

SUMMARY
Hepatitis B virus (HBV) causes acute and chronic necroinflammatory liver diseases and hepatocellular carcinoma (HCC). HBV replicates noncytopathically in the hepatocyte, and most of the liver injury associated with this infection reflects the immune response. While the innate immune response may not contribute significantly to the pathogenesis of liver disease or viral clearance, the adaptive immune response, particularly the cytotoxic T lymphocyte (CTL) response, contributes to both. Recent observations also reveal that antigen-nonspecific inflammatory cells enhance CTL-induced liver pathology and, more surprisingly, that platelets facilitate the intrahepatic accumulation of CTLs, suggesting that the host response to HBV infection is a highly complex but coordinated process. The notion that platelets contribute to liver disease and viral clearance by promoting the recruitment of virus-specific CTLs into the liver is a new concept in viral pathogenesis, which may prove useful to implement treatments of chronic HBV infection in man.
Keywords: Hepatitis B virus, viral hepatitis, cytotoxic T cells, platelets, cytokines, chemokines
INTRODUCTION
HBV is an enveloped, noncytopathic and hepatotropic DNA virus that causes a liver disease of variable duration and severity (1). Over 95% of acutely infected adults completely and spontaneously recover from the infection, while most neonatally transmitted infections become persistent (2). Chronic HBV infection often progresses to the development of life-threatening complications such as cirrhosis and hepatocellular carcinoma (HCC) (3). On a worldwide basis, over 350 million people are chronically infected by HBV and about 1 million of them die each year from the complications of chronic infection (2). As many of these patients do not have a sustained response to currently available therapies (nucleoside analogues and/or interferon) (2), it is very important to improve our understanding of HBV pathogenesis if we are to develop better treatments.
The experimental approaches examining HBV pathogenesis have been difficult because the host range of HBV is limited to man and chimpanzees, and because in vitro systems for the propagation of HBV do not exist. Studies of HBV pathogenesis using models of HBV-related hepadnavirus infections in the woodchuck, ground squirrel and Pekin duck have also been difficult because the immune systems of these outbred species have not been characterized. To overcome these limitations, we developed transgenic mice that express the viral genes individually (4–7), and also mice that express all of the viral gene products and replicate HBV at high levels in the primary hepatocyte in vivo (8).
The availability of these small animal models with a well-defined immune system and the experimental use of HBV-infected chimpanzees have helped to elucidate in recent years many immunological mechanisms involved in HBV pathogenesis (1). In the course of these studies several other previously unknown aspects of viral pathogenesis have been discovered, and they represent the main subject of this review. In particular, we will examine current evidence pertaining to the pathogenetic and antiviral role of innate and adaptive immune responses, with particular emphasis on the cellular and molecular mechanisms responsible for liver damage and the role that platelets play in these processes.
Innate immune responses
As viruses infect target cells, the host rapidly triggers early innate defense mechanisms in order to contain viral spread. These mechanisms comprise the induction of apoptosis by the virus (9), the production of antiviral cytokines such as IFN-α̃β by the infected cells (10), and the activation of effector functions of cellular components of the innate immune system (such as NK and NKT cells) (11).
There is currently no evidence that HBV can trigger apoptosis. During the early phase of HBV infection in chimpanzees (i.e. before virus-specific T cells enter the liver) there is no histological or biochemical evidence of hepatocellular injury (12, 13). In addition, HBV is able to replicate at high levels in the liver of both patients and transgenic mice noncytopathically, when cellular immune responses are pharmacologically suppressed, anergic or deleted (1, 8). The evidence that HBV induces the production of IFN-α̃β by the infected cells is also lacking. Indeed, global gene expression profiling performed on liver RNA samples from HBV-infected chimpanzees and transgenic mice indicate that no IFN-α̃β or IFN-α̃β-responsive genes are induced in the organ before the entry of adaptive immune responses (14). Although activation of NK cells, NKT cells or engagement of Toll-like receptors have been shown to inhibit viral replication in HBV transgenic mice (15–17), there is still no evidence that these cells or pathways of the innate immune system play a role in disease pathogenesis or viral clearance during the initial phase of HBV infection. Indeed, activated NK cells and NKT cells are an abundant source of IFN-γ (18, 19), but neither IFN-γ nor IFN-γ-inducible genes are detected in the liver of chimpanzees as HBV spreads throughout the organ noncytopathically (12, 13). Collectively, these results indicate that early innate defense mechanisms may not significantly contribute to the pathogenesis of liver injury or to viral clearance, and that HBV remains quite undetected until the adaptive immune response enters the liver.
Adaptive immune responses
Virus-specific CD4+ T helper cells and CD8+ CTLs participate in tissue damage and viral clearance either by killing infected cells or by producing soluble factors such as cytokines and chemokines that contribute to the inflammatory process and/or inhibit viral replication (20). T-cell derived cytokines and chemokines also promote the shaping of antiviral antibody responses that take part in viral clearance, mainly by blocking virus entry into susceptible cells and by removing infectious virions from the circulation.
As mentioned earlier, we lack cell culture systems capable of being productively infected by HBV. Therefore, the kinetics and function of neutralizing Abs in the resolution or prevention of HBV is still poorly understood. Evidence that Abs with neutralizing activity emerge following a self-limited HBV infection is supported by the observation that chimpanzees that resolved a previous infection are completely protected from rechallenge (21). The appearance of neutralizing Abs, however, is thought to occur relatively late after HBV exposure and, thus, it is unlikely to contribute to the early phase of viral clearance during acute infection (21).
It is also unlikely that CD4+ T helper cells play an important pathogenetic role in HBV infection, despite the fact that they have been shown to have cytolytic activity in other infection systems (22). Indeed, recent observations in an acutely HBV-infected chimpanzee that was depleted of CD4+ T cells at the peak of infection indicated that the liver disease in this animal was comparable to that detected in immunologically unmanipulated controls (13). Thus, CD4+ T helper cells may contribute to the control of HBV infection mainly by facilitating the induction and maintenance of virus-specific CTLs, as has been suggested for hepatitis C virus (HCV) (23). In keeping with this, relatively vigorous HBV-specific T helper responses are always associated with quantitatively and qualitatively significant CTL responses in humans and chimpanzees that resolve HBV infection (21).
In contrast to CD4+ T helper cells that are primed within secondary lymphoid organs by antigen presenting cells (APCs) that have internalized soluble viral antigens, priming of CTLs requires the processing of viral proteins that are either endogenously produced within or phagocytosed by professional APCs (24, 25). For viruses like HBV that do not considerably infect professional APCs, tissue-derived dendritic cells that have internalized apoptotic virus-infected cells and debris are expected to migrate to the regional lymph nodes to permit T cell priming to occur (24, 25). The recognition of antigen by naïve CD8+ CTL precursors circulating through secondary lymphoid organs initiates the development of effector CTLs, which clonally expand and leave the lymph nodes, due to the altered expression of surface molecules (26). Indeed, developing effector CTLs modify the expression of selectin ligands such as P-selectin glycoprotein ligand (PSGL)-1 (27), as well as distinct chemokine receptors and integrins, which direct their recruitment to nonlymphoid vascular beds, like those of the liver (28). It is also noteworthy that activated platelets express abundant P-selectin (see below) and functional PSGL-1 is expressed at particularly high levels in effector T cells polarized in the direction of type 1 (29, 30), like the murine HBV-specific CTL lines and clones we have used over the years in many studies involving HBV transgenic mice.
Effector CD8+ CTLs are thought to play a fundamental role in the pathogenesis of liver disease and viral clearance during HBV infection, and this is supported by the following data. First, the beginning of liver injury kinetically corresponds with the influx of virus-specific CTLs into the liver of chimpanzees infected by HBV, and depletion of these cells at the peak of viremia delays the onset of biochemical, histological and clinical evidence of viral hepatitis as well as viral clearance (12, 13). Second, the association between the magnitude of virus-specific CTL responses, liver disease severity and viral clearance has been reported not only in infected chimpanzees but also in many studies of patients infected with HBV. Indeed, patients with acute viral hepatitis, who successfully clear HBV, mount a relatively vigorous multispecific polyclonal CTL response to several HBV-encoded antigens that is usually associated with a relatively severe degree of hepatocyte damage (21, 31). In contrast, liver cell injury is rather limited in chronically infected patients in whom the CTL response is extremely weak and narrowly focused (21, 31). Third, the adoptive transfer of HBV-specific CTL lines and clones into immunologically tolerant HBV transgenic mice triggers a necroinflammatory liver disease that shares the same histologic features of acute viral hepatitis in man and results in the inhibition of HBV replication (32).
The latter studies in mice have also taught us that the antiviral potential of virus-specific CTLs is largely mediated by noncytolytic mechanisms involving the local production of IFN-γ by these cells early after antigen recognition (32). Indeed, it has been reported that IFN-γ (mostly via its capacity to induce nitric oxide in the liver (33)) prevents the assembly of replication-competent HBV RNA-containing capsids in the hepatocyte (34) in a proteasome- (35) and kinase-dependent (36) manner. During this process, the viral nucleocapsids disappear from the cytoplasm of the hepatocytes (37) and the viral RNAs are destabilized by a SSB/La-dependent mechanism in the nucleus (38–40), yet the hepatocytes remain perfectly healthy. The notion that IFN-γ produced by activated CTLs plays a direct role in viral clearance is corroborated by studies in chimpanzees acutely infected with HBV. It was shown in those animals that most of the viral DNA disappeared from the liver before the peak of liver disease, concomitant with the initial intrahepatic appearance of IFN-γ (12). Moreover, neither intrahepatic IFN-γ induction nor viral clearance occurred in HBV-infected chimpanzees that were depleted of CTLs at the peak of infection (13).
Interestingly, recent work in the HBV transgenic mouse model indicates that, upon entry into the liver, the capacity of virus-specific effector CTLs to secrete IFN-γ rapidly subsides, and this phenotype is maintained until HBV antigens are cleared from the liver (41). The results suggest that sustained antigen stimulation (as it occurs during chronic infection) is responsible for the lack of IFN-γ production by CTLs. Even more interesting is the observation that this process is kinetically followed by the intrahepatic expansion of IFN-γ-non-producing CTLs with increased cytotoxic capabilities (41), suggesting that the antiviral (i.e. production of IFN-γ), but not pathogenetic (i.e. killing of hepatocytes) potential of intrahepatic CTLs is likely to be impaired in chronically infected patients. According to this scenario, chronic HBV infection may be characterized by a numerically deficient CTL response that contributes more to liver damage than to viral clearance.
Based on the aforementioned results, is apparent that HBV replicates noncytopathically in the hepatocyte, and that most of the liver damage associated with this infection reflects the immune response. It is also evident that the innate immune response does not contribute significantly to the pathogenesis of liver disease or viral clearance, while the adaptive immune response, especially the virus-specific CTL response, contributes to both. Although hepatocellular injury is initiated and mediated by the CTLs, however, it is becoming increasingly clear that antigen-nonspecific inflammatory cells exacerbate CTL-induced immunopathology. Results pertaining to this subject are summarized below.
The pathogenetic role of antigen non-specific inflammatory cells
As mentioned above, we produced HBV-replicating transgenic mice that show no signs of liver disease (8) until the adoptive transfer of virus-specific CTLs, whereupon they develop a necroinflammatory liver disease that is histologically similar to acute viral hepatitis in man (32). As CTLs reach the liver parenchyma, the first step in the disease process is antigen recognition by these cells, which rapidly induces hepatocellular apoptosis (42). The initial apoptotic process, however, involves a relatively small number of hepatocytes. As time progresses, many host-derived, antigen non-specific inflammatory cells are recruited into the liver, thereby contributing to the formation of necroinflammatory foci scattered throughout the liver parenchyma, in which apoptotic hepatocytes and virus-specific CTLs are outnumbered by host derived mononuclear and polymorphonuclear inflammatory cells (43).
The recruitment process of antigen non-specific mononuclear cells is a chemokine-dependent event, since blocking the chemokines CXCL9 and CXCL10 reduces the trafficking of these cells into the liver without affecting the homing capacity of virus-specific CTLs and polymorphonuclear neutrophils (PMNs) (44). The association of reduced liver disease with reduced recruitment of antigen non-specific mononuclear cells implies that these cells can amplify the liver damage initiated by the CTLs. Similar mechanisms may contribute to the pathogenesis of viral hepatitis in man, where, like as in our system, the number of HBV-specific T cells detected in the liver is outnumbered by recruited non-virus-specific T cells and other mononuclear inflammatory cells (45).
Further studies also demonstrated that the severity of CTL-induced liver disease in this model is ameliorated by the depletion of Gr-1+ cells (Gr-1 is an antigen highly expressed by PMNs), which, secondarily, abolishes the intrahepatic recruitment of all antigen non-specific Gr-1− mononuclear cells (NK and NKT cells, T and B lymphocytes, monocytes and, macrophages, dendritic cells) despite the strong induction of chemokine gene expression (46). Those results suggest that in addition to chemokine expression, other CTL-induced functions are necessary for mononuclear cell recruitment to occur. These functions likely include the production of matrix-degrading metalloproteinases (MMPs) by PMNs (such as MMP-8 and MMP-9), since these enzymes are rapidly activated into the liver after CTL transfer, and their functional inhibition reduces the intrahepatic recruitment of antigen non-specific mononuclear cells and much of the attendant liver disease (47). The results are compatible with the hypothesis that PMNs are the first cell type to be recruited into the liver following antigen recognition by the CTLs. According to this, the production of MMPs by PMNs may cleave components of the extracellular matrix and, thus, facilitate the trafficking of mononuclear cells into the liver parenchyma in response to their own chemoattractants (i.e. CXCL9 and CXCL10).
Recent data also indicates that hepatocellular damage itself is may be involved in the initial CTL-induced recruitment of PMNs in our model. Studies by others have shown that high-mobility group box 1 (HMGB1) protein, an abundant nuclear protein acting as an architectural chromatin-binding factor, can be passively released by necrotic or damaged cells and chemoattract PMNs (48, 49). Following transfer of virus-specific CTLs into HBV transgenic mice, HMGB1 translocates from the nucleus to the cytoplasm of injured hepatocytes, and this phenotype likely leads to the release/secretion of HMGB1 into the extracellular space (50). Treatment of CTL-injected HBV transgenic mice with HMGB1 inhibitors decreases the intrahepatic recruitment of PMNs and, secondarily, the homing of antigen non-specific mononuclear cells that amplify the CTL-induced liver damage (50). A cartoon summarizing the results we described thus far in this section is provided in Figure 1.
Figure 1. Pathogenesis of liver disease and viral clearance in HBV transgenic mice.
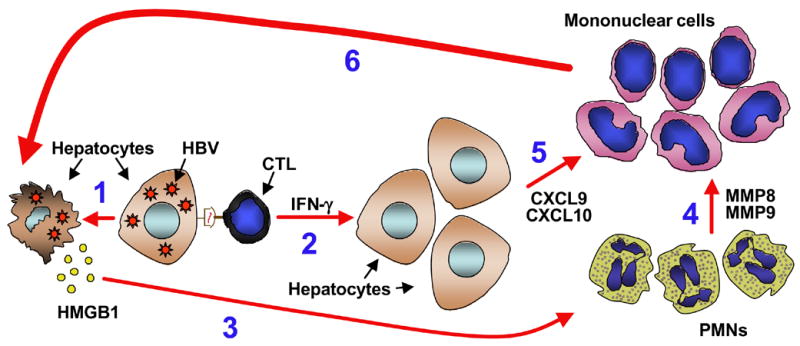
Following antigen recognition, HBV-specific CTLs (CTL) get activated and kill a small number of hepatocytes (1). Activated CTLs also secrete IFN-γ (2), which inhibits viral replication noncytopathically. Damaged or necrotic hepatocytes release HMGB1 (3), which attracts antigen non-specific polymorphonuclear cells (PMNs) into the liver. Production of MMPs by these cells (4) remodels the extracellular matrix and facilitate the intrahepatic migration of antigen non-specific mononuclear cells (i.e. NK cells, T and B cells and monocytes). The migration of these mononuclear cells also requires CXCL9 and CXCL10 (5), two chemokines produced by parenchymal and nonparenchymal cells of the liver in response to IFN-γ. Once they reach the liver parenchyma, antigen non-specific mononuclear cells amplify the liver disease initiated by the CTLs (6).
It is also noteworthy that HMGB1 inhibitors did not impair the homing capacity of HBV-specific CTLs (50), similar to what occurred when we blocked CXCL9 and CXCL10, we depleted PMNs or we inhibited the function of PMN-dependent MMPs (44, 46, 47). These results indicate that antigen-specific CTLs can enter the liver parenchyma and recognize antigen independently of HMGB1, CXCL9, CXCL10 or PMNs, suggesting that other processes may mediate their intrahepatic recruitment.
As mentioned in our Summary, recent observations pertaining to the role of platelets in the trafficking of CTLs within the liver provided new insight regarding this issue. Interestingly, this work (which will be described at length in the following section) was preceded by the observation that the intrahepatic production of nitric oxide (a potent inhibitor of platelet activation (51)) is responsible for limiting CTL recruitment and liver disease severity in our HBV transgenic mouse system as well as in normal inbred mice infected with hepatotropic viruses (33).
The role of platelets in HBV pathogenesis
It is long known that by their capacity to adhere to areas of vascular injury, platelets become activated, aggregate and contribute to the formation of vessel-repairing clots (52, 53). Besides being cellular effectors of hemostasis, platelets are rapidly deployed to sites of injury or infection and potentially modulate inflammatory processes by interacting with leukocytes and by secreting cytokines, chemokines and other inflammatory mediators (52, 53).
Using two different models of acute viral hepatitis, HBV transgenic mice as recipients of HBV-specific CTLs and normal inbred mice acutely infected with adenovirus, we recently showed that platelets are detectable within CTL-containing intrahepatic necroinflammatory foci, alongside apoptotic hepatocytes and inflammatory cells (including virus-specific CTLs) (54). Importantly, platelet depletion greatly ameliorates the severity of liver disease (54). The profound reduction in liver disease severity observed in platelet-depleted animals is associated with a nearly proportional reduction in the intrahepatic accumulation of virus-specific effector CTLs, both of which are restored upon reconstitution with normal platelets, but not upon reconstitution with platelets treated with prostaglandin (PG) E1, (a known inhibitor of platelet activation) (54). Although reduced in number, CTLs recovered from the liver of animals whose platelets are either absent or incapable of becoming activated are intact in their capacity to kill target cells, produce cytokines and chemokines and recruit antigen-nonspecific inflammatory cells into the infected organ (54).
The mechanisms through which platelets facilitate the intrahepatic accumulation of CTLs are poorly understood, and efforts to elucidate these processes are currently pursued. In vitro findings suggest that, under the low shear rate flow conditions likely to occur in the venous circulation of the liver, virus-specific effector CTLs tightly interact with activated platelet and, again, this process is inhibited when platelets are treated with PGE1 (54). Thus, the results of in vivo and in vitro studies are in agreement with the hypothesis that an initial inflammatory response within the liver may result in changes of the vessel wall that promote platelet activation and activation-dependent events resulting in interaction with CTLs. This interaction may eventually facilitate CTLs to egress from the bloodstream, enter the liver parenchyma and perform pathogenetic and/or antiviral functions. A cartoon summarizing this concept is provided in Figure 2.
Figure 2. Platelets facilitate the accumulation of CTLs in the infected liver.
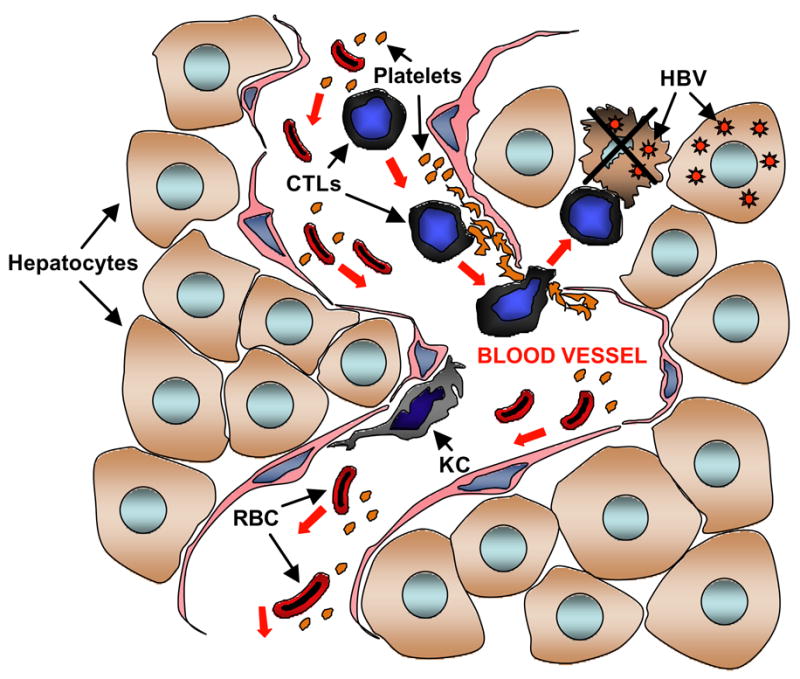
Inflammation-induced changes of the vessel wall may promote platelet adhesion and activation, which in turn favor the exit of virus-specific CTLs from the bloodstream and their accumulation within the liver parenchyma where HBV is replicating. RBC, red blood cells; KC, Kupffer cells.
Platelet activation is a complex process that includes cytoskeletal assembly and shape changes, secretion of agonists such as thromboxane A2 (TXA2) and ADP that promote further activation and aggregation, and functional expression of molecules such as P-selectin, GPIIbIIIa or PSGL-1 (52, 53, 55) that can mediate leukocyte interaction. Pertinent to this, it is noteworthy that platelet P-selectin has been shown to interact with PSGL-1 on leukocytes (including T cells) and promote their rolling along the endothelium of lymph nodes (56). Upon interaction with platelets (via platelet P-selectin), leukocytes are also thought to roll on the endothelium of cutaneous post-capillary venules thanks to platelet expression of PSGL-1 and GPIIbIIIa (57). While platelet PSGL-1 may facilitate this process by directly binding endothelial P-selectin, platelet GPIIbIIIa may do so by binding fibrinogen, which secondarily interacts with endothelial ICAM-1 ((57). Along these lines, intravital microscopy studies in mesenteric venules have recently suggested that, after directly supporting an initial rolling of leukocytes in a P-selectin-dependent manner, platelets (again through P-selectin) stimulate endothelial cells to become activated, express P-selectin themselves, and further sustain leukocyte rolling (58). Following ischemia-reperfusion liver injury, the rolling of polymorphonuclear neutrophils (PMNs) on the endothelium of post-sinusoidal venules has also been shown to require P-selectin (59).
The aforementioned results suggest that molecules such as P-selectin, PSGL-1 and/or GPIIbIIIa could be involved in mediating platelet/CTL interactions within the inflamed liver. Through the use of intravital microscopy studies, platelets, virus-specific CTLs, HBV transgenic mice, adenovirus-infected normal inbred mice and mice genetically deficient for molecules such as P-selectin, PSGL-1 and GPIIbIIIa, we plan to address this hypothesis in the future. Defining the molecular mechanisms whereby platelets interact with CTLs and mediate liver disease and/or viral clearance may shed new light on previously unknown and important aspects of the pathogenesis of HBV and possibly other viruses (i.e. HCV) which infect the liver and whose clearance depends on CTLs (23, 60).
In addition to P-selectin, PSGL-1 and GPIIbIIIa, activated platelets express CD154 (or CD40L), cytokines such as interleukin (IL)-1-β and chemokines such as CCL5 (i.e. RANTES), all of which have the potential to induce endothelial cells to become activated and facilitate leukocyte rolling through the expression of integrins and intercellular (ICAMs) or vascular (VCAM-1) adhesion molecules (52, 53). Although it has been reported that platelet CD154 induces (at least under non-flow conditions in vitro) endothelial expression of adhesion molecules (61), its involvement in leukocyte rolling has been denied by intravital microscopic studies within mesenteric venules (58). Notably, both IL-1-β and CCL5 are rapidly and strongly expressed in the liver of CTL-injected HBV transgenic mice (44, 62). Passive neutralization of IL-1-β in these animals, however, affects neither the pathogenetic nor the antiviral potential of CTLs (62), indicating that this cytokine is likely not involved in our system. As per CCL5, it is worth mentioning that its secretion and/or deposition on the microvasculature occurs in a platelet P-selectin-dependent manner (63).
While the identification of the molecules involved in platelet/CTL interaction remains to be determined, the aforementioned data suggest that pharmacologic intervention targeting the pro-activating functions of platelet agonists may result in the inhibition of CTL recruitment into the liver and the amelioration of CTL-induced liver disease. Indeed, preliminary experiments indicate that the combined administration of aspirin and clopidogrel drastically inhibited both events in our mouse models of CTL-mediated acute viral hepatitis without causing bleeding side effects (Iannacone et al., unpublished data). Aspirin, the most widely used inhibitor of platelet function, affects TXA2 production by irreversibly inactivating cyclooxigenase-1 (COX-1), thus blocking a feedback activation mechanism (64). Clopidogrel is a prodrug that needs to be converted into an active metabolite by the liver and irreversibly inactivates the ADP-receptor P2Y12, which is required for stable platelet aggregation (65, 66). Of note, aspirin and clopidogrel not only inhibit platelet activation and aggregation in an additive manner (64), but also reduce P-selectin expression, GPIIbIIIa activation and the formation of platelet-leukocyte aggregates (67–69).
The notion that anti-platelet treatment diminishes the severity of CTL-induced liver disease has therapeutic potential for the treatment of chronic HBV infection in man. Indeed, chronic HBV infection is characterized by an inefficient CTL response that is unable to completely eradicate the virus from the liver, often resulting in continuous cycles of low-level liver cell destruction. Persistence of these events for long periods of time is a major cause for the development of life-threatening complications, including HCC. As such, the CTL response in chronically infected patients may cause more harm than good, and, therefore, it is of some interest determining whether continuous administration of aspirin/clopidogrel may reduce its detrimental impact. For this reason, we plan to define in the future whether this aspirin/clopidogrel treatment may prevent the onset or decrease the incidence of HCC in a HBV transgenic mouse model of CTL-mediated chronic liver injury (70, 71). We believe, that findings emerging from these studies may set an important proof of principle for a pharmacologically-based anti-platelet treatment that would control unfavorable immunopathology without causing generalized immune suppression. This knowledge may also pave the road for the design of new and more specific platelet inhibitors (like small molecules inhibiting platelet/CTL interactions).
Concluding remarks
Our comprehension of the pathogenesis of HBV infection has significantly advanced in recent years, particularly because of the experimental use of chimpanzees and transgenic mice. It is becoming increasingly apparent that HBV replicates noncytopathically within the hepatocyte, and that the adaptive immune response, mainly the virus-specific CTL response, plays a crucial role in both liver disease and viral clearance. Recent studies also indicate that antigen-nonspecific inflammatory cells enhance CTL-induced immunopathology in the liver, and that platelets are required for virus-specific CTