then immunoprecipitates were resuspended in Raf kinase buffer and incubated for 30 minutes at room temperature with 10μM ATP, 0.2 μl 32P-γ-ATP (3000Ci/mmol), and 50 ng kinase competent MEK plus 150 ng kinase negative ERK (knERK) expressed in E.coli as described [13]. The reaction was resolved on 10% SDS-PAGE and blotted. The blots were autoradiographed and subsequently stained for Raf-1 and RKIP.
Phosphorylation of Raf-1 by v-SRC or PAK in vitro
Activated mutants of PAK, Myc-Pak (T423E), or Src, HA-v-Src, were overexpressed in COS-1 cells and purified by immunoprecipitation with 9E10 or 12CA5 antibodies, respectively. The immunoprecipitates were incubated with increasing amounts of RKIP (1, 3 and 9 μM) or 9 μM bovine serum albumin (BSA) as control at room temperature for 10 min. Then 9 μM of the purified recombinant GST-Raf-1 peptide (325-349) or GST-Raf-1 peptide (325-349SS338/339AA), 2 μM ATP, and 10 μCi of [32P] ATP were added and incubated in a reaction volume of 30 μl. After 30 minutes incubation at room temperature the reaction was terminated with 10 μl of SDS sample buffer. Samples were separated by SDS-PAGE, transferred to a PVDF membrane (Millipore Corp.), and subjected to autoradiography. PAK and v-SRC proteins were detected by Western blotting.
Peptide arrays
Peptide arrays were manufactured by the Cancer Research UK core facility. Overlapping synthetic Raf-1 peptides (23-mers offset by four amino acids) immobilized on a nitrocellulose membrane were probed with [35S]-methionine labeled RKIP protein that had been produced using a coupled in vitro transcription/translation kit (TNT T7 quick coupled transcription/translation kit, Promega) according to the manufacturers instructions. Membranes were blocked with 5% BSA for 1 hour and incubated with the radioactive RKIP solution for 1 hour. Afterwards, they were washed three times for 15 min with TBST and autoradiographed using a phosphoimager. Signals were quantitated using ImageJ 1.34s software.
Surface Plasmon Resonance
Experiments were performed with a BIAcore T100 instrument using Sensor Chip SA (BIAcore, Uppsala, Sweden) which permits the immobilization of biotinylated peptides to a streptavidin surface. Biotinylated peptides of >95% purity were purchased from piCHEM, Graz, Austria and Cancer Research UK, London, United Kingdom:
Raf-1 NR1: RPRGQRDSSYYWEIEASEV;
Raf-1 NR2: RPRGQRDpSpSYYWEIEASEV;
Raf-1 NR3: RPRGQRDSSpYpYWEIEASEV;
Raf-1 NR4: RPRGQRDpSpSpYpYWEIEASEV;
Raf-1 NR5: RPRGQRDpSpSYpYWEIEASEV;
Sensor chips were primed with running buffer (10 mM Hepes, pH 7.4, 0.15 M NaCl, 3.4 mM EDTA, and 0.05% surfactant P20; BIAcore), followed by 40% glycerol, and conditioned with 1 M NaCl and 50 mM NaOH. The peptides were immobilized to the surface in running buffer to give approximately 850 RU (resonance units). Free streptavidin on the chip was inactivated with biotin solution (Pierce, United Kingdom). RKIP was expressed and purified from E.coli as previously described [4], and prepared to a 2mg/ml solution. Different concentrations of purified RKIP were applied to the sensor chip at a flow rate of 30 μl/min allowing 4 minutes for association and 5 minutes for dissociation. Between applications the chip was regenerated with 1M NaCl followed by running buffer, which was used as a blank for the subsequent measurement. All experiments were performed in duplicates at 25 °C. The results were plotted as RU versus time, and analyzed with the BIA-evaluation Software 4.1 (BIAcore, Uppsala, Sweden). For each binding curve, the response obtained using control surfaces (no peptide or irrelevant peptide) and the response obtained from an injection of buffer immediately prior to each analyte injection was subtracted (double referencing). Binding of all the peptides fitted a 1:1 Langmuir binding model, which describes a reversible interaction of two molecules in a 1:1 complex.
