

Abstract
The term apoptosis often has been used interchangeably with the term programmed cell death. Here we describe a form of programmed cell death that is distinct from apoptosis by the criteria of morphology, biochemistry, and response to apoptosis inhibitors. Morphologically, this alternative form of programmed cell death appears during development and in some cases of neurodegeneration. Despite its lack of response to caspase inhibitors and Bcl-xL, we show that this form of cell death is driven by an alternative caspase-9 activity that is Apaf-1-independent. Characterization of this alternative form of programmed cell death should lead to new insight into cell death programs and their roles in development and degeneration.
The original definition of apoptosis as a form of cell death distinct from necrosis was based on morphological criteria (1). Genetic studies in Caenorhabditis elegans led to the identification of key components of the apoptotic cell death pathway, including the protease Ced-3 (2–5). However, it has become clear that the morphological descriptions of apoptosis and necrosis are not adequate to describe all cell deaths; examples that do not conform to the criteria for either of these forms of cell death include: (i) certain developmental cell deaths, such as autophagic cell death (6–10) and cytoplasmic cell death (7, 9, 11–14); (ii) some neurodegenerative cell deaths, such as those in a transgenic mouse model of amyotrophic lateral sclerosis (15); and (iii) some ischemia-related cell deaths featuring cell swelling, referred to as oncosis (16).
The biochemical basis for these alternative morphological forms of cell death remains unknown. Understanding the mechanisms for these forms has implications for the understanding of evolutionary aspects of cell death programs, developmental cell death, neurodegeneration, and cancer therapeutics and for the design of novel therapeutic agents for diseases featuring these alternative forms of cell death.
Here we describe a form of nonapoptotic programmed cell death (paraptosis, from para = next to or related to, and apoptosis) that fails to fulfill the criteria for apoptosis (Table 1). We also show that this form of cell death is programmatic, because it requires gene expression. Despite its lack of response to caspase inhibitors and Bcl-xL, however, this alternative form of cell death nonetheless is inhibited by a catalytic mutant of caspase-9 zymogen. Furthermore, in complementary studies we show that the expression of caspase-9 zymogen leads to both apoptotic and nonapoptotic programmed cell death; block of the apoptotic arm of this response by caspase inhibitors (17) converts the cell death to the nonapoptotic form. Finally, the alternative, nonapoptotic pathway mediated by caspase-9 is Apaf-1-independent and is not inhibited by mutation of the sites of zymogen processing to the apoptotically active forms (D315 and D330).
Table 1.
Comparison of apoptosis, necrosis and paraptosis
| Apoptosis | Necrosis | Paraptosis | |
|---|---|---|---|
| Morphology | |||
| Nuclear fragmentation | + | − | − |
| Chromatin condensation | + | − | ± |
| Apoptotic bodies | + | − | − |
| Cytoplasmic vacuolation | − | + | + |
| Mitochondrial swelling | Sometimes | + | Late |
| Genomic effect | |||
| TUNEL | + | Usually − | − |
| Internucleosomal DNA fragmentation | + | − | − |
| Caspase activity | |||
| DEVD-cleaving activity | + | − | − |
| Caspase-3 processing | + | − | − |
| PARP cleavage | + (85-kDa fragment) | + (50- to 62-kDa fragments) | − |
| Inhibition by: | |||
| zVAD.fmk | + | − | − |
| BAF | + | − | − |
| p35 | + | − | − |
| xiap | + | − | − |
| Bcl-xL | + | Usually − | − |
| Actinomycin D | Sometimes | − | + |
| Cycloheximide | Sometimes | − | + |
DEVD, Asp-Glu-Val-Asp; PARP, poly(ADP-ribose) polymerase.
Methods
Cells and Culture Conditions.
293T cells and Apaf-1 null mouse embryonic fibroblasts were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin. Transient transfections used Lipofectamine (GIBCO/BRL) according to the manufacturer's instructions. A total of 1 × 106 293T cells or 2 × 105 Apaf-1 null cells were seeded in 6-cm dishes and transfected the next day by using a ratio of DNA/Lipofectamine of 1 μg/5 μl. Transfection efficiency was 60–80% for 293T and 10–20% for the Apaf-1 KO cells, determined by 5-bromo-4-chloro-3-indolyl β-d-galactoside staining after transfection of a β-galactosidase construct.
T-butyloxycarbonyl-Asp(O-methyl)-fluoromethyl ketone (BAF) and benzyloxycarbonyl-valyl-alanyl-aspartyl fluoromethyl ketone (zVAD.fmk) were obtained from Enzyme Systems Products (Livermore, CA). Actinomycin D and cycloheximide were from Sigma.
