

Abstract
Corticotropin-releasing factor (CRF), a neuropeptide released during stress, has been reported to modulate startle behavior, including reducing the threshold for acoustic startle responding and reducing prepulse inhibition (PPI). The central mechanisms mediating CRF system regulation of startle and PPI are still unclear. Some antipsychotic drugs attenuate CRF-induced deficits in PPI in rats and mice. Here we tested the hypothesis that indirect activation of DA1-receptors (D1) and DA2-receptors (D2) contributes to the effects of CRF on PPI. We compared the effect of central administration of h/r-CRF (0.2-0.6 nmol) on PPI in mice with either a D1 or D2 receptor null mutation (knockout, KO) or in mice pretreated with D1 or D2 receptor antagonists SCH23390 (1 mg/kg) or haloperidol (1 mg/kg). D1 and D2 KO mice exhibited no significant differences in their sensitivity to CRF-induced disruptions of PPI. Similarly, neither SCH23390 nor haloperidol pretreatment altered the CRF-induced disruption in PPI, although both increased PPI at baseline. CRF-induced increases in startle also remained unchanged by any of the DA receptor manipulations. These results indicate that neither D1- nor D2-receptor activation is necessary for CRF to exert its effects on acoustic startle and PPI in mice.
Keywords: CRH (corticotropin-releasing hormone), CRF, dopamine, haloperidol, SCH23390, prepulse inhibition, startle, knockout mice
Introduction
Prepulse inhibition (PPI) is a cross-species phenomenon used as an operational measure of sensorimotor gating (Braff et al. 2001; Geyer et al. 2001; Swerdlow et al. 2001). PPI is the inhibition of the acoustic startle reflex (ASR), a contraction of the skeletal and facial muscles in response to a sudden, intense auditory stimulus, when the startling stimulus is preceded 30-300 ms by a non-startling stimulus or “prepulse” (Graham, 1975). PPI is suggested to measure pre-attentional filtering mechanisms that filter or “gate” internal and external stimuli during critical periods of information processing (Braff and Geyer, 1990). Many neuropsychiatric disorders including schizophrenia, obsessive compulsive disorder (OCD) and Huntington's disease exhibit disrupted PPI, as do panic disorder and possibly post traumatic stress disorder (PTSD) subjects (Braff et al. 2001; Grillon et al. 1998; Grillon et al. 1996; Ludewig et al. 2002; Swerdlow et al. 2001). Over 20 years of studies support a modulatory role of dopaminergic and serotonergic signaling mechanisms in both human and rodent PPI (for review see Geyer et al. 2001). Recently, the neuropeptide corticotropin releasing factor (CRF) has also been shown to modulate PPI in rats and mice (Conti et al 2002; Risbrough et al 2004).
CRF and the related urocortin ligand family (Urocortin 1-3) mediate behavioral, autonomic, and endocrine responses to stress (Bale and Vale, 2004; Heinrichs and Koob, 2004; Dautzenberg and Hauger, 2002; Hauger et al. 2006). In addition to hypothalamic sites, CRF-containing neurons are found in a number of brainstem, limbic, and cortical nuclei (Asan et al. 2005; Hatalski et al. 1998; Hsu et al. 1998; Merali et al. 1998; Swanson, et al.1983; for review see Reul and Holsboer, 2002). In rodents and non-human primates, CRF receptors are expressed in neural circuits that modulate startle and mediate PPI, including nucleus accumbens, amygdala, and brain stem regions such as peduncular pontine tegemental nucleus and inferior colliculus (Van Pett et al 2000). CRF system abnormalities have been implicated in patients with certain neuropsychiatric disorders where PPI is deficient (panic disorder, PTSD, OCD, Tourette's syndrome) (Altemus et al 1992; Baker et al 1999; Bremner et al 1997; Castellanos et al 1996; Chappell et al 1996; Grillon et al 1998; Grillon et al 1996; Hoenig et al 2005; Holsboer et al 1987; Kellner and Yehuda 1999; Ludewig et al 2005; Ludewig et al 2002; Roy-Byrne et al 1986; Smoller et al 2005). CRF and urocortins acts in the brain at two distinct G-protein-coupled receptors, CRF1 and CRF2 (Bale and Vale 2004; Hauger et al. 2006). In mice, transgenic-induced overexpression or exogenous administration of CRF reduces PPI (Dirks et al. 2003; Risbrough et al. 2004). CRF-induced activation of the CRF1 receptor alone or concomitant activation of both CRF1 and CRF2 receptors increases startle magnitude and reduces startle threshold (Risbrough et al. 2003). However, PPI was differentially regulated whereby selective CRF1 receptor activation decreased PPI while selective CRF2 receptor activation increased PPI (Risbrough et al. 2004). The downstream signaling processes and neurotransmission mechanisms required for CRF receptor modulation of PPI are unknown.
There is ample evidence supporting a role for dopamine (DA) receptors in PPI (Geyer et al. 2001). Systemic administration of both direct and indirect DA receptor agonists (e.g. apomorphine and amphetamine respectively) reduces PPI (Dulawa and Geyer, 1996; Mansbach et al. 1988; Swerdlow et al. 1990). Similarly, direct infusions of DA into the nucleus accumbens decrease PPI (Swerdlow et al. 1992). Mice with excess synaptic DA levels via genetic deletion of the DA transporter exhibit robust PPI deficits (Ralph et al. 2001). Most of the above manipulations are reversed by blockade of DA1-receptors (D1) or DA2-receptors (D2) in mice (Ralph-Williams et al. 2002, 2003, Ralph et al. 2001). Hence increased synaptic release of DA can produced significant PPI impairment, presumably via D1 and D2 receptor activation.
Stressors are reported to stimulate synaptic DA release and activate the DA system (Abercrombie et al. 1989; Coco et al. 1992; Deutch and Roth, 1990; Dunn, 1988; Pani et al. 2000), which may be mediated by stress-induced CRF receptor signaling (Dunn, 1988; but see Dunn, 2000). Indeed several recent reports suggest that CRF receptor activation may modulate DA release. Intracerebroventricular (ICV) CRF-administration leads to increased DA catabolites and utilization (Lavicky and Dunn, 1993; Matsuzaki et al. 1989). CRF1 and CRF2 receptors are localized in the ventral tegmental area (VTA) where CRF receptor agonists modulate DA release (Sauvage and Steckler 2001; Ungless et al 2003; Wang et al 2005). CRF1 receptor antagonist administration reduces cocaine-induced DA release and behavioral effects of cocaine (Lodge and Grace 2005; Lu et al 2003) as well as regulates dopamine D2 receptor expression in the brain (Lawrence et al 2005). D2 receptors are co-localized with CRF immunoreactive neurons in the amygdala (Eliava et al. 2003). Interestingly, D2-family receptor antagonists have been reported to attenuate CRF-induced deficits in learning (Radulovic et al. 2000) and PPI (Conti et al 2005), suggesting that some behavioral effects of CRF may require DA receptor activation.
Thus, the primary goal of this study was to test the hypothesis that downstream DA receptor activation may be necessary for CRF-induced deficits in PPI. Because D1 and D2 receptors appear to be necessary for direct and indirect DA agonist induced disruption of PPI in mice, we also hypothesized that D1 and D2 receptors would contribute to any putative DA effects downstream of CRF system activation (Geyer et al. 2001; Ralph-Williams et al. 2003; Ralph-Williams et al. 2002). To test our hypotheses, we utilized a complementary pharmacological and genetic approach by examining the CRF effects on startle plasticity in D1 and D2 receptor null mutation mice (knockout mice, KO) and after pharmacological blockade of D1 and D2 receptors in wild-type (WT) mice.
Materials and methods
Subjects
Male DA receptor D1 and D2 WT and KO mice (constitutive gene deletion background mice; 3-6 months of age at testing) were bred and shipped from the Oregon Health & Science University. The D2 mice (B6.129S2-Drd2tm1Low/J) were originally generated at the Oregon Health & Science University (Kelly et al. 1998) and backcrossed onto the C57BL/6J background strain for 17 generations. Stocks of D1 mice (B6.129S4-Drd1atm1Lcd/J) (Drago et al. 1994) were obtained from the mutant mouse repository at the Jackson Laboratory (Bar Harbor, ME) and were backcrossed onto the C57BL/6J background for 10-12 generations. All mice were housed one per cage after surgery in a temperature (21–22°C) controlled room under a reverse 12 h/12 h light cycle (lights off at 8:00 a.m.). 129T2/SVEmsJ mice (2-3 months of age at testing) from Jackson Laboratories (Bar Harbor, Maine, USA) were used for DA antagonist studies (haloperidol and SCH23390). This strain was chosen because unlike C57BL/6J mice, this strain exhibits similar PPI performance and CRF sensitivity to the D1 and D2 receptor WT mice (Risbrough et al. 2004; Crawley et al. 1997). A reverse light/dark cycle was used to minimize interactions with the stress of disruptions in diurnal cycles associated with testing during the sleep phase. The mice were allowed a 1-week period of acclimation to the animal room before cannulation surgery. All animal testing occurred from 10:00 a.m. to 6:00 p.m. and was conducted in accordance with the “Principles of Laboratory Animal Care” NIH guidelines, as approved by the University of California, San Diego and Veterans Affairs Medical Center animal care committees.
Surgery
Mice were anesthetized using a 90 mg/kg ketamine-2 mg/kg acepromazine cocktail and prepared with a 23 gauge 7-mm-length unilateral guide cannula in the lateral ventricle (flat skull; anteroposterior, ±0.1 mm; mediolateral, −1 mm; dorsoventral, −1.5 mm below dura). Cannulae were secured with one skull screw and dental cement (Den-Mat Corp., Santa Maria, CA) and closed with a removable stylet. Mice were allowed a 5 to 7 days recovery period before testing.
Apparatus
Startle chambers (SR-LAB, San Diego Instruments, San Diego, CA, USA) consisted of nonrestrictive Plexiglas cylinders 5 cm in diameter resting on a Plexiglas platform in a ventilated chamber. High frequency speakers mounted 33 cm above the cylinders produced all acoustic stimuli, which were controlled by SR-LAB software. Piezoelectric accelerometers mounted under the cylinders transduced the movements of the animal, which were digitized and stored by an interface and computer assembly. Beginning at startling stimulus onset, 65 consecutive 1-ms readings were recorded to obtain the average amplitude of the animal's startle response. A dynamic calibration system was used to ensure comparable sensitivities across chambers. Sound levels were measured as described elsewhere (Mansbach et al., 1988) using the A weighting scale in units of dBA sound pressure level. The chamber house-light remained off throughout all testing.
Drugs
Peptide infusions
ICV injections of human/rat-corticotropin-releasing factor (h/r-CRF) and subsequent histologies were as previously described (Spina et al. 2000). In brief, injections were conducted in unanesthetized mice using a 30 gauge 8 mm injector (1 mm below the tip of the guide cannulae). Injection volume was 5 μl using gravity flow. Within one week after testing was completed, mice were anesthetized and 2 μl of dye was injected via the 8 mm injector. Mice were immediately killed, and the brains were removed. As the brains were removed, presence of the dye in the fourth ventricle was noted. A coronal cut was made along the guide tract to reveal lateral and third ventricles, which were also noted for presence of dye, and brains were digitally scanned with the cut side on slides. Eight of the total 77 mice used in these studies were removed from the analysis due to incorrect cannula placement.
Experiment 1: h/r-CRF in D2 WT and KO mice
Mice received either 0.6 nmol (3 μg/5μl, ICV; Bachem, Torrance, CA; n=8-10) of human/rat-CRF (h/r-CRF) or artificial cerebral spinal fluid (aCSF) vehicle and were tested 1 h after injection. Pilot studies in female D2 WT mice indicated that the 0.6 nmol dose of h/r-CRF had maximal efficacy on PPI in these mice. Mice were tested in a within-subject cross over design, with half receiving h/r-CRF and half receiving aCSF on the first test, and vice versa on the second test. Tests were separated by 1 week.
Experiment 2: h/r-CRF in D1 WT and KO mice
Mice received 0, 0.2, and 0.6 nmol h/r-CRF in a within-subject design (n=9/group), receiving each dose and vehicle once, in a latin square design. Tests were separated from each other by at least 1 week. The lower dose (0.2 nmol) was included as we had no pilot studies on which to base our dose selection.
Experiment 3: Haloperidol versus h/r-CRF
Mice received either 0.6 nmol h/r-CRF or aCSF vehicle 1 h before testing (n=8-9/group) and either sterile saline vehicle or 1 mg/kg of the D2/D3 receptor antagonist haloperidol (injectable preparation, Novaplus, Bedford, OH). Haloperidol was delivered by intraperitoneal (IP) injection in a 10 ml/kg volume 30 min before testing. This dose and time point was chosen because it was the most effective in attenuating PPI deficits in CRF-overexpressing (CRFOE) mice (Dirks et al. 2003).
Experiment 4: SCH23390 versus h/r-CRF
h/r-CRF at 0.6 nmol or aCSF vehicle was administered 1 h before startle testing (n=7-9/group). Sterile water vehicle or 1 mg/kg of the selective D1 receptor antagonist SCH23390 (Tocris, Ellisville, MO) was administered by subcutaneous (SC) injection in a 10 ml/kg volume 10 min before testing. This dose and time point was chosen as it was the most effective in blocking apomorphine-induced deficits in PPI in mice (Ralph-Williams et al. 2003).
Behavioral testing
All experiments used the same acoustic startle session. The intertrial interval averaged 15 sec (range of 7-23 sec). During each inter-trial interval, the movements of the mice were also recorded once to measure responding when no stimulus was present. A 65 dB background was presented continuously throughout the session. After placement into the startle chambers, a 5 min acclimation period preceded testing. Startle pulses were 40 ms in duration, prepulses were 20 ms in duration, and prepulses preceded the pulse by 100 ms (onset-onset). The acoustic startle session included two blocks of different trial types. The first block tested acoustic startle response only and included 9 each of three different acoustic stimulus intensities: 90, 105, and 120 dB. The second block consisted of six startle pulse intensities (each of 105 or 120 dB) and five prepulse+pulse trials (73 and 81 dB preceding either a 105 or 120 dB pulse). In this second block, the interstimulus interval between prepulse and pulse onset was 100 ms.
Mice used in experiments 3 and 4 were tested 5-7 days after surgery for baseline startle and PPI performance using the session described above. These data were used to counterbalance startle and PPI performance across drug groups. An initial test using 0.2 nmol h/r-CRF vs. haloperidol was conducted; however this dose of CRF did not produce a significant disruption of PPI (data not shown). Mice were reassigned to drug groups (equal distribution of previous treatment across groups) for experiment 3 and again for experiment 4. There was a 1 week washout period between drug tests.
Data analysis
The average startle magnitude over the record window (65 ms) was used for all data analysis. Percentage of PPI was calculated using the following formula: 100−((average startle of the prepulse + pulse trial)/average startle in the pulse alone trial)*100.
PPI analyses
In initial analyses, h/r-CRF effects on PPI were consistently independent of pulse intensity (105 or 120 dB) in all 4 experiments; hence all data and analyses shown are collapsed across pulse intensity. For experiments 1-2, a 3 ANOVA was used, with genotype (WT or KO) as a between subject factor and treatment (h/r-CRF) and prepulse intensity (73 and 81 dB) as within-subject factors. For experiments 3 and 4, the ANOVA included treatment (h/r-CRF) and pretreatment (Haloperidol or SCH23390) as between subject factors and prepulse intensity as within-subject factors. To assess order effects on PPI a 3 way ANOVA model with order, prepulse intensity and gene was used.