RESULTS AND DISCUSSION
We have previously shown that in response to mitogenic stimulation RKIP quickly dissociated from Raf-1 but re-associated later, and that the window of RKIP dissociation from Raf-1 coincided with the peak of ERK activation [4]. These results suggested that Raf-1 only can efficiently activate ERK if RKIP is released. In Fig. 1a we have examined the kinase activity of Raf-1 and RKIP binding. COS1 cells were co-transfected with Raf-1 and HA-RKIP, serum starved overnight and stimulated with 100ng/ml EGF for the timepoints indicated. Raf-1 immunoprecipitates were examined for associated RKIP and also tested for kinase activity using a linked kinase assay, where Raf-1 is incubated with recombinant MEK and kinase negative recombinant ERK (knERK). The readout is phosphorylation of knERK. RKIP co-precipitated with Raf-1 from serum starved cells, and transiently dissociated upon EGF treatment. At 10 minutes RKIP re-associated again and stayed associated for the rest of the timecourse. Interestingly, Raf-1 kinase activity was still high when RKIP re-associated and stayed high for at least another 30 minutes. In addition, Raf-1 kinase activity tested in vitro was similar in cells overexpressing RKIP or not (Supplementary Fig. 1). These results are not easily explainable by a simple model where the role of RKIP is to inhibit Raf-1 activation by binding to and interfering with the phosphorylation of the N-region [11], which is required for Raf-1 activity. Further, this model makes it difficult to rationalize how RKIP mutants that cannot bind to Raf-1 still can suppress ERK signaling [5]. These observations suggested that RKIP can interfere with Raf-1 signaling at several levels, including Raf-1 activation and substrate phosphorylation.
Figure 1.
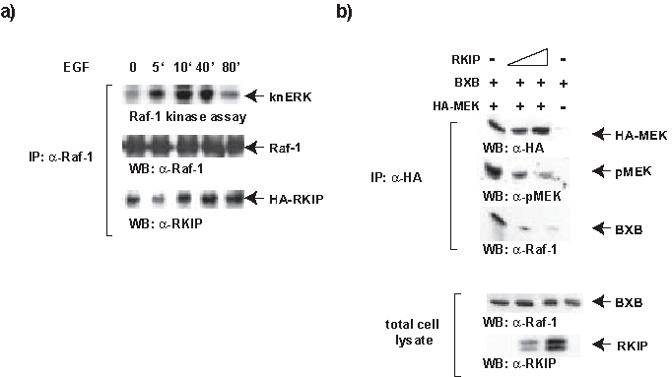
RKIP can bind to activated Raf-1 and compete for MEK binding. (a) Raf-1 and HA-RKIP were co-expressed in COS-1 cells. Serum starved cells were stimulated as indicated using 30ng/ml EGF. Raf-1 immunoprecipitates were subjected to coupled kinase assay using recombinant MEK and kinase negative ERK (knERK) as substrates. Associated HA-RKIP was detected by immunoblotting the Raf-1 immunoprecipitates with RKIP antibodies. (b) COS-1 cells were co-transfected with HA-MEK, RKIP and BXB (corresponding to the Raf-1 kinase domain) as indicated. Three days post-transfection growing cells were lysed in TBST and HA-immunoprecipitates were examined for MEK phosphorylation using a phospho-specific antibodies and associated BXB using Raf-1 antiserum.
It is difficult to conclusively distinguish between these possibilities. Since MEK is the only bona fide Raf-1 substrate, both inhibition of Raf-1 activation and interference with substrate phosphorylation have to be assayed using MEK as substrate. This poses a “Boolean dilemma” where the boundary between “AND” and “OR” options is blurred. Thus, we need to build up evidence from correlative data from different types and sources of experiments. In order to assess inhibitory contributions we first re-examined whether RKIP can interfere with the ability of activated Raf-1 to phosphorylate MEK in vitro. Previous studies [4,5] had analyzed RKIP binding to Raf-1 and shown that this will reduce the binding of MEK. However, this still leaves the possibility that a high turnover could permit MEK to become phosphorylated despite its binding to Raf-1 being reduced. Therefore, we immunoprecipitated MEK and analyzed the effects of RKIP on the phosphorylation of its activating sites and Raf-1 association. To ensure efficient Raf-1 activity not dependent on growth factor stimulation, which may confound the results, we used BXB, a Raf-1 mutant rendered constitutively active by deletion of the N-terminal regulatory domain [14]. BXB still can bind RKIP with similar efficiency as Raf-1 [4]. BXB could be coimmunoprecipitated with HA-MEK, which exhibited readily detectable phosphorylation of the activating sites (Fig. 1b). Coexpression of increasing amounts of RKIP led to a loss of BXB binding to MEK and also to a concomitant reduction of MEK phosphorylation. These data show that RKIP can competitively dissociate the BXB-MEK complex and that this also prevents the phosphorylation of MEK. Consistent results were obtained with full length Raf-1. When subjected to an in vitro kinase assay in the presence of increasing amounts of purified recombinant RKIP, a dose dependent inhibition of Raf-1 kinase activity was observed (Supplementary Fig. 2)