Constructs.
The human insulin-like growth factor I receptor (IGFIR) β subunit intracellular domain was subcloned from a NT2 (human teratocarcinoma) cDNA library by PCR amplification and inserted into pcDNA3 (Invitrogen). Fifteen amino acids containing the Src myristylation signal sequence were incorporated into the construct.
Mutants of IGFIR intracellular domain (IGFIR-IC) were obtained by QuikChange mutagenesis (Stratagene) from the wild-type construct. The sequences of all constructs were confirmed, and Western blot analyses were performed to verify protein expression.
Assessment of Cell Death and Apoptosis.
Cell death was assessed by trypan blue exclusion. Floating cells were collected at the indicated times after transfection, by centrifugation for 10 min at 1,500 rpm. The cell pellet was resuspended in 50 μl of PBS, to which 50 μl of trypan blue was added. Dead cells were counted by four independent hemocytometer counts.
For the Apaf-1 KO cells, both floating and adherent cells were collected by trypsinization, and the cell death was determined as the percentage of dead cells over the total number of cells.
For the assessment of cell death after treatment with actinomycin D and cycloheximide, 293T cells were transfected as described above; 8.5 h after the beginning of transfection the drugs were added at the indicated concentrations and subsequently removed after 12 h, at which time the cells were fed with fresh medium. Floating cells were collected 24 h after the removal of the drugs, and dead cells were counted in the presence of trypan blue.
CPP32 (caspase-3)-like activity was determined by using the ApoAlert Caspase-3 Assay Kit (CLONTECH), following the manufacturer's instructions.
Terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) staining was performed by using the Apoptosis Detection System (Fluorescein) from Promega, following the manufacturer's instructions.
Oligonucleosomal DNA fragmentation was assessed by extraction of soluble DNA from 3 × 106 transfected cells, by the method of Frisch and Francis (18).
Coimmunoprecipitation Assay.
Cell lysis and immunoprecipitation were performed as described in Ye et al. (19).
Electron Microscopy.
Samples for electron microscopy were prepared as described (20) and examined on a Hitachi 600 electron microscope.
Results
The IGFIR Induces Cell Death.
During studies of dependence receptors (21) [i.e., receptors such as the common neurotrophin receptor (22), the androgen receptor (23), and DCC (deleted in colorectal cancer) (24) that induce cell death unless blocked by ligand binding], we observed that the expression of the IGFIR induced cell death that did not fit the criteria for apoptosis (Table 1 and Figs. 1 and 2).
Figure 1.

The IGFIR-IC induces cell death. (A) Comparison between the cell death induced by pIGFIR-IC and nonmyristylated form of the IC domain and deletion mutants. Asterisks indicate a highly significant difference from the control as determined by one-way ANOVA (F = 63.3; P < 0.0001); P < 0.0001 for pIGFIR-IC and pIGFIR-ICΔ1264–1276, Bonferroni/Dunn posthoc test. Error bars represent the SEM from three independent experiments. (B) Light microscopy, using Hoffman optics, demonstrating morphological changes induced by IGFIR-IC. IGFIR-IC expression caused a rounding of the cells and intracellular vacuoles. (C) Ultrastructural characteristics of IGFIR-induced cell death. 293T cells transfected with control empty vector pcDNA3 (a), pC9 (b), or pIGFIR-IC (c and d). Although caspase-9-transfected cells displayed characteristic features of apoptosis, including chromatin condensation, IGFIR-IC-transfected cells did not. In contrast, extensive cytoplasmic vacuolization was observed in the absence of nuclear fragmentation, cellular blebbing, or apoptotic body formation. Note that the cell shown in b displays both the chromatin condensation characteristic of apoptosis and membrane disruption characteristic of necrosis, suggesting secondary necrosis after apoptosis. (Original magnification: ×6,000.)
Figure 2.