Startle analyses
For experiments 1-2, a 3 way ANOVA was used, with genotype (WT or KO) as a between subject factor and treatment (h/r-CRF) and pulse intensity (90, 105 and 120 dB) as within-subject factors. For experiments 3 and 4, the ANOVA included treatment (h/r-CRF) and pretreatment (Haloperidol or SCH23390) as between subject factors and pulse intensity as within-subject factors. To assess order effects on startle a 3 way ANOVA model with order, pulse intensity and gene was used. Post hoc analyses followed significant main or interaction effects as appropriate.
Results
Experiment 1: h/r-CRF in D2 WT and KO mice
Administration of h/r-CRF (0.6 nmol) significantly increased acoustic startle responding in all mice at all stimulus intensities regardless of genotype (Table 1) [h/r-CRF: F(1,16)=14.88, p<0.001; h/r-CRF × Gene: F(1,16)=0.43, n.s.]. There were no main or interactive effects of order [e.g. Order × Gene × Intensity (2,28)=2.72, n.s.]. CRF also induced significant disruption of PPI in both KO and WT mice (Figure 1) (h/r-CRF: F(1,16)=28,18, p<0.001). PPI effects were independent of gene, although there was a trend for KO mice to exhibit greater CRF-induced PPI disruption compared to WT mice [h/r-CRF × gene, F(1,16)=3.08, p=0.09]. On closer inspection of the data, the main effect of CRF was largely due to a significant disruption of PPI at the 8 dB but not 16 dB above background trials (73 and 81 dB respectively) (Figure 1) [h/r-CRF × prepulse, F(1,16)=28.32, p<0.001]. There were no main or interactive effects of order [e.g. Order × Gene × Prepulse F(1,14)<1, n.s.]
Table 1.
Effect of h/r-CRF administration on acoustic startle magnitude (mean±SEM) during D1 or D2 receptor blockade or in D1 and D2 receptor null mutant mice.
| Vehicle | CRF* 1 μg |
3 μg | |||
|---|---|---|---|---|---|
| D2 | WT | p90 p105 p120 |
37 ± 11 64 ± 15 92 ± 13 |
- - - |
75 ± 19 138 ± 42 209 ± 48 |
| KO | p90 p105 p120 |
23 ± 6 43 ± 5 63 ± 11 |
- - - |
43 ± 14 93 ± 30 156 ± 30 |
|
| D1 | WT | p90 p105 p120 |
41 ± 11 74 ± 11 91 ± 15 |
52 ± 13 99 ± 11 168 ± 51 |
72 ± 25 175 ± 58 293 ± 76 |
| KO | p90 p105 p120 |
47 ± 10 95 ± 11 132 ± 13 |
74 ± 26 221 ± 53 324 ± 52 |
83 ± 13 213 ± 39 331 ± 47 |
|
| Haloperidol | Vehicle | p90 p105 p120 |
18 ± 3 67 ± 12 163 ± 25 |
- - - |
133 ± 23 273 ± 30 407 ± 89 |
| Haloperidol | p90 p105 p120 |
19 ± 3 59 ± 7 169 ± 20 |
- - - |
138 ± 29 239 ± 48 488 ± 120 |
|
| SCH23390** | Vehicle | p90 p105 p120 |
29 ± 9 87 ± 18 228 ± 54 |
- - - |
209 ± 28 338 ± 46 693 ± 90 |
| SCH23390 | p90 p105 p120 |
15 ± 2 51 ± 8 177 ± 30 |
- - - |
110 ± 29 242 ± 34 484 ± 60 |
|
Main effect of CRF in all experiments (p<0.01).
Main effect of SCH23390 (p<0.05).
Figure 1.
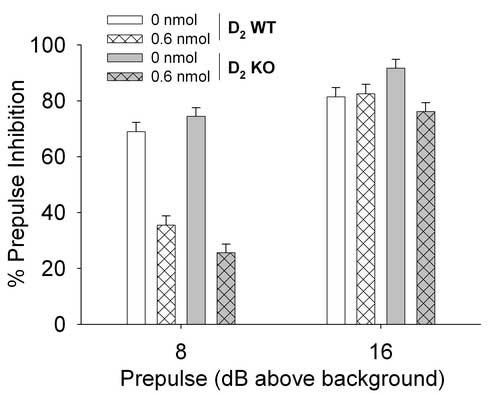
Effects of h/r-CRF on PPI in D2 wild-type (WT) and knockout (KO) mice. D2 WT and KO mice were administered artificial cerebral spinal fluid (aCSF) and 0.6 nmol h/r-CRF (ICV, 5 μl) over 2 test sessions (1 week apart). Mice were tested for startle and PPI 1 h after CRF administration. Data are presented as mean± pooled SEM % prepulse inhibition. CRF significantly decreased PPI across genotype at 8 but not 16 dB above background prepulse trials (see results for details).
Experiment 2: h/r-CRF in D1 WT and KO mice
Overall CRF treatment increased startle [h/r-CRF: F(2,32)=10.39, p<0.001] independently of the presence of the D1-receptor [Table 1; h/r-CRF × gene: F(2,32)=1.43, n.s.]. Again, PPI was significantly reduced in response to h/r-CRF treatment at the 8 dB above background prepulse trials (Figure 2) [h/r-CRF × prepulse: F(2,32)=3.41, p<0.05]. CRF-induced disruption of PPI was not found to depend on D1 genotype [h/r-CRF × gene: F(2,32)<1, n.s.]. Similarly to experiment 1, there were no main or interactive effects of order for either PPI analyses [e.g. Order × Gene × Intensity (2,12)<1, n.s.] or startle analyses [e.g. Order × Gene × Intensity (4, 24)<1, n.s.].
Figure 2.
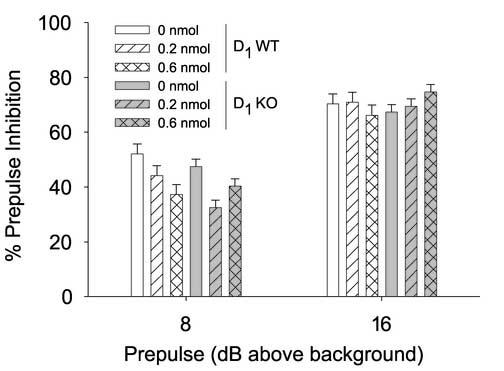
Effects of h/r-CRF on PPI in D1 wild-type (WT) and knockout (KO) mice. D1 WT and KO mice were administered artificial cerebral spinal fluid (aCSF), 0.2 and 0.6 nmol h/r-CRF (ICV, 5 μl) over 3 test sessions (1 week apart). Mice were tested for startle and PPI 1 h after CRF administration. Data are presented as mean± pooled SEM % prepulse inhibition. CRF significantly decreased PPI across genotype at 8 but not 16 dB above background prepulse trials (see results for details).
Experiment 3: Haloperidol versus h/r-CRF
The data from experiment 1 led to the hypothesis that the D2 receptor is not essential for the effects of CRF. We investigated whether we could replicate these data with a pharmacological D2 receptor blockade. Haloperidol, an antagonist at D2 and D3 receptors, was chosen as it had previously been reported to attenuate the PPI deficits in CRFOE mice (Dirks et al. 2003). The 0.6 nmol dose of h/r-CRF increased startle magnitude at all intensities [h/r-CRF: F(1,29) = 19.11, p<0.001]. CRF treatment groups exhibited decreased PPI at the 8 dB above background prepulse trials (Table 2) [h/r-CRF × prepulse, F(1,29) = 7.33, p<0.05]. As seen in D2 KO mice, D2 blockade via haloperidol had no effect on CRF-induced decreases in PPI (Table 2) [h/r-CRF × haloperidol: F(1,29)<1, n.s.], although haloperidol did significantly increase PPI independent of CRF at some trial types [Haloperidol × prepulse × pulse intensity: F(1,29) = 6.82, p<0.05], confirming previous reports (Ouagazzal et al 2001).
Table 2.