Second, we tested whether RKIP can interfere with the activation of Raf-1 by phosphorylation of the N-region as suggested by Trakul et al. [11]. To this end we repeated the experiments done by Trakul et al. [11] where they showed that transfection of an activated PAK and activated Src mutant induced the phosphorylation of Raf-1 on Ser338/339 and Tyr341, and that the co-expression of increasing amounts of RKIP interfered with these phosphorylations. We obtained entirely consistent results that confirm these data (Supplementary Fig. 3). However, as co-expression is a rather indirect assay for phosphorylation, we also tested whether RKIP could interfere with the phosphorylation of Raf-1 at Ser338/339 and Tyr341 using purified proteins in vitro (Fig. 2). As full length Raf-1 is notoriously difficult to produce and purify in a functional form and in the quantities required for use as substrate in in vitro kinase assays, we used a fragment (aa 325-349) encompassing the N-region instead. This construct could be expressed and purified from E.coli, and was readily phosphorylated by either PAK or v-Src in vitro. Mutation of SS338/9AA completely abolished phosphorylation by PAK, but did not affect phosphorylation by Src, assuring that the correct sites are targeted. Surprisingly, the addition of increasing amounts of purified RKIP protein to the in vitro kinase assays did not have any effect on the phosphorylation of either site by PAK or Src. It has to be noted that the Raf-1 fragments used as substrates have low affinity to RKIP, and hence RKIP may not protect efficiently. The same purified RKIP protein used, however, was able to suppress the activation of MEK-ERK by Raf-1 in vitro (Supplementary Figure 2). In summary, these results suggested that the role of RKIP in Raf-1 regulation is multifaceted, and that as a prerequisite to elucidate it fully it is necessary to understand the detailed regulation of RKIP binding to Raf-1.
Figure 2.
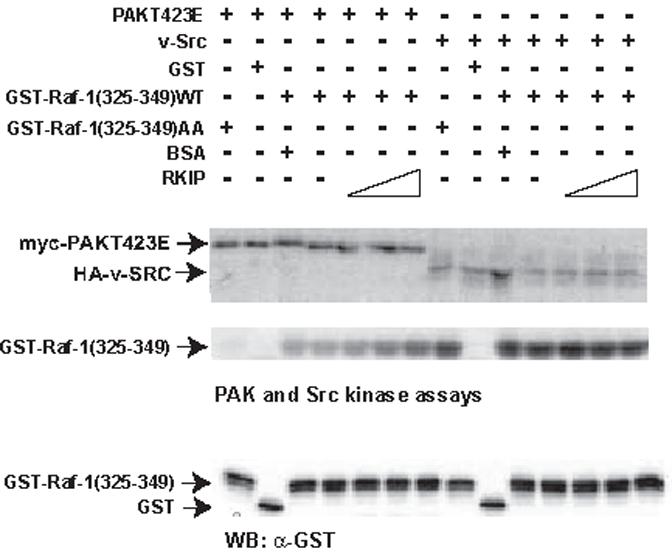
RKIP has no effect on the phosphorylation of Raf-1 by v-SRC or PAK in vitro. Purified recombinant GST-Raf-1(325-349) and GST-Raf-1(325-349SS338/339AA) peptides were tested for phosphorylation by Myc-Pak (T423E) or HA-v-Src in the presence of increasing amounts of RKIP (1, 3 and 9 μM) or BSA (9 μM) as control. Phosphorylated proteins were detected by autoradiography. PAK and v-SRC proteins were detected by Western blotting.
Therefore, we decided to fine map the binding sites of RKIP to Raf-. For this purpose we co-expressed full length HA-tagged RKIP with GST-fusion proteins of Raf-1 deletion mutants in 293FT cells, and assayed RKIP recovery in GST-pulldowns (Fig. 3). Our original study had mapped the RKIP binding region in Raf-1 to kinase subdomains I and II, a region of approximately 100 amino acids [5]. To map the RKIP binding region more precisely, we constructed additional deletions starting from the amino end of the Raf-1 catalytic domain (amino acids 325-648) (Fig. 3a). Different GST-Raf-1 deletion mutants were coexpressed with HA-tagged RKIP in 293FT cells and examined for their ability to bind full-length wild type RKIP (Fig. 3b). Consistent with our previously published results [5], the Raf-1 catalytic domain, GST-Raf-1325-648, was sufficient to bind RKIP. Removal of the N-terminal 13 amino acids from this construct (GST-Raf-1338-648) did not affect RKIP binding, while removing four more amino acids which contain the N-region phosphorylation sites (338-341) completely abolished its ability to bind to RKIP. To determine whether RKIP also interacts with regions in CR1 and CR2, additional GST fusion proteins containing the first 324 amino acids of Raf-1 were constructed. Detectable amounts of RKIP were pulled down with GST-Raf-11-324, and the binding of RKIP increased as the construct was extended towards the C-terminus with the GST-Raf-11-339 construct showing similar RKIP binding as the GST-Raf-1338-648 construct. Our results therefore suggested that beside the region in CR3 located between amino acids 325-349 additional regions in the CR1 and CR2 of Raf-1 might also bind to RKIP. To determine whether the first 24 amino acids (325-349) of the Raf-1 catalytic domain are sufficient to bind to RKIP, we constructed GST-Raf-1325-349 and analyzed its binding to RKIP in the pull-down assay. Concurring with the aforementioned deletion mutation studies, detectable amounts of RKIP were found to bind to the Raf-1 24-amino-acid peptide (Fig. 3b, right panel)
Figure 3.