IGFIR-IC-induced cell death is nonapoptotic. (A) Internucleosomal DNA fragmentation of cells transfected as in Fig 1. Soluble DNA was extracted from cells 24 or 48 h after transfection. Whereas the Bax-transfected cells demonstrated internucleosomal DNA fragments, the chromatin in IGFIR-IC transfected cells remained intact at 24 and 48 h after transfection. (B) TUNEL staining of cells transfected as in A. Cells were assayed 48 h after transfection. (Upper) FITC staining of the TUNEL labeling. Bax, but not IGFIR-IC, transfected cells show TUNEL-positive staining. (Lower) The same fields of cells stained with propidium iodide (PI). Cells were permeabilized before staining. Similar results were obtained at 24 h. (C) Transcription and protein synthesis are required for IGFIR-induced cell death. Actinomycin D (Act.D) or cycloheximide (CHX) (numbers represent μg/ml) were added 8.5 h after the transfections, and the medium was replaced after 12 h with fresh medium without drugs. Floating cells were collected after 24 h and counted in the presence of trypan blue. Control samples were treated with solvent only (ethanol 0.05%). The expression of IGFIR-IC at the time of administration of the drugs was comparable to the expression at 24 h, as assessed by Western blot analysis. The error bars represent the SEM of four independent experiments. Within the samples transfected with pIGFIR-IC, the asterisks indicate a highly significant difference (P = 0.0011 for 0.25 μg/ml CHX, and P < 0.0001 for 0.5 μg/ml CHX, and 0.5 μg/ml and 1 μg/ml Act.D; Bonferroni/Dunn posthoc test) from the pIGFIR-IC transfected sample treated with solvent only (EtOH), as determined by one-way ANOVA (F = 20.727; P < 0.0001).
This was observed after expression of IGFIR (the IC domain was used to prevent ligand-induced signaling) in 293T cells (Figs. 1 and 2), 293, MCF-7, Cos-7, and primary cultures of mouse embryonic fibroblasts (data not shown), demonstrating that the ability of IGFIR to induce this alternate type of cell death was not restricted to a specific cell type. The overexpression of the full-length IGFIR in 293 cells also induced cell death reproducing the same morphology, although the expression of the IC domain alone was a stronger inducer of cell death (not shown).
IGFIR initially was evaluated as a potential dependence receptor because its targeted disruption in transgenic mice was shown to lead to an increase in neuronal density in the spinal cord and brainstem (25) and because its overexpression was shown to inhibit tumor formation and induce cell death (26–28).
Membrane targeting was required for cell death induction by IGFIR, because omission of the myristylation site in the IGFIR-IC construct destroyed its ability to induce cell death (Fig. 1A). To exclude the possibility that the cell death induction was a nonspecific effect of the overexpression of a membrane-targeted protein, we performed mutagenic analyses. The presence of both the kinase domain and noncatalytic carboxyl terminus proved to be necessary to induce cell death (Fig. 1A). These results suggested that the cell death was a specific effect of membrane-targeted IGFIR.
IGFIR-Induced Cell Death Is Nonapoptotic.
Ultrastructurally, cell death induced by IGFIR-IC in 293T cells lacked the features of apoptosis, including nuclear fragmentation, apoptotic body formation, and chromatin condensation (Fig. 1C). Instead of these characteristics, cytoplasmic vacuolation was observed (Fig. 1 B and C). Vacuoles were derived predominantly from the endoplasmic reticulum, but mitochondrial swelling also was observed. Autophagic vacuoles were not observed.
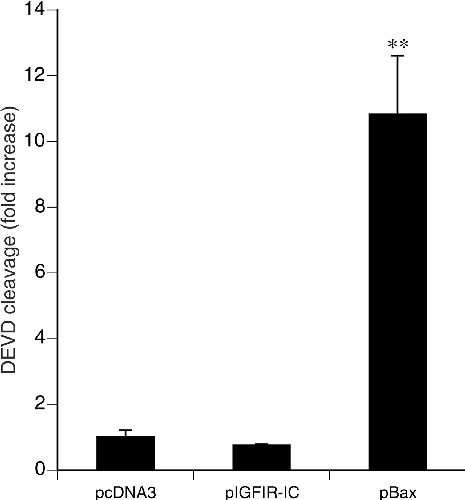
Caspase enzymatic assays showed that, whereas the proapoptotic protein Bax induced cell death with associated caspase activation, IGFIR-IC induced cell death without associated caspase activation (Fig. 5, which is published as supplemental data on the PNAS web site, www.pnas.org). Similar results were obtained with immunoblot assays for caspase-3 cleavage (not shown).
One of the defining features of apoptotic cell death is the fragmentation of chromatin (29, 30). We examined genome integrity by agarose gel electrophoresis and observed fragments suggestive of internucleosomal DNA cleavage in the control cells transfected with a Bax expression construct, but not in the IGFIR-IC-transfected cells (Fig. 2A). We also performed TUNEL staining of cells transfected with either IGFIR-IC or Bax. The Bax-transfected cells were labeled in the TUNEL assay, but those transfected with IGFIR-IC were not (Fig. 2B).