Effect of pharmacological blockade of D1 and D2 receptors on CRF-induced deficits in %PPI (data shown as mean±SEM for 8 dB and 16 dB above background prepulse trials).
| Prepulse Intensity | 8 dB | 16 dB | ||
|---|---|---|---|---|
| Vehicle | CRF* | Vehicle | CRF | |
| Vehicle | 47±7 | 39±9 | 62±6 | 64±7 |
| Haloperidol | 58±6 | 38±7 | 67±4 | 64±4 |
| Vehicle | 47±9 | 33±6 | 62±7 | 59±6 |
| SCH23390** | 63±6 | 49±6 | 76±3 | 73±3 |
Main effect of CRF on PPI at 8 dB above background trials in both Haloperidol and SCH23390 studies.
Main effect of SCH23390 treatment (see text for details).
Experiment 4, SCH23390 versus h/r-CRF
CRF administration increased startle significantly at all startle intensities (Table 1) [h/r-CRF, F(1,29)=63,35, p<0.001], independently of SCH23390 treatment [h/r-CRF × SCH23390, F(1,29)=2.63, n.s.]. SCH23390 treatment decreased startle independently of the presence of CRF [SCH23390, F(1,29)=7.30, p<0.05]. CRF disrupted PPI at the 73 dB prepulse trials (Table 2) [h/r-CRF × prepulse, F(1,29)=4.64, p<0.05]. SCH23390 administration did not reverse the CRF-induced PPI deficits [h/r-CRF × SCH23390, F(1,29)<1, n.s.] although it did increase overall PPI-performance in both aCSF and CRF treatment groups [SCH23390, F(1,29)=10.49, p<0.01]
Discussion
The objective of the present study was to investigate the role of D1 and D2 receptors in CRF-induced deficits in PPI. The present studies used a complementary approach to address this question, by determining the effects of CRF in mice with either genetic null mutation of the D1 or D2 receptor, and in mice treated with selective D1-family and D2-family receptor antagonists. We found that acute CRF administration increased startle reactivity and reduced PPI when either the D1 or the D2 receptor was genetically deleted or pharmacologically blocked. These results indicate that neither D1 nor D2-receptor activation is necessary for CRF to exert its effects on acoustic startle and PPI.
Several studies have shown that central CRF administration and stress can influence synaptic DA release in rodents (see introduction). In addition, D2 receptors appear to contribute to the PPI disruption induced by DA release in mice. For example, D2 KO mice or mice treated with the D2-family antagonist raclopride are insensitive to amphetamine-induced PPI disruption, while raclopride administration reverses the PPI deficits observed in DA transporter KO mice (Ralph-Williams et al. 2003; Ralph-Williams et al. 2002). Recent data indicate that haloperidol attenuates the PPI deficits observed in CRFOE mice with constitutive CRF-overexpression (Dirks et al. 2003). Haloperidol treatment can also reverse CRF-induced disruption in PPI in Wistar Kyoto but not Brown Norway rats (Conti et al. 2005). Hence it was surprising that neither haloperidol nor D2 receptor gene deletion attenuated the effects of acute CRF on PPI in mice. The haloperidol dose used here is the same dose shown to be effective in reducing the magnitude of PPI disruption observed in CRFOE mice and CRF-treated rats (Conti et al. 2005; Dirks et al. 2003). The discrepancy between our findings and previous studies showing that haloperidol attenuates CRF-dependent PPI disruption may be explained by the following: (1) haloperidol is only effective in cases were CRF is chronically released or (2) haloperidol's effect is dependent upon strain and/or species. Although CRFOE mice have been shown to exhibit alterations in the CRF system, it is unknown if these mice have abnormal functioning of their DA systems (Kozicz et al. 2004; Peeters et al. 2004; Weninger et al.2000). A very recent study has indicated that D2 receptors are upregulated after chronic CRF1 receptor antagonism (Lawrence et al 2005), suggesting that CRF receptor activity can modulate D2 receptor signaling. Haloperidol has also been reported to attenuate acute CRF effects on PPI in rats (Conti et al. 2005), although this effect was strain dependent. Furthermore, other evidence suggests that D2 receptor agonist and antagonist effects on PPI depend on strain in mice (McCaughran et al. 1997; Ralph and Caine, 2005). In the present studies we used 129T2/SVEmsJ mice, whereas CRFOE mice were on a C57BL/6J background. Thus, haloperidol effects on CRF-induced deficits in PPI may be dependent upon mouse strain. It should also be noted that in the present studies we did not see a deficit in PPI in the D2 KO mice, which has been observed in some (Ralph-Williams et al. 2002) but not all (Ralph et al. 1999) studies. It is not clear if this inconsistent phenotype is due to continuous backcrossing to C57BL/6J background over the years, however our experience is that these mice generally do not show a deficit across cohorts (unpublished observations).
Although the dose of haloperidol chosen in the present studies has been shown to be efficacious in CRFOE mice and CRF-treated rats, it is possible that higher doses of haloperidol would have been efficacious. In the present studies, we observed a slight but significant increase in PPI in the haloperidol treatment groups. Hence, increasing the dose would likely increase PPI regardless of CRF treatment, as well as reduce startle responding (Dirks et al. 2003; Ouagazzal et al. 2001), reducing the interpretability of such a study. In agreement with the present haloperidol data, we found that D2 KO mice exhibit normal or even slightly potentiated effects of CRF on PPI. These data would indicate that D2 receptor signaling is not necessary for CRF effects on PPI. Although haloperidol attenuated CRF-OE PPI deficits, clozapine and risperidone, also D2–family antagonists (among other activity), did not (Dirks et al. 2003). Taking these data together, we suggest that haloperidol effects on CRF-induced disruptions in PPI may be via activity at other receptors.
In mice, activation of D1 receptors could also contribute to putative DA-mediated disruptions of PPI (Ralph-Williams et al. 2003; Ralph-Williams et al. 2002; Ralph and Caine, 2005). In the present studies, CRF significantly reduced PPI in D1 KO mice and in the presence of behaviorally active doses of the D1 antagonist SCH23390. These data support the conclusion that D1 receptors are not required for CRF effects on startle or PPI. D1 KO mice did appear to be less sensitive to the effects of the high dose (0.6 nmol) of h/r-CRF (Figure 2). In contrast, pharmacological blockade of D1 with SCH23390 had no specific effects on this dose of CRF in WT mice. This 1 mg/kg dose of SCH23390 significantly increased PPI and reduced startle in WT mice, which we have not observed in other strains (Ralph et al. 2001). We chose this dose as it has been shown to attenuate the locomotor phenotype in DAT KO mice as well as reverse apomorphine- and cocaine-induced decreases in PPI (Ralph-Williams et al. 2003, Geyer unpublished observations; Ralph et al. 2001). While our study shows that blockade of either D1 or D2 receptors does not reverse CRF-induced PPI deficits, it could be argued that either D1 or D2 receptors may still be sufficient for CRF-induced deficits in PPI, hence blocking only one receptor at a time will have no effect. In such a case, simultaneous blockade of both receptor subtypes would reverse CRF-induced reductions in PPI. We have found however, that when given in combination, haloperidol and SCH23390 do not reverse CRF effects on PPI (Risbrough, Caldwell, Geyer unpublished observations). Thus, taken together, it seems that CRF effects on PPI and startle do not require activation of D1 or D2 receptors.
Previously, we and others have shown that h/r-CRF decreases PPI independent of prepulse and pulse intensity (Risbrough et al. 2004). In our experiments in mice and in studies with certain rat strains, however, h/r-CRF had consistent effects on PPI only at the lower prepulse-intensity trials (Conti et al. 2005; Conti et al. 2002). The lack of effect of h/r-CRF at the higher prepulse intensities in these studies may be due to differences in baseline performance. In the present studies, we found an average of 70% inhibition across all trials, whereas using the same testing session Risbrough et al. (2004) showed a 40-50% average across the trial types. The high intensity prepulse trials (81 dB) in the present studies may thus be less vulnerable (inhibition was up to 80% at these trials) to disruption than the lower intensity and presumably less salient 73 dB prepulse trials (Figure 1 and 2). Thus lower prepulse intensities may produce a “threshold” of inhibition that is more easily disrupted by CRF administration. The dose response of h/r-CRF effects on PPI was also slightly shifted compared to previous reports in mice and in rats (Conti et al. 2002; Risbrough et al. 2004), with the present studies requiring higher doses of h/r-CRF to induce significant disruption of PPI (Risbrough et al. 2004). It is unclear if this pattern is due to potential batch differences in the peptide, or in the strains used (C57BL/6J from Jackson and 129SvEv from Taconic vs. a congenic C57BL/6J bred at OHSU and 129T2/SVEmsJ mice in the present studies). Despite these small discrepancies in parameters and doses, however, the CRF-induced reduction in PPI and increase in startle appears to be reasonably replicable across mouse strains.
The administration of CRF robustly increased startle in all of our experiments regardless of treatment. Administration of haloperidol or SCH23390 as well as DA receptor gene manipulation did not affect CRF-induced increases in startle (Table 1). These results confirm earlier experiments in which CRF induces large increases in startle magnitude in both rats and mice (Liang et al 1992; Risbrough et al 2003; Risbrough et al 2004; Swerdlow et al 1989; Swerdlow et al 1986). In rats, CRF effects on startle have been localized to a hippocampal-septal-bed nucleus stria terminalis circuit (for review see Davis et al 1997), which contains both CRF1 and CRF2 receptors (Van Pett et al 2000). CRF-induced increases in startle are blocked by steroids such as progesterone treatment and alphaxalone (Swerdlow and Britton 1994; Toufexis et al 2004), blocked by GABAergic activation via chlordiazepoxide administration (Swerdlow et al 1989), and enhanced by corticosterone and vasopressin treatment (Lee et al 1994; Pelton et al 1997). Recently, Meloni et al. (2006) reported that SCH23390 attenuated CRF-induced increases in startle in rats (PPI was not tested). It is unclear if the discrepancy between our findings (no effect of SCH23390 or genetic deletion of D1) and those of Meloni et al. (2006) are due to species (rat vs. mice) or methods of administration of SCH23390. In rats, SCH23390 induced a U-shaped dose response curve, with the high dose of SCH23390 (0.5 mg/kg) having no effect, while lower doses reduced CRF effects on startle (Meloni et al. 2006). It is thus possible that lower doses of SCH23390 would have been more effective in the present studies. A difference in the timepoint of administration before CRF administration may also account for the discrepancies seen across these studies. Nevertheless, in light of our present finding in mice that full blockade of D1 via null mutation has no effect on CRF induced alterations in startle behaviors, a positive result of SCH23390 might have called into question the selectivity of SCH23390 for D1 rather than indicate that D1 receptors are required for CRF effects on startle in mice. SCH23390 has been shown to be active at other receptors (e. potent efficacy at 5-HT2C (Millan et al.2001) and possibly D5 receptors (Centonze et al.2003)). Indeed, the 0.05 and 0.1 mg/kg doses of SCH23390 that were effective in blocking CRF-induced increases in startle in rats (Meloni et al. 2006) have been shown to have behavioral effects in D1 KO mice, indicating that at least in mice, SCH23390 has functional activity at receptors other than D1 (Centonze et al.2003). A more general explanation of why D1 and D2 blockade had no effect on CRF-induced effects on startle in the present studies, but has been shown to be effective in rats (Meloni et al. 2006; Conti et al. 2005) may be due to species differences in dopamine system control of startle behaviors. We have previously shown that D1 and D2 agonist effects on PPI in mice do not match the pattern of effects seen in rats (Ralph-Williams et al. 2003; Ralph-Williams et al. 2002; Wan et al.1996). Hence, it is possible that our discrepant findings are due to differences in DA control of startle behaviors across species.
Besides the enervation of dopaminergic systems, CRF also interacts with other monoamine systems that modulate sensorimotor gating (Geyer et al. 2001; Sauvage and Steckler, 2001). CRF fibers innervate brain structures such as the serotonergic dorsal raphe nucleus (DRN) (Kirby et al. 2000; Lowry et al. 2000; Pernar et al. 2004) and the noradrenergic locus coeruleus (LC-NE) (Dunn et al. 2004; Emoto et al. 1993; Lavicky and Dunn, 1993; Matsuzaki et al. 1989; Pernar et al. 2004; Valentino et al. 2001). ICV administration of CRF increases NE turnover and utilization in the medial frontal cortex and hippocampus (Lavicky and Dunn, 1993; Matsuzaki et al. 1989; Zhang et al. 1998), and β-adrenergic receptors have been shown to be involved with stress- and CRF-induced effects on defensive behavior (Gorman and Dunn, 1993; Yang and Dunn, 1990). CRF modulation of glutamate neurotransmission, perhaps at the amygdala, may also be a potential mechanism for CRF effects on startle behavior (Liu et al 2004; Liu et al 2005; Swerdlow et al 2001). Additional research is required to explore these alternative mechanisms for CRF induced alterations in startle behavior.
In summary, we found that neither gene null mutation nor pharmacological blockade of DA D1 or D2 receptors significantly affected CRF-induced decreases in PPI and increases in startle in mice. The present studies indicate that CRF-induced disruptions of PPI do not require D1 or D2 receptor activation in mice.
Acknowledgements
The authors thank V. Otero-Corchon and R. Kruse for valuable technical assistance breeding and genotyping mice. These studies were funded by the National Institute of Health grants MH074697 (MAG) and MH076850 (VBR) and the Department of Veterans Affairs VISN 22 Mental Illness Research, Education, and Clinical Center (MAG and RLH); and a VA Merit Review grant (RLH). M.A. Geyer holds an equity interest in San Diego Instruments, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem. 1989;52:1655–8. doi: 10.1111/j.1471-4159.1989.tb09224.x. [DOI] [PubMed] [Google Scholar]
- Altemus M, Pigott TA, Kalogeras KT, Demitrack M, Dubbert B, Murphy DL, Gold PW. Abnormalities in the regulation of vasopressin and corticotropin releasing factor secretion in obsessive compulsive disorder. Arch Gen Psychiatry. 1992;49:9–20. doi: 10.1001/archpsyc.1992.01820010009002. [DOI] [PubMed] [Google Scholar]
- Asan E, Yilmazer-Hanke DM, Eliava M, Hantsch M, Lesch KP, Schmitt A. The corticotropin-releasing factor (CRF)-system and monoaminergic afferents in the central amygdala: investigations in different mouse strains and comparison with the rat. Neuroscience. 2005;131:953–7. doi: 10.1016/j.neuroscience.2004.11.040. [DOI] [PubMed] [Google Scholar]
- Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN, Geracioti TD., Jr Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry. 1999;156:585–8. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–57. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–58. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry. 1990;47:181–8. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, Nemeroff CB, Charney DS. Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry. 1997;154:624–9. doi: 10.1176/ajp.154.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos FX, Fine EJ, Kaysen D, Marsh WL, Rapoport JL, Hallett M. Sensorimotor gating in boys with Tourette's syndrome and ADHD: preliminary results. Biol Psychiatry. 1996;39:33–41. doi: 10.1016/0006-3223(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Centonze D, Grande C, Saulle E, Martin AB, Gubellini P, Pavon N, et al. Distinct roles of D1 and D5 dopamine receptors in motor activity and striatal synaptic plasticity. J Neurosci. 2003;23:8506–12. doi: 10.1523/JNEUROSCI.23-24-08506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell P, Leckman J, Goodman W, Bissette G, Pauls D, Anderson G, Riddle M, Scahill L, McDougle C, Cohen D. Elevated cerebrospinal fluid corticotropin-releasing factor in Tourette's syndrome: comparison to obsessive compulsive disorder and normal controls. Biol Psychiatry. 1996;39:776–83. doi: 10.1016/0006-3223(95)00221-9. [DOI] [PubMed] [Google Scholar]
- Coco ML, Kuhn CM, Ely TD, Kilts CD. Selective activation of mesoamygdaloid dopamine neurons by conditioned stress: attenuation by diazepam. Brain Res. 1992;590:39–47. doi: 10.1016/0006-8993(92)91079-t. [DOI] [PubMed] [Google Scholar]
- Conti LH, Costill JE, Flynn S, Tayler JE. Effects of a typical and an atypical antipsychotic on the disruption of prepulse inhibition caused by corticotropin-releasing factor and by rat strain. Behav Neurosci. 2005;119:1052–60. doi: 10.1037/0735-7044.119.4.1052. [DOI] [PubMed] [Google Scholar]
- Conti LH, Murry JD, Ruiz MA, Printz MP. Effects of corticotropin-releasing factor on prepulse inhibition of the acoustic startle response in two rat strains. Psychopharmacology (Berl) 2002;161:296–303. doi: 10.1007/s00213-002-1025-2. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N, Hitzemann RJ, Maxson SC, Miner LL, Silva AJ, Wehner JM, Wynshaw-Boris A, Paylor R. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology (Berl) 1997;132:107–24. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- Dautzenberg FM, Hauger RL. The CRF peptide family and their receptors: yet more partners discovered. Trends Pharmacol Sci. 2002;23:71–7. doi: 10.1016/s0165-6147(02)01946-6. [DOI] [PubMed] [Google Scholar]
- Davis M, Walker DL, Lee Y. Amygdala and bed nucleus of the stria terminalis: differential roles in fear and anxiety measured with the acoustic startle reflex. Philos Trans R Soc Lond B Biol Sci. 1997;352:1675–87. doi: 10.1098/rstb.1997.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutch AY, Roth RH. The determinants of stress-induced activation of the prefrontal cortical dopamine system. Prog Brain Res. 1990;85:367–402. doi: 10.1016/s0079-6123(08)62691-6. [DOI] [PubMed] [Google Scholar]
- Dirks A, Groenink L, Westphal KG, Olivier JD, Verdouw PM, van der Gugten J, Geyer MA, Olivier B. Reversal of startle gating deficits in transgenic mice overexpressing corticotropin-releasing factor by antipsychotic drugs. Neuropsychopharmacology. 2003;28:1790–8. doi: 10.1038/sj.npp.1300256. [DOI] [PubMed] [Google Scholar]
- Drago J, Gerfen CR, Lachowicz JE, Steiner H, Hollon TR, Love PE, Ooi GT, Grinberg A, Lee EJ, Huang SP, et al. Altered striatal function in a mutant mouse lacking D1A dopamine receptors. Proc Natl Acad Sci U S A. 1994;91:12564–8. doi: 10.1073/pnas.91.26.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulawa SC, Geyer MA. Psychopharmacology of prepulse inhibition in mice. Chin J Physiol. 1996;39:139–46. [PubMed] [Google Scholar]
- Dunn AJ. Footshock-induced changes in brain catecholamines and indoleamines are not mediated by CRF or ACTH. Neurochem Int. 2000;37:61–9. doi: 10.1016/s0197-0186(99)00163-1. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH, Palamarchouk V. Brain circuits involved in corticotropin-releasing factor-norepinephrine interactions during stress. Ann N Y Acad Sci. 2004;1018:25–34. doi: 10.1196/annals.1296.003. [DOI] [PubMed] [Google Scholar]
- Dunn AJ. Stress-related activation of cerebral dopaminergic systems. Ann N Y Acad Sci. 1988;537:188–205. doi: 10.1111/j.1749-6632.1988.tb42106.x. [DOI] [PubMed] [Google Scholar]
- Eliava M, Yilmazer-Hanke D, Asan E. Interrelations between monoaminergic afferents and corticotropin-releasing factor-immunoreactive neurons in the rat central amygdaloid nucleus: ultrastructural evidence for dopaminergic control of amygdaloid stress systems. Histochem Cell Biol. 2003;120:183–197. doi: 10.1007/s00418-003-0557-9. [DOI] [PubMed] [Google Scholar]
- Emoto H, Tanaka M, Koga C, Yokoo H, Tsuda A, Yoshida M. Corticotropin-releasing factor activates the noradrenergic neuron system in the rat brain. Pharmacol Biochem Behav. 1993;45:419–22. doi: 10.1016/0091-3057(93)90259-v. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156:117–54. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Gorman AL, Dunn AJ. Beta-adrenergic receptors are involved in stress-related behavioral changes. Pharmacol Biochem Behav. 1993;45:1–7. doi: 10.1016/0091-3057(93)90078-8. [DOI] [PubMed] [Google Scholar]
- Graham FK. Presidential Address, 1974. The more or less startling effects of weak prestimulation. Psychophysiology. 1975;12:238–48. doi: 10.1111/j.1469-8986.1975.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Grillon C, Morgan CA, 3rd, Davis M, Southwick SM. Effects of experimental context and explicit threat cues on acoustic startle in Vietnam veterans with posttraumatic stress disorder. Biol Psychiatry. 1998;44:1027–36. doi: 10.1016/s0006-3223(98)00034-1. [DOI] [PubMed] [Google Scholar]
- Grillon C, Morgan CA, Southwick SM, Davis M, Charney DS. Baseline startle amplitude and prepulse inhibition in Vietnam veterans with posttraumatic stress disorder. Psychiatry Res. 1996;64:169–78. doi: 10.1016/s0165-1781(96)02942-3. [DOI] [PubMed] [Google Scholar]
- Hatalski CG, Guirguis C, Baram TZ. Corticotropin releasing factor mRNA expression in the hypothalamic paraventricular nucleus and the central nucleus of the amygdala is modulated by repeated acute stress in the immature rat. J Neuroendocrinol. 1998;10:663–9. doi: 10.1046/j.1365-2826.1998.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauger RL, Risbrough VB, Brauns O, Dautzenberg FM. Corticotropin Releasing Factor (CRF) Receptor Signaling in the Central Nervous System: New Molecular Targets. Current Drug Targets. 2006;5:453–79. doi: 10.2174/187152706777950684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenig K, Hochrein A, Quednow BB, Maier W, Wagner M. Impaired prepulse inhibition of acoustic startle in obsessive-compulsive disorder. Biol Psychiatry. 2005;57:1153–8. doi: 10.1016/j.biopsych.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Holsboer F, von Bardeleben U, Buller R, Heuser I, Steiger A. Stimulation response to corticotropin-releasing hormone (CRH) in patients with depression, alcoholism and panic disorder. Horm Metab Res Suppl. 1987;16:80–8. [PubMed] [Google Scholar]
- Hsu DT, Chen FL, Takahashi LK, Kalin NH. Rapid stress-induced elevations in corticotropin-releasing hormone mRNA in rat central amygdala nucleus and hypothalamic paraventricular nucleus: an in situ hybridization analysis. Brain Res. 1998;788:305–10. doi: 10.1016/s0006-8993(98)00032-8. [DOI] [PubMed] [Google Scholar]
- Kellner M, Yehuda R. Do panic disorder and posttraumatic stress disorder share a common psychoneuroendocrinology? Psychoneuroendocrinology. 1999;24:485–504. doi: 10.1016/s0306-4530(99)00012-8. [DOI] [PubMed] [Google Scholar]
- Kelly MA, Rubinstein M, Phillips TJ, Lessov CN, Burkhart-Kasch S, Zhang G, Bunzow JR, Fang Y, Gerhardt GA, Grandy DK, Low MJ. Locomotor activity in D2 dopamine receptor-deficient mice is determined by gene dosage, genetic background, and developmental adaptations. J Neurosci. 1998;18:3470–9. doi: 10.1523/JNEUROSCI.18-09-03470.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby LG, Rice KC, Valentino RJ. Effects of corticotropin-releasing factor on neuronal activity in the serotonergic dorsal raphe nucleus. Neuropsychopharmacology. 2000;22:148–62. doi: 10.1016/S0893-133X(99)00093-7. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Koob GF. Corticotropin-Releasing Factor in Brain: A Role in Activation, Arousal, and Affect Regulation. J Pharmacol Exp Ther. 2004;311:427–40. doi: 10.1124/jpet.103.052092. [DOI] [PubMed] [Google Scholar]
- Kozicz T, Korosi A, Korsman C, Tilburg-Ouwens D, Groenink L, Veening J, van Der Gugten J, Roubos E, Olivier B. Urocortin expression in the Edinger-Westphal nucleus is down-regulated in transgenic mice over-expressing neuronal corticotropin-releasing factor. Neuroscience. 2004;123:589–94. doi: 10.1016/j.neuroscience.2003.10.042. [DOI] [PubMed] [Google Scholar]
- Lavicky J, Dunn AJ. Corticotropin-releasing factor stimulates catecholamine release in hypothalamus and prefrontal cortex in freely moving rats as assessed by microdialysis. J Neurochem. 1993;60:602–12. doi: 10.1111/j.1471-4159.1993.tb03191.x. [DOI] [PubMed] [Google Scholar]
- Lawrence AJ, Parish CL, Chen F, et al. Chronic corticotropin-releasing factor type 1 receptor antagonism with antalarmin regulates the dopaminergic system of Fawn-Hooded rats. J Neurochem. 2005;94:1523–34. doi: 10.1111/j.1471-4159.2005.03300.x. [DOI] [PubMed] [Google Scholar]
- Lee Y, Schulkin J, Davis M. Effect of corticosterone on the enhancement of the acoustic startle reflex by corticotropin releasing factor (CRF) Brain Res. 1994;666:93–8. doi: 10.1016/0006-8993(94)90286-0. 1994. [DOI] [PubMed] [Google Scholar]
- Liang KC, Melia KR, Miserendino MJ, Falls WA, Campeau S, Davis M. Corticotropin-releasing factor: long-lasting facilitation of the acoustic startle reflex. J Neurosci. 1992;12:2303–12. doi: 10.1523/JNEUROSCI.12-06-02303.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yu B, Neugebauer V, Grigoriadis DE, Rivier J, Vale WW, Shinnick-Gallagher P, Gallagher JP. Corticotropin-releasing factor and Urocortin I modulate excitatory glutamatergic synaptic transmission. J Neurosci. 2004;24:4020–9. doi: 10.1523/JNEUROSCI.5531-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yu B, Orozco-Cabal L, Grigoriadis DE, Rivier J, Vale WW, Shinnick-Gallagher P, Gallagher JP. Chronic cocaine administration switches corticotropin-releasing factor2 receptor-mediated depression to facilitation of glutamatergic transmission in the lateral septum. J Neurosci. 2005;25:577–83. doi: 10.1523/JNEUROSCI.4196-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Acute and chronic Corticotropin-Releasing Factor 1 receptor blockade inhibits cocaine-induced dopamine release: Correlation with dopamine neuron activity. J Pharmacol Exp Ther. 2005;314:201–6. doi: 10.1124/jpet.105.084913. [DOI] [PubMed] [Google Scholar]
- Lowry CA, Rodda JE, Lightman SL, Ingram CD. Corticotropin-releasing factor increases in vitro firing rates of serotonergic neurons in the rat dorsal raphe nucleus: evidence for activation of a topographically organized mesolimbocortical serotonergic system. J Neurosci. 2000;20:7728–36. doi: 10.1523/JNEUROSCI.20-20-07728.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Liu Z, Huang M, Zhang Z. Dopamine-dependent responses to cocaine depend on corticotropin-releasing factor receptor subtypes. J Neurochem. 2003;84:1378–86. doi: 10.1046/j.1471-4159.2003.01635.x. [DOI] [PubMed] [Google Scholar]
- Ludewig S, Ludewig K, Geyer MA, Hell D, Vollenweider FX. Prepulse inhibition deficits in patients with panic disorder. Depress Anxiety. 2002;15:55–60. doi: 10.1002/da.10026. [DOI] [PubMed] [Google Scholar]
- Ludewig S, Geyer MA, Ramseier M, Vollenweider FX, Rechsteiner E, Cattapan-Ludewig K. Information-processing deficits and cognitive dysfunction in panic disorder. J Psychiatry Neurosci. 2005;30:37–43. [PMC free article] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology (Berl) 1988;94:507–14. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- Matsuzaki I, Takamatsu Y, Moroji T. The effects of intracerebroventricularly injected corticotropin-releasing factor (CRF) on the central nervous system: behavioural and biochemical studies. Neuropeptides. 1989;13:147–55. doi: 10.1016/0143-4179(89)90085-1. [DOI] [PubMed] [Google Scholar]
- McCaughran J, Jr., Mahjubi E, Decena E, Hitzemann R. Genetics, haloperidol-induced catalepsy and haloperidol-induced changes in acoustic startle and prepulse inhibition. Psychopharmacology (Berl) 1997;134:131–9. doi: 10.1007/s002130050434. [DOI] [PubMed] [Google Scholar]
- Meloni EG, Gerety LP, Knoll AT, Cohen BM, Carlezon WA., Jr Behavioral and anatomical interactions between dopamine and corticotropin-releasing factor in the rat. J Neurosci. 2006;26:3855–63. doi: 10.1523/JNEUROSCI.4957-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merali Z, McIntosh J, Kent P, Michaud D, Anisman H. Aversive and appetitive events evoke the release of corticotropin-releasing hormone and bombesin-like peptides at the central nucleus of the amygdala. J Neurosci. 1998;18:4758–66. doi: 10.1523/JNEUROSCI.18-12-04758.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Newman-Tancredi A, Quentric Y, Cussac D. The “selective” dopamine D1 receptor antagonist, SCH23390, is a potent and high efficacy agonist at cloned human serotonin2C receptors. Psychopharmacology (Berl) 2001;156:58–62. doi: 10.1007/s002130100742. [DOI] [PubMed] [Google Scholar]
- Ouagazzal AM, Jenck F, Moreau JL. Drug-induced potentiation of prepulse inhibition of acoustic startle reflex in mice: a model for detecting antipsychotic activity? Psychopharmacology (Berl) 2001;156:273–83. doi: 10.1007/s002130100763. [DOI] [PubMed] [Google Scholar]
- Pani L, Porcella A, Gessa GL. The role of stress in the pathophysiology of the dopaminergic system. Mol Psychiatry. 2000;5:14–21. doi: 10.1038/sj.mp.4000589. [DOI] [PubMed] [Google Scholar]
- Peeters PJ, Fierens FL, van den Wyngaert I, Goehlmann HW, Swagemakers SM, Kass SU, Langlois X, Pullan S, Stenzel-Poore MP, Steckler T. Gene expression profiles highlight adaptive brain mechanisms in corticotropin releasing factor overexpressing mice. Brain Res Mol Brain Res. 2004;129:135–50. doi: 10.1016/j.molbrainres.2004.06.038. [DOI] [PubMed] [Google Scholar]
- Pelton GH, Lee Y, Davis M. Repeated stress, like vasopressin, sensitizes the excitatory effects of corticotropin releasing factor on the acoustic startle reflex. Brain Res. 1997;778:381–7. doi: 10.1016/s0006-8993(97)00669-0. [DOI] [PubMed] [Google Scholar]
- Pernar L, Curtis AL, Vale WW, Rivier JE, Valentino RJ. Selective activation of corticotropin-releasing factor-2 receptors on neurochemically identified neurons in the rat dorsal raphe nucleus reveals dual actions. J Neurosci. 2004;24:1305–11. doi: 10.1523/JNEUROSCI.2885-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic J, Fischer A, Katerkamp U, Spiess J. Role of regional neurotransmitter receptors in corticotropin-releasing factor (CRF)-mediated modulation of fear conditioning. Neuropharmacology. 2000;39:707–10. doi: 10.1016/s0028-3908(99)00185-9. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Caine SB. Dopamine D1 and D2 Agonist Effects on Prepulse Inhibition and Locomotion: Comparison of Sprague-Dawley Rats to Swiss-Webster, 129X1/SvJ, C57BL/6J, and DBA/2J Mice. J Pharmacol Exp Ther. 2005;312:733–41. doi: 10.1124/jpet.104.074468. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Fumagalli F, Caron MG, Geyer MA. Prepulse inhibition deficits and perseverative motor patterns in dopamine transporter knock-out mice: differential effects of D1 and D2 receptor antagonists. J Neurosci. 2001;21:305–13. doi: 10.1523/JNEUROSCI.21-01-00305.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph RJ, Varty GB, Kelly MA, Wang YM, Caron MG, Rubinstein M, Grandy DK, Low MJ, Geyer MA. The dopamine D2, but not D3 or D4, receptor subtype is essential for the disruption of prepulse inhibition produced by amphetamine in mice. J Neurosci. 1999;19:4627–33. doi: 10.1523/JNEUROSCI.19-11-04627.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph-Williams RJ, Lehmann-Masten V, Geyer MA. Dopamine D1 rather than D2 receptor agonists disrupt prepulse inhibition of startle in mice. Neuropsychopharmacology. 2003;28:108–18. doi: 10.1038/sj.npp.1300017. [DOI] [PubMed] [Google Scholar]
- Ralph-Williams RJ, Lehmann-Masten V, Otero-Corchon V, Low MJ, Geyer MA. Differential effects of direct and indirect dopamine agonists on prepulse inhibition: a study in D1 and D2 receptor knock-out mice. J Neurosci. 2002;22:9604–11. doi: 10.1523/JNEUROSCI.22-21-09604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reul JM, Holsboer F. Corticotropin-releasing factor receptors 1 and 2 in anxiety and depression. Curr Opin Pharmacol. 2002;2:23–33. doi: 10.1016/s1471-4892(01)00117-5. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Hauger RL, Pelleymounter MA, Geyer MA. Role of corticotropin releasing factor (CRF) receptors 1 and 2 in CRF-potentiated acoustic startle in mice. Psychopharmacology (Berl) 2003;170:178–87. doi: 10.1007/s00213-003-1535-6. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Hauger RL, Roberts AL, Vale WW, Geyer MA. Corticotropin-releasing factor receptors CRF1 and CRF2 exert both additive and opposing influences on defensive startle behavior. J Neurosci. 2004;24:6545–52. doi: 10.1523/JNEUROSCI.5760-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy-Byrne PP, Uhde TW, Post RM, Gallucci W, Chrousos GP, Gold PW. The corticotropin-releasing hormone stimulation test in patients with panic disorder. Am J Psychiatry. 1986;143:896–9. doi: 10.1176/ajp.143.7.896. [DOI] [PubMed] [Google Scholar]
- Sauvage M, Steckler T. Detection of corticotropin-releasing hormone receptor 1 immunoreactivity in cholinergic, dopaminergic and noradrenergic neurons of the murine basal forebrain and brainstem nuclei--potential implication for arousal and attention. Neuroscience. 2001;104:643–52. doi: 10.1016/s0306-4522(01)00137-3. [DOI] [PubMed] [Google Scholar]
- Smoller JW, Yamaki LH, Fagerness JA, Biederman J, Racette S, Laird NM, Kagan J, Snidman N, Faraone SV, Hirshfeld-Becker D, Tsuang MT, Slaugenhaupt SA, Rosenbaum JF, Sklar PB. The corticotropin-releasing hormone gene and behavioral inhibition in children at risk for panic disorder. Biol Psychiatry. 2005;57:1485–92. doi: 10.1016/j.biopsych.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Spina MG, Basso AM, Zorrilla EP, Heyser CJ, Rivier J, Vale W, Merlo-Pich E, Koob GF. Behavioral effects of central administration of the novel CRF antagonist astressin in rats. Neuropsychopharmacology. 2000;22:230–9. doi: 10.1016/S0893-133X(99)00108-6. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–86. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Britton KT. Alphaxalone, a steroid anesthetic, inhibits the startle-enhancing effects of corticotropin releasing factor, but not strychnine. Psychopharmacology (Berl) 1994;115:141–6. doi: 10.1007/BF02244764. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Britton KT, Koob GF. Potentiation of acoustic startle by corticotropin-releasing factor (CRF) and by fear are both reversed by alpha-helical CRF (9-41) Neuropsychopharmacology. 1989;2:285–92. doi: 10.1016/0893-133x(89)90033-x. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Caine SB, Geyer MA. Regionally selective effects of intracerebral dopamine infusion on sensorimotor gating of the startle reflex in rats. Psychopharmacology (Berl) 1992;108:189–95. doi: 10.1007/BF02245306. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Vale WW, Koob GF. Corticotropin-releasing factor potentiates acoustic startle in rats: blockade by chlordiazepoxide. Psychopharmacology (Berl) 1986;88:147–52. doi: 10.1007/BF00652231. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Mansbach RS, Geyer MA, Pulvirenti L, Koob GF, Braff DL. Amphetamine disruption of prepulse inhibition of acoustic startle is reversed by depletion of mesolimbic dopamine. Psychopharmacology (Berl) 1990;100:413–6. doi: 10.1007/BF02244616. [DOI] [PubMed] [Google Scholar]
- Toufexis DJ, Davis C, Hammond A, Davis M. Progesterone attenuates corticotropin-releasing factor-enhanced but not fear-potentiated startle via the activity of its neuroactive metabolite, allopregnanolone. J. Neurosci. 2004;24:10280–7. doi: 10.1523/JNEUROSCI.1386-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Singh V, Crowder TL, Yaka R, Ron D, Bonci A. Corticotropin-releasing factor requires CRF binding protein to potentiate NMDA receptors via CRF receptor 2 in dopamine neurons. Neuron. 2003;39:401–7. doi: 10.1016/s0896-6273(03)00461-6. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Rudoy C, Saunders A, Liu XB, Van Bockstaele EJ. Corticotropin-releasing factor is preferentially colocalized with excitatory rather than inhibitory amino acids in axon terminals in the peri-locus coeruleus region. Neuroscience. 2001;106:375–84. doi: 10.1016/s0306-4522(01)00279-2. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Taaid N, Swerdlow NR. Do D1/D2 interactions regulate prepulse inhibition in rats? Neuropsychopharmacology. 1996;14:265–74. doi: 10.1016/0893-133X(95)00133-X. [DOI] [PubMed] [Google Scholar]
- Wang B, Shaham Y, Zitzman D, Azari S, Wise RA, You ZB. Cocaine experience establishes control of midbrain glutamate and dopamine by corticotropin-releasing factor: a role in stress-induced relapse to drug seeking. J Neurosci. 2005;25:5389–96. doi: 10.1523/JNEUROSCI.0955-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weninger SC, Peters LL, Majzoub JA. Urocortin expression in the Edinger-Westphal nucleus is up-regulated by stress and corticotropin-releasing hormone deficiency. Endocrinology. 2000;141:256–63. doi: 10.1210/endo.141.1.7277. [DOI] [PubMed] [Google Scholar]
- Yang XM, Dunn AJ. Central beta 1-adrenergic receptors are involved in CRF-induced defensive withdrawal. Pharmacol Biochem Behav. 1990;36:847–51. doi: 10.1016/0091-3057(90)90088-y. [DOI] [PubMed] [Google Scholar]
- Zhang JJ, Swiergiel AH, Palamarchouk VS, Dunn AJ. Intracerebroventricular infusion of CRF increases extracellular concentrations of norepinephrine in the hippocampus and cortex as determined by in vivo voltammetry. Brain Res Bull. 1998;47:277–84. doi: 10.1016/s0361-9230(98)00117-8. [DOI] [PubMed] [Google Scholar]