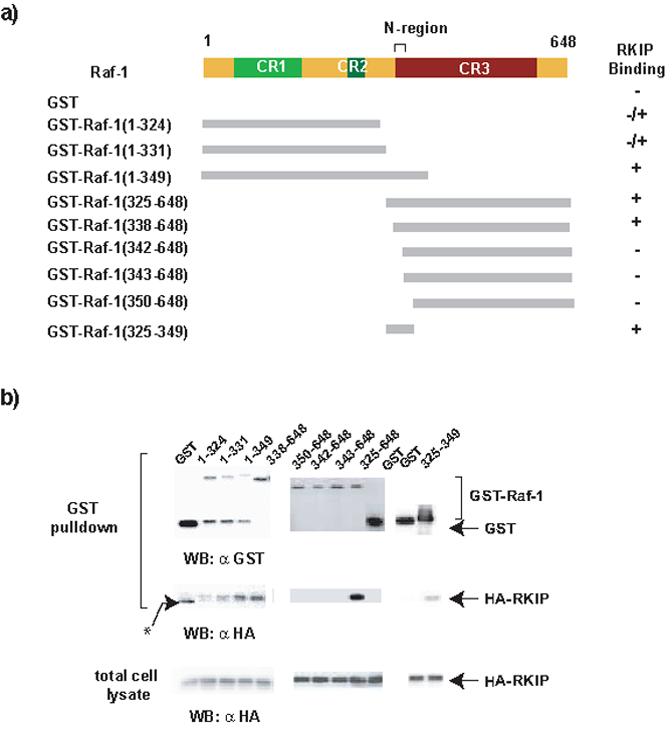
Deletion mapping of the minimal RKIP binding domain in Raf-1. (a) Schematic of the GST-Raf-1 deletion constructs used in GST pull down assays. The upper top panel is the schematic of the Raf-1 protein showing the location of the three conserved regions (CR) and the N-region (negative-charge regulatory region). Summary of RKIP binding is shown on the right. (b) Western blots of the GST-pull-down assay. GST-Raf-1 deletion mutants were co-expressed with HA-RKIP. Total cell lysates and GST pulldowns were Western blotted with anti-GST and anti-HA antibodies as indicated. The results shown are representative of at least three independent experiments.
The minimal 24-amino-acid RKIP binding region encompasses the Ser338 and Tyr341 phosphorylation sites. We and others have previously shown that the binding of RKIP to Raf-1 is dynamically regulated by phosphorylation of both RKIP [15] and Raf-1 [4,5]. Therefore, we investigated whether the Raf-1 N-region phosphorylation sites are important for RKIP binding using mutational analysis, where we conservatively substituted Ser338/339 with Ala, and Tyr340/341 with Phe (Fig. 4a). These mutants were co-expressed in COS-1 cells and analyzed by co-immunoprecipitation. As shown previously, HA-RKIP associated with myc-Raf-1 in quiescent cells and dissociated upon EGF stimulation [4,5]. These interactions were specific, as RKIP was not found in the anti-myc immunoprecipitates when myc-Rab24 was co-transfected as control. While the Raf-1 Y340F had no effect, mutation of Y341F abolished detectable RKIP binding in quiescent cells, and so did the double mutation YY340/341FF. Similarly, mutation of S338A or S339A reduced binding of Raf-1 to RKIP, and the double mutation SS338/339AA abolished it (Fig. 4b). This result was surprising as Trakul et al. [11] had suggested that the phosphorylation of these site provides a dissociation signal, and one would have expected that replacing them by Ala should stabilize RKIP binding to Raf-1. Therefore, we tested the effects of replacing these sites with phosphomimetic residues (Fig. 4c). Mutation of either Ser338/9 to Asp and Glu, respectively, and Tyr340 or Tyr341 to Asp also reduced binding of Raf-1 to RKIP in quiescent cells. A plausible interpretation for why both conservative and phosphomimetic mutations in the N-region interfered with RKIP binding is that the phosphate is physically involved in the binding.
Figure 4.
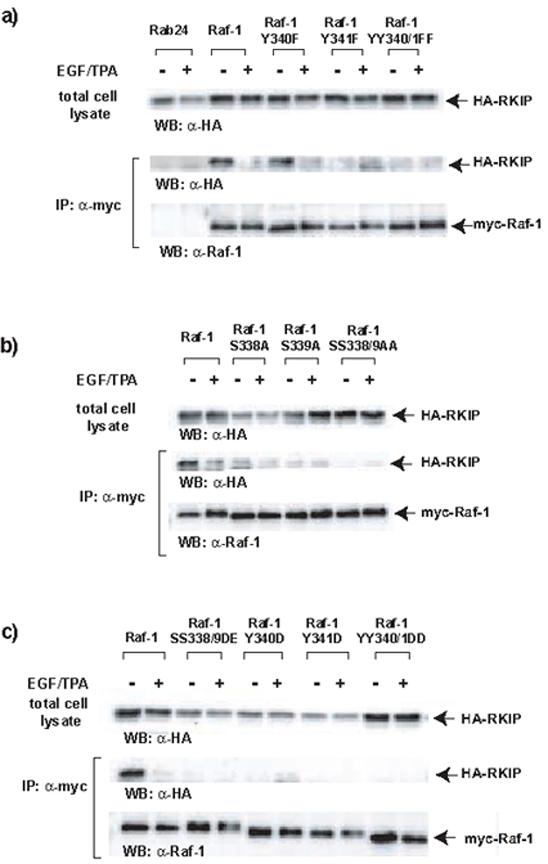
Ser338-339 and Tyr341 in the N-region of Raf-1 kinase domain are important for RKIP binding. (a,b) Conservative substitutions in the N-region drastically reduced the affinity of Raf-1 for RKIP in quiescent cells. COS-1 cells were cotransfected with HA-RKIP and different myc-Raf-1 mutant constructs as indicated. Myc-tagged Rab24 was used as a co-immunoprecipitation control. Twenty-four hours post-transfection cells were serum starved for 24 h and stimulated with EGF plus TPA for 10 minutes. Lysates were immunoprecipitated (IP) with the anti-myc tag monoclonal antibody 9E10 and associated HA-RKIP proteins were detected by Western blotting. (c) Phosphomimetic substitutions in the N-region also decrease the affinity of Raf-1 for RKIP. COS-1 cells were cotransfected with HA-RKIP and different myRaf-1 constructs mutant as indicated. Cells were treated and lysates were immunoprecipitated with 9E10 ant-myc antibody and examined as above. The results shown are representative of at least three independent experiments.
Therefore, we investigated the role of Raf-1 N-region phosphorylation for RKIP binding in more detail. We performed binding experiments with synthetic peptides representing this region in Raf-1 (aa 331-349), determining the equilibrium binding affinities as well as association and dissociation rates by plasmon surface resonance (BIAcore). To assess the effects of phosphorylation, phospho-amino acids were incorporated at the crucial positions in various combinations. The Raf-1 peptides were immobilized on the chip and assayed for binding of recombinant purified RKIP protein (Table 1 and Supplementary Fig. 4). The non-phosphorylated Raf-1 peptide bound to RKIP with low affinity (KD=3.6x10-3) and fast association and dissociation kinetics indicating that RKIP binding to the unphosphorylated N-region is unstable and turns over fast. Phosphorylation of Ser338 and Ser339 slowed down the association rate, but decreased the dissociation rate even more, resulting in an approximately 6-fold net enhancement of the equilibrium binding. The phosphorylation of Tyr340/1 decreased both the association and dissociation rates compared to the non phosphorylated peptide, resulting in an overall increase of equilibrium binding similar to the peptide phosphorylated at Ser338/9. The phosphorylation of all four residues cooperated to further enhance the association rate and reduce the dissociation rate leading to a ~30 fold gain in equilibrium binding. Interestingly, omitting the phosphate at Tyr340 further increased the binding of the triple phosphorylated peptide. These data support the notion that Tyr340 may not be a physiological phosphorylation site [16], and that the phosphomimetic replacement at this site behaved different from the phosphomimetic replacements at the other three sites. Taken together these data suggest that the phosphorylation of the N-region actually provides a docking signal for RKIP binding rather than a dissociation signal.
Table 1.
Kinetic analysis of RKIP binding to immobilized Raf-1 peptides by surface plasmon resonance. Binding was performed at 25°C as described in Materials and Methods. Shown are the affinity (KD), association (ka), and dissociation constants (kd) with their standard errors (SE) and chi2 values; RU, resonance units.
| Peptide | ka (1/Ms) | SE(ka) | kd (1/s) | SE(kd) | RUmax | SE (Rmax) | KD | chi2 |
|---|---|---|---|---|---|---|---|---|
| NR1 SSYY | 46.1 | 3.98 | 0.168 | 2.56×10-3 | 3.2×10-3 | 258 | 3.62×10-3 | 0.281 |
| NR2 pSpSYY | 16.8 | 1.52 | 9.71×10-3 | 2.82×10-5 | 242 | 16.8 | 5.77×10-4 | 0.873 |
| NR3 SSpYpY | 23.5 | 2.87 | 0.0127 | 2.65×10-4 | 206 | 21.6 | 5.42×10-4 | 0.187 |
| NR4 pSpSpYpY | 321 | 4.33 | 3.25 ×10-3 | 2.00×10-5 | 302 | 3.48 | 1.01×10-5 | 0.397 |
| NR5 pSpSYpY | 2218 | 54 | 0.04518 | 3.3 ×10-4 | 105.6 | 2.0 | 2.04×10-5 | 0.366 |
Using the Biacore T-100 we also undertook a detailed thermodynamic analysis of the transitional state thermodynamics of the binding of the triple phosphorylated peptide NR5 (pSer338-pSer339-Tyr-pTyr341) (Table 2). The results suggested that the interaction between RKIP and NR5 is largely enthalpically driven, as the enthalpy change (ΔH) for the interaction is -87.91, whereas the entropy change is unfavorable (TΔS = -62.45). However, the enthalpic favorability of the interaction outweighs the entropic penalty of the interaction, giving a negative Gibbs free energy value, ΔG= -25.46 (ΔG= ΔH-TΔS), which drives the association. Interestingly, these measurements indicated that heat is given out during the transition state, and that for the complex to form a very specific orientation of the molecules may be needed, which is highly ordered and not favored by the rather high negative entropic value ΔS. Thus, these data suggested that steric constraints operate at the binding surface.
Table 2.
Thermodynamic analysis of Raf-1 peptide NR5 (pS338-pS339-Y-pY341) binding. Binding of RKIP to the immobilized NR5 peptide was performed at the indicated temperatures. Shown are the affinity (KD), association (ka), and dissociation constants (kd) with their standard errors (SE) and chi2 values; RU, resonance units. SD, standard deviation; H, enthalpy; S, entropy; T, absolute temperature (Kelvin); G, Gibb’s free energy.
| NR5 | ka (1/Ms) | SE(ka) | kd (1/s) | SE(kd) | RUmax | SE(Rmax) | KD | Chi2 |
|---|---|---|---|---|---|---|---|---|
| 37°C | 219.5 | 21 | 0.03911 | 5.2×10-4 | 203.0 | 19 | 1.782×10-4 | 2.34 |
| 25°C | 2218 | 54 | 0.04518 | 3.3 ×10-4 | 105.6 | 2.0 | 2.04×10-5 | 0.366 |
| 15°C | 2007 | 11 | 0.02496 | 5.8 ×10-4 | 158.2 | 0.62 | 1.24×10-5 | 0.179 |
| 8°C | 4142 | 17 | 0.01765 | 4.1 ×10-5 | 112.1 | 0.27 | 4.26×10-6 | 0.329 |
| Parameter Name | Parameter Value | SD |
|---|---|---|
| ΔH° [kJ/mol] | -87.91 | 14.81 |
| ΔS° [J/(K*mol)] | -209.5 | 50.42 |
| TΔS° [kJ/mol] | -62.45 | 15.03 |
| ΔG [kJ/mol] at 25°C | -25.46 | 0.2193 |
Therefore, we fine mapped the RKIP binding site in the N-region using peptide arrays (Fig. 5). 23-mer peptides offset by four amino acids spanning the N-region plus the upstream and downstream sequences were immobilized on nitrocellulose filters and probed with [35S]-labeled RKIP protein produced by coupled in vitro transcription/translation. The results show that RKIP binds upstream and downstream of the Ser338Ser339Tyr340Tyr341 sequence much like a clamp (Fig. 5). The binding to the Ser338Ser339Tyr340Tyr341 sequence itself is below the detection threshold of the peptide array filter assay, however could be clearly detected in the Biacore experiments showing that the peptide array possesses lower sensitivity than the Biacore. These results suggest a binding epitope that consists of flanking sequences that exhibit constitutive binding and a central core where binding is regulated by phosphorylation and actually requires phosphate residues for efficient docking. Thus, taken together these data suggest that RKIP binds to the N-region of Raf-1 with rather loose affinity and high turnover rates. Phosphorylation of Ser338/9 and Tyr341 raises the affinity of the N-region for RKIP providing a binding signal that may recruit RKIP after dissociation from Raf-1.
Figure 5.
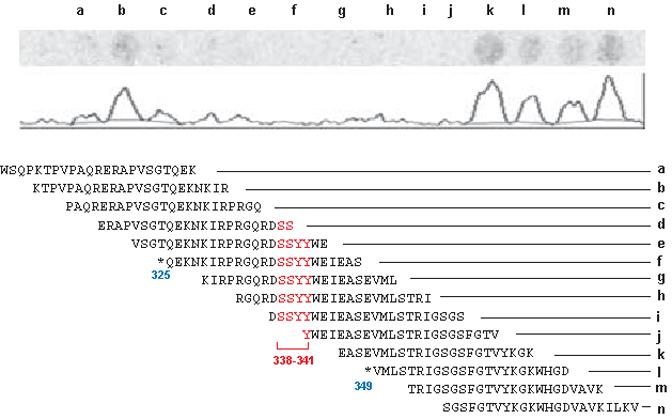
Peptide array mapping of RKIP binding to the N-region of Raf-1. A peptide array was probed with [35S]-methionine labeled RKIP. The autoradiograph and quantitation trace are shown above the peptide sequences. Serines338/339 and tyrosines340/341 are shown in red, and the start and end points of selected deletion constructs from Fig. 3 are indicated by blue arrows.
This model would predict that phosphorylation of the N-region still should persist when RKIP re-binds to Raf-1. Therefore, we monitored RKIP binding and S338 phosphorylation over a time course of TPA stimulation (Fig. 6a). Y341 phosphorylation could not be examined because the sensitivity of this phosphospecific antibody was too low to detect proteins expressed in cells. After TPA stimulation RKIP dissociated when S338 phosphorylation was rising. RKIP rebinding ensued while S338 phosphorylation was still high. To directly test whether S338 phosphorylated Raf-1 can bind to RKIP during later phases of growth factor stimulation we examined S338 phosphorylation of RKIP bound Raf-1 over a timecourse of EGF stimulation (Fig. 6b). Raf-1 phosphorylated on S338 was readily detectable in RKIP immunoprecipitates during later phases of stimulation showing that S338 phosphorylation is compatible with RKIP binding.
Figure 6.
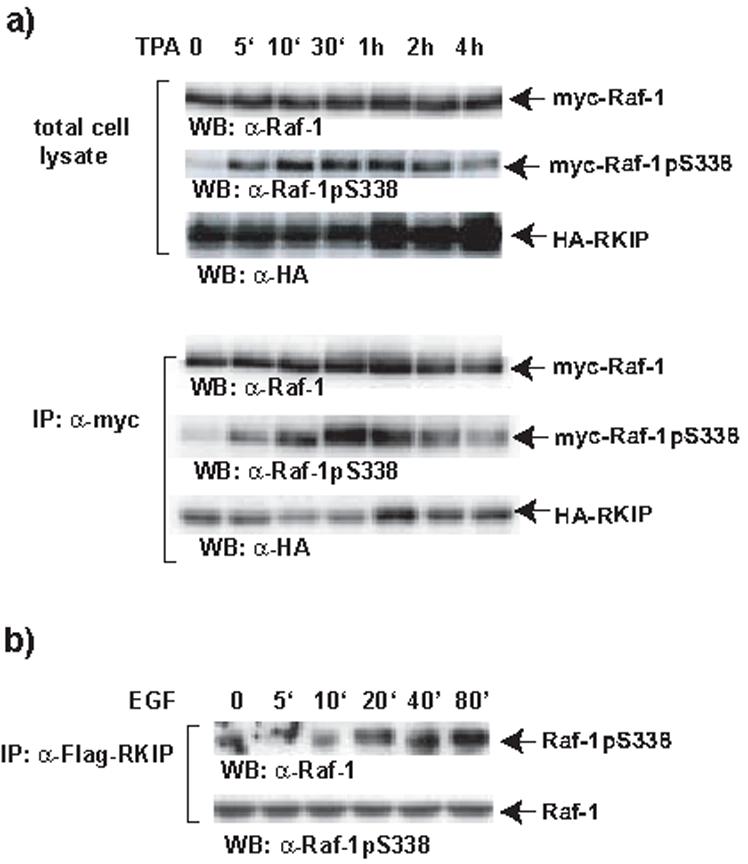
Kinetic analysis of Raf-1 S338 phosphorylation and RKIP rebinding. (a) HA-RKIP and myc-Raf-1 and were co-expressed in COS-1 cells. Cells were stimulated with TPA for different time points as indicated. Myc-Raf-1 immunoprecipitates were examined for S338 phosphorylation by blotting with a phospho-specific antibody, and HA-RKIP binding using anti-HA antibody. (b) MCF-7 cells were transfected with FLAG-RKIP and GFP-Raf-1. Serum starved cells were stimulated with 100 ng/ml EGF for the timepoints indicated. RKIP was immunoprecipitated and co-precipitated Raf-1 adjusted for equal loading was blotted with anti-phospho-S338 and Raf-1 antibodies.
These data are consistent with the hypothesis that the initial dissociation of RKIP is caused by mitogen induced phosphorylation of RKIP [15], but that the rebinding of RKIP involves phosphorylation of the Raf-1 N-region. This working hypothesis is difficult to prove directly. A mutational approach is inconclusive, as it prevents the association - dissociation - re-association cycle. For instance, mutation of RKIP phosphorylation sites was reported to prevent the initial RKIP dissociation from Raf-1 [15], so re-association cannot be measured; and mutation of N-region phosphorylation sites prevents RKIP binding and hence dissociation cannot be measured. Thus, experimental proof hinges on the correlation of biochemical kinetic data such as we have presented here. Our working model makes predictions that open new avenues of research and also will be valuable to validate the model. For instance, RKIP rebinding induced by N-region phosphorylation presumably will protect these sites from dephosphorylation and hence should prolong Raf-1 activation. However, this may be futile as the N-region also is one part of the bipartite MEK-binding site identified in the Raf-1 kinase domain [12]. Thus, binding of RKIP will also dissociate Raf-1 from its substrate and thereby terminate signaling. An attractive speculation is that RKIP may redirect Raf-1 from MEK to an alternative substrate. The existence of alternative Raf-1 effectors is suggested by multiple lines of evidence including the phenotype of Raf-1 knockout mice [3] and the recent discovery of new MEK independent apoptosis pathways controlled by Raf-1 [17] although the latter does not depend on Raf-1 kinase activity. Given the known complexity of Raf-1 regulation [2,18] it may not surprise that regulation or Raf-1 by RKIP also turns out to be more complicated and involve multiple layers.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Richard Marais and William Maltese for plasmid constructs. We also thank David Dignam for stimulating discussions. This work was supported by NIH grants to KCY (R01 GM64767) and ZL (R01 GM057959). We also acknowledge the funding support to WK by the European Community as part of the FP6 COSBICS Project (512060) and Cancer Research UK. We thank John Butler from Biacore for valuable advice and giving us the opportunity to perform measurements on the new Biacore T100 instrument.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19:2435–44. doi: 10.1128/mcb.19.4.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Neill E, Kolch W. Conferring specificity on the ubiquitous Raf/MEK signalling pathway. Br J Cancer. 2004;90:283–8. doi: 10.1038/sj.bjc.6601488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–85. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 4.Yeung K, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 5.Yeung K, et al. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–85. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odabaei G, et al. Raf-1 kinase inhibitor protein: structure, function, regulation of cell signaling, and pivotal role in apoptosis. Adv Cancer Res. 2004;91:169–200. doi: 10.1016/S0065-230X(04)91005-6. [DOI] [PubMed] [Google Scholar]
- 7.Trakul N, Rosner MR. Modulation of the MAP kinase signaling cascade by Raf kinase inhibitory protein. Cell Res. 2005;15:19–23. doi: 10.1038/sj.cr.7290258. [DOI] [PubMed] [Google Scholar]
- 8.Park S, Yeung ML, Beach S, Shields JM, Yeung KC. RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene. 2005 doi: 10.1038/sj.onc.1208435. [DOI] [PubMed] [Google Scholar]
- 9.Yeung KC, et al. Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol. 2001;21:7207–17. doi: 10.1128/MCB.21.21.7207-7217.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426:574–9. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- 11.Trakul N, Menard RE, Schade GR, Qian Z, Rosner MR. Raf kinase inhibitory protein regulates Raf-1 but not B-Raf kinase activation. J Biol Chem. 2005;280:24931–24940. doi: 10.1074/jbc.M413929200. [DOI] [PubMed] [Google Scholar]
- 12.Xiang X, Zang M, Waelde CA, Wen R, Luo Z. Phosphorylation of 338SSYY341 regulates specific interaction between Raf-1 and MEK1. J Biol Chem. 2002;277:44996–5003. doi: 10.1074/jbc.M203953200. [DOI] [PubMed] [Google Scholar]
- 13.Hafner S, et al. Mechanism of inhibition of Raf-1 by protein kinase A. Mol Cell Biol. 1994;14:6696–703. doi: 10.1128/mcb.14.10.6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heidecker G, et al. Mutational activation of c-raf-1 and definition of the minimal transforming sequence. Mol Cell Biol. 1990;10:2503–12. doi: 10.1128/mcb.10.6.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corbit KC, et al. Activation of Raf-1 signaling by protein kinase C through a mechanism involving Raf kinase inhibitory protein. J Biol Chem. 2003;278:13061–8. doi: 10.1074/jbc.M210015200. [DOI] [PubMed] [Google Scholar]
- 16.Morrison DK, Heidecker G, Rapp UR, Copeland TD. Identification of the major phosphorylation sites of the Raf-1 kinase. J Biol Chem. 1993;268:17309–16. [PubMed] [Google Scholar]
- 17.O’Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 2004;306:2267–70. doi: 10.1126/science.1103233. [DOI] [PubMed] [Google Scholar]
- 18.Dhillon AS, Kolch W. Untying the regulation of the Raf-1 kinase. Arch Biochem Biophys. 2002;404:3–9. doi: 10.1016/s0003-9861(02)00244-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.