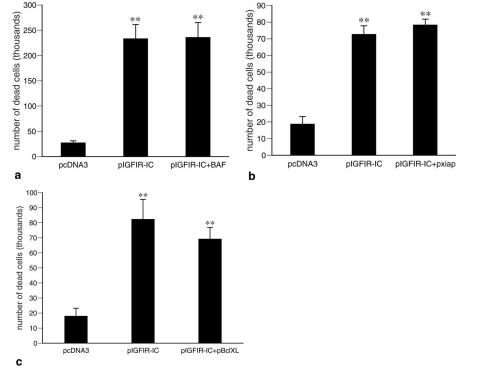
Results obtained with apoptosis inhibitors supported the ultrastructural and biochemical results described above: neither the caspase inhibitors zVAD.fmk, BAF, p35, and X-chromosome-linked inhibitor of apoptosis (xiap), nor the Bcl-2 family member Bcl-xL inhibited 293T cell death induced by IGFIR-IC (Table 1, and supplemental Fig. 6).
Cell Death Induced by IGFIR Requires Protein Synthesis.
In some paradigms de novo protein synthesis is required for programmed cell death. As shown in Fig. 2C, actinomycin D restored cell viability to control levels after transfection with IGFIR-IC in 293T cells. Furthermore, treatment with cycloheximide induced a dose-dependent block of IGFIR-induced cell death. These results suggest that IGFIR-driven cell death requires transcription and translation. Further support that this phenotype is distinct from necrosis was provided by a difference in cleavage of poly(ADP-ribose) polymerase (PARP), which results in 50- to 62-kDa fragments in necrosis (31) but not in paraptosis (Table 1), and by the finding that oligomycin (100 ng/ml), which blocks Bax-induced apoptosis (32), blocked the phenotype (data not shown).
Caspase-9 Mediates IGFIR-Induced Cell Death.
Paradoxically, despite the lack of effect of caspase inhibitors on nonapoptotic programmed cell death induced by IGFIR, a catalytic mutant of caspase-9 zymogen, which acts as a dominant negative form of pro-caspase-9 (5, 33–35), blocked IGFIR-IC-induced cell death (Fig. 3A). This effect was specific for caspase-9, because similar forms of caspases-2, -3, -6, -7, and -8 had no effect (Fig. 3 A and B, and data not shown). Caspase-9 also coimmunoprecipitated with IGFIR-IC (Fig. 3C), suggesting that the recruitment of caspase-9 by IGFIR may trigger the cell death stimulus.
Figure 3.

IGFIR-IC-induced cell death is blocked by a catalytic mutant of pro-caspase-9. (A and B) 293T cells were transfected with 2 μg of DNA consisting of either pcDNA3, 0.5 μg pIGFR-IC + 1.5 μg pcDNA3, or 0.5 μg pIGFR-IC + 1.5 μg dominant negative caspase-9 (pC9DN, in A) or dominant negative caspase-7 (pC7DN, in B) expression constructs. Forty-eight hours after transfection, dead cells were counted. Asterisks denote a highly significant difference from the control as determined by one-way ANOVA. In A, F = 25.2, P = 0.0002 with P = 0.0001 for pIGFIR-IC (Bonferroni/Dunn posthoc test). In B, F = 118.487, P < 0.0001, with P < 0.0001 for pIGFIR-IC and pIGFIR-IC + pC7DN (Bonferroni/Dunn posthoc test). Error bars represent the SEM from three independent experiments. (C) Protein extracts from cells cotransfected with pIGFR-IC wild type or mutants, as indicated, and FLAG-tagged C9DN expression constructs were subjected to immunoprecipitation (IP) using an anti-IGFIR polyclonal antibody. Immunoprecipitates then were resolved by 15% SDS/PAGE, and Western blotting was performed by using a mAb against the FLAG epitope to detect caspase-9. WCE = whole-cell extract.
Consistent with the possibility for a functional role of caspase-9 in this alternative form of cell death, neither the IGFIR-IC construct missing the membrane recruitment signal, nor the truncated forms previously described that failed to induce cell death, coimmunoprecipitated with caspase-9 (Fig. 3C), whereas the deletion mutant in the C terminus (Δ1264–1276), which retained cell death-promoting activity, also coimmunoprecipitated with caspase-9.
Caspase-9 Induces Both Apoptosis and Nonapoptotic Cell Death.
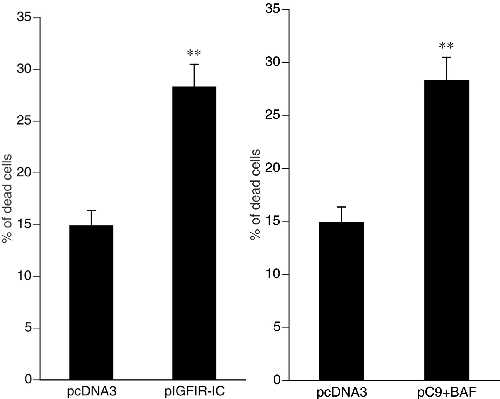
The results obtained with expression studies of IGFIR-IC implied that caspase-9 (or caspase-9 zymogen) mediates both an apoptotic and a nonapoptotic form of programmed cell death. To test this hypothesis, we expressed caspase-9 (in the absence of IGFIR-IC expression) and found that both apoptosis and nonapoptotic programmed cell death were induced (Fig. 4), with apoptosis representing approximately 80–90% of the cell death. However, when apoptosis was inhibited by BAF, virtually all of the apoptosis was eliminated, revealing the nonapoptotic cell death more clearly (Fig. 4 A and B). Furthermore, all of the features of cell death induced by IGFIR-IC were reproduced by the expression of caspase-9 in the presence of BAF (Fig. 4 and Table 1). In contrast, cell death induced by the expression of caspase-7 was completely eliminated by BAF (Fig. 4A).
Figure 4.

Caspase-9 induces both apoptosis and nonapoptotic cell death. (A) 293T cells were transfected with the indicated pcDNA3-based caspase zymogen expression constructs. The general caspase inhibitor BAF was added at 50 μM final concentration. Cell death was assayed 48 h after transfection. Although BAF was able to inhibit caspase-7-induced cell death completely, it only inhibited caspase-9-induced cell death partially (30%). Asterisks indicate a highly significant difference from the control as determined by one-way ANOVA analysis (F = 34.624, P < 0.0001), P < 0.0001 for pC9, P = 0.0002 for pC7, and P = 0.0001 for pC9 + BAF (Bonferroni/Dunn posthoc test). Error bars represent the SEM from three independent experiments. (B) Light microscopy of 293T cells transfected with control vector (pcDNA3), pro-caspase-9 alone (pC9), or in the presence of 50 μM BAF (pC9 + BAF). Cells were photographed 24 h after transfection. Expression of pro-caspase-9 resulted in the induction of a typical apoptotic phenotype, including cell shrinkage and the formation of apoptotic bodies (Middle). BAF suppressed the apoptotic phenotype, but cells displayed a vacuolated morphology similar to that observed with IGFIR-IC-induced cell death (Bottom, arrowheads). (C) Internucleosomal DNA fragmentation of 293T cells transfected with the indicated pro-caspase expression constructs alone or in the presence of 50 μM BAF. Internucleosomal DNA fragmentation was completely inhibited by BAF. (D) TUNEL staining of 293T cells 48 h after transfection with the indicated constructs. Cells were stained as described in Fig. 2B. (E) Electron micrograph of 293T cells transfected with an expression construct for pro-caspase-9 in the presence of zVAD.fmk (40 μM). Morphology similar to IGFIR-IC-induced cell death was observed, including cytoplasmic vacuolation and the absence of chromatin condensation, nuclear fragmentation, and apoptotic body formation. (Original magnification: ×6,000.)
In addition to separating the proapoptotic and nonapoptotic effects of caspase-9 by adding BAF, these effects could be distinguished by expression in Apaf-1 null cells [which have been shown previously to be resistant to a variety of apoptotic stimuli (36)], in which case cell death was induced both by IGFIR-IC and caspase-9 expression in the presence of BAF (supplemental Fig. 7). The morphological features of nonapoptotic cell death described above also were reproduced in Apaf-1 null cells.
A third distinction between the proapoptotic and nonapoptotic effects of caspase-9 occurred with mutation of the zymogen processing sites within caspase-9 zymogen, D315 and D330. These mutations, which block processing of caspase-9, reduced the percentage of dead cells caused by apoptosis from 85 ± 6% in the wild type to 36 ± 8% in the double mutant, with a concomitant increase in nonapoptotic cell death.
Discussion
Despite the widespread occurrence of apoptosis in physiological and pathological cell death, the occurrence of cell deaths that fulfill neither the criteria for apoptosis nor necrosis has been well documented. However, the biochemical mechanisms involved in these alternative forms of cell death are unknown. Therefore, modulation of the apoptotic pathway genetically or pharmacologically may prove ineffective in situations in which nonapoptotic cell death occurs, such as in at least some cases of neurodegeneration (15).
The studies described here demonstrate that IGFIR, as well as the IGFIR intracytoplasmic domain, induces a form of nonapoptotic programmed cell death characterized by cytoplasmic vacuolation and resistance to apoptosis inhibitors. This form of cell death requires transcription and de novo protein synthesis. Preliminary results from microarray screening comparing gene expression profiles between IGFIR-induced cell death and apoptotic cell death were consistent with distinct cell death programs. Among the 7,075 genes evaluated, fewer than 2% of those differentially expressed were found to be shared between the two cell death programs (two of 116) (S. Castro-Obregon, G. del Rio, M. Eshoo, and D. B., unpublished results).
Induction of cell death by IGFIR has been documented previously: overexpression of fragments of the IGFIR-IC in cancer cells was shown to reduce tumorigenicity in nude mice and to induce cell death (27, 37). Furthermore, the expression of IGFIR was decreased in prostate cancer (38), and its reexpression in immortalized human prostate cells inhibited the malignant phenotype (26). A potential role for IGFIR in developmental cell death was suggested by the phenotype of IGFIR-null mice, which includes a higher neuronal density in the brainstem and spinal cord (25).
We have shown here that IGFIR may induce a form of programmed cell death with features distinctive from apoptosis. This form of programmed cell death may have been assumed to be necrosis in some cases because of its morphological features of cytoplasmic vacuolation and mitochondrial swelling, along with the lack of effect of caspase inhibitors. We have dubbed this previously uncharacterized form of cell death paraptosis.
The current studies show that paraptosis induced by IGFIR is mediated by caspase-9, despite the initially paradoxical finding that caspase inhibitors failed to inhibit the process. Mutants of IGFIR that failed to coimmunoprecipitate with caspase-9 also failed to induce paraptosis; furthermore, a catalytic mutant of caspase-9 that functions as a dominant negative caspase-9 blocked this nonapoptotic cell death induction. Finally, the expression of caspase-9 in the presence of the apoptosis inhibitor BAF (or zVAD.fmk) induced nonapoptotic cell death that was indistinguishable from that induced by IGFIR.
Although the only known function of caspase-9 to date has been as an inducer of apoptosis via cleavage and activation of caspase-3 zymogen (and possibly other caspase zymogens such as caspase-7), we found that the proapoptotic and the proparaptotic effects of caspase-9 could be distinguished by the lack of effect of caspase inhibitors (BAF, zVAD.fmk, p35, and xiap), lack of requirement for Apaf-1, and lack of suppression by mutation of D315 and D330 of paraptosis. Thus caspase-9 displays at least two distinct activities, one that is proapoptotic and one that induces nonapoptotic cell death.
These data suggest a reconsideration of previously published conclusions in which a lack of effect of caspase inhibitors on cell death has been interpreted as indicating caspase-independent cell death (e.g., ref. 39). Furthermore, the conclusion that an Apaf-1 independent type of cell death that resembles necrosis in some morphological aspects may in fact be a form of programmed cell death involved in development is supported by the finding that in transgenic mice null for Apaf-1, the deletion of the interdigital webs is not blocked (although it is slightly delayed) and features a necrosis-like morphology with negative TUNEL staining (40).
It is noteworthy that morphologically similar forms of cell death have been described previously in neural development (7, 9, 11) and degeneration (15) (Table 2). Terms applied to this form include cytoplasmic death (11) and type 3B death (9). This form has been shown to occur in developing chick spinal cord motor neurons and neonatal rat superior colliculus, among other locations (41–44).
Table 2.
Examples of nonapoptotic cell death featuring cytoplasmic vacuolation
| Location | Description | Characteristics | Reference |
|---|---|---|---|
| Neuronal development | “Cytoplasmic” | ER swelling, later mitochondrial swelling | (11) |
| Neuronal development | “Type 3B” | ER swelling, later mitochondrial swelling | (9) |
| S. pombe | Bax-induced death | Cytoplasmic vacuolation | (45) |
| D. discoideum | DIF-induced cell death | Cytoplasmic vacuolation | (46) |
| ALS model G93A transgenic mouse | Motor neuron degeneration | ER swelling, later mitochondrial swelling | (15) |
ALS, amyotrophic lateral sclerosis; DIF, differentiation-inducing factor; ER, endoplasmic reticulum.
It is of interest to ask what the evolutionary relationship between apoptosis and nonapoptotic programmed cell death is. Reed's laboratory (45) reported that Bax expression in Schizosaccaromyces pombe induced cell death that featured massive cytoplasmic vacuolation and was not inhibited by p35. Although that form of cell death is at least superficially similar to what we have reported here, additional studies are required to determine whether or not S. pombe, which has not been shown to undergo apoptosis, has an alternative program for cell death. In addition, programmed cell death in Dictyostelium discoideum features cytoplasmic vacuolation without nuclear fragmentation or DNA cleavage (46).
Understanding the biochemical pathway(s) for nonapoptotic programmed cell death has potential implications for the understanding of neurodegeneration, cancer therapeutics, development, and evolutionary aspects of cell death programs.
Supplementary Material
Acknowledgments
We thank Guy Salvesen for the caspase zymogen cDNAs, Paul Friesen for the p35 cDNA, John Reed for the Bax, Bcl-xL, and xiap cDNAs, Peter Gruss for the Apaf-1 null cells, Jerry Olefsky for the full-length IGFIR cDNA, Ed Monosov for supervising the electron microscopy, Glenn Otero for the 293T cells, and Xin Ye for the catalytic mutant caspase constructs. This work was supported by Grants NS33376, NS35155, and AG12282 from the National Institutes of Health, Grant DAMD17–98-1–8613 from the U.S. Army, and a grant from the Amyotrophic Lateral Sclerosis Association. S.S. was supported by a fellowship from the North Atlantic Treaty Organization and the American-Italian Cancer Foundation. I.d.B. was supported by a fellowship from the Department of Defense (DAMD17–99-1–9092).
Abbreviations
- zVAD.fmk
benzyloxycarbonyl-valyl-alanyl-aspartyl fluoromethyl ketone
- BAF
t-butyloxycarbonyl-Asp(O-methyl)-fluoromethyl ketone
- IGFIR
insulin-like growth factor I receptor
- IGFIR-IC
IGFIR intracellular domain
- TUNEL
terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling
- xiap
X-chromosome-linked inhibitor of apoptosis
References
- 1.Kerr J F, Wyllie A H, Currie A R. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellis H M, Horvitz H R. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 3.Hengartner M O, Horvitz H R. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 4.Hengartner M O, Horvitz H R. Nature (London) 1994;369:318–320. doi: 10.1038/369318a0. [DOI] [PubMed] [Google Scholar]
- 5.Yuan J, Shaham S, Ledoux S, Ellis H M, Horvitz H R. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 6.Schweichel J U. Z Anat Entwicklungsgesch. 1972;136:192–203. [PubMed] [Google Scholar]
- 7.Schweichel J U, Merker H J. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz L M. BioEssays. 1991;13:389–395. doi: 10.1002/bies.950130805. [DOI] [PubMed] [Google Scholar]
- 9.Clarke P G. Anat Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 10.Lockshin R A, Williams C M. J Insect Physiol. 1964;10:643–649. [Google Scholar]
- 11.Pilar G, Landmesser L. J Cell Biol. 1976;68:339–356. doi: 10.1083/jcb.68.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oppenheim R W. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 13.Oppenheim R W. Trends Neurosci. 1985;17:487–493. [Google Scholar]
- 14.Cunningham T J. Int Rev Cytol. 1982;74:163–186. doi: 10.1016/s0074-7696(08)61172-9. [DOI] [PubMed] [Google Scholar]
- 15.Dal Canto M C, Gurney M E. Am J Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- 16.Majno G, Joris I. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 17.Ekert P G, Silke J, Vaux D L. Cell Death Differ. 1999;6:1081–1086. doi: 10.1038/sj.cdd.4400594. [DOI] [PubMed] [Google Scholar]
- 18.Frisch S M, Francis H. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye X, Mehlen P, Rabizadeh S, VanArsdale T, Zhang H, Shin H, Wang J J, Leo E, Zapata J, Hauser C A, et al. J Biol Chem. 1999;274:30202–30208. doi: 10.1074/jbc.274.42.30202. [DOI] [PubMed] [Google Scholar]
- 20.Bozzola J J, Russell L D. Electron Microscopy. Boston: Jones and Bartlett; 1992. [Google Scholar]
- 21.Bredesen D E, Ye X, Tasinato A, Sperandio S, Wang J J, Assa-Munt N, Rabizadeh S. Cell Death Differ. 1998;5:365–371. doi: 10.1038/sj.cdd.4400378. [DOI] [PubMed] [Google Scholar]
- 22.Rabizadeh S, Oh J, Zhong L T, Yang J, Bitler C M, Butcher L L, Bredesen D E. Science. 1993;261:345–348. doi: 10.1126/science.8332899. [DOI] [PubMed] [Google Scholar]
- 23.Ellerby L M, Hackam A S, Propp S S, Ellerby H M, Rabizadeh S, Cashman N R, Trifiro M A, Pinsky L, Wellington C L, Salvesen G S, et al. J Neurochem. 1999;72:185–195. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- 24.Mehlen P, Rabizadeh S, Snipas S J, Assa-Munt N, Salvesen G S, Bredesen D E. Nature (London) 1998;395:801–804. doi: 10.1038/27441. [DOI] [PubMed] [Google Scholar]
- 25.Liu J P, Baker J, Perkins A S, Robertson E J, Efstratiadis A. Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 26.Plymate S S, Bae V L, Maddison L, Quinn L S, Ware J L. Endocrine. 1997;7:119–124. doi: 10.1007/BF02778078. [DOI] [PubMed] [Google Scholar]
- 27.Hongo A, Yumet G, Resnicoff M, Romano G, O'Connor R, Baserga R. Cancer Res. 1998;58:2477–2484. [PubMed] [Google Scholar]
- 28.Reiss K, Yumet G, Shan S, Huang Z, Alnemri E, Srinivasula S M, Wang J Y, Morrione A, Baserga R. J Cell Physiol. 1999;181:124–135. doi: 10.1002/(SICI)1097-4652(199910)181:1<124::AID-JCP13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 29.Gavrieli Y, Sherman Y, Ben-Sasson S A. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wyllie A H. Nature (London) 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- 31.Casiano C A, Ochs R L, Tan E M. Cell Death Differ. 1998;5:183–190. doi: 10.1038/sj.cdd.4400336. [DOI] [PubMed] [Google Scholar]
- 32.Matsuyama S, Xu Q, Velours J, Reed J C. Mol Cell. 1998;1:327–336. doi: 10.1016/s1097-2765(00)80033-7. [DOI] [PubMed] [Google Scholar]
- 33.Rehemtulla A, Hamilton C A, Chinnaiyan A M, Dixit V M. J Biol Chem. 1997;272:25783–25786. doi: 10.1074/jbc.272.41.25783. [DOI] [PubMed] [Google Scholar]
- 34.Friedlander R M, Gagliardini V, Hara H, Fink K B, Li W, MacDonald G, Fishman M C, Greenberg A H, Moskowitz M A, Yuan J. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li P, Nijhawan D, Budihardjo I, Srinivasula S M, Ahmad M, Alnemri E S, Wang X. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida H, Kong Y Y, Yoshida R, Elia A J, Hakem A, Hakem R, Penninger J M, Mak T W. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Lehar S, Corvi C, Payne G, O'Connor R. Cancer Res. 1998;58:570–576. [PubMed] [Google Scholar]
- 38.Tennant M K, Thrasher J B, Twomey P A, Drivdahl R H, Birnbaum R S, Plymate S R. J Clin Endocrinol Metab. 1996;81:3774–3782. doi: 10.1210/jcem.81.10.8855837. [DOI] [PubMed] [Google Scholar]
- 39.Kitanaka C, Kuchino Y. Cell Death Differ. 1999;6:508–515. doi: 10.1038/sj.cdd.4400526. [DOI] [PubMed] [Google Scholar]
- 40.Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. Curr Biol. 1999;9:967–970. doi: 10.1016/s0960-9822(99)80425-4. [DOI] [PubMed] [Google Scholar]
- 41.Chu-Wang I W, Oppenheim R W. J Comp Neurol. 1978;177:33–57. doi: 10.1002/cne.901770105. [DOI] [PubMed] [Google Scholar]
- 42.Giordano D L, Murray M, Cunningham T J. J Neurocytol. 1980;9:603–614. doi: 10.1007/BF01205028. [DOI] [PubMed] [Google Scholar]
- 43.Sohal G S, Weidman T A. Exp Neurol. 1978;61:53–64. doi: 10.1016/0014-4886(78)90180-2. [DOI] [PubMed] [Google Scholar]
- 44.Cunningham T J, Mohler I M, Giordano D L. Brain Res. 1981;254:203–215. doi: 10.1016/0165-3806(81)90032-8. [DOI] [PubMed] [Google Scholar]
- 45.Jurgensmeier J M, Krajewski S, Armstrong R C, Wilson G M, Oltersdorf T, Fritz L C, Reed J C, Ottilie S. Mol Biol Cell. 1997;8:325–339. doi: 10.1091/mbc.8.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cornillon S, Foa C, Davoust J, Buonavista N, Gross J D, Golstein P. J Cell Sci. 1994;107:2691–2704. doi: 10.1242/jcs.107.10.2691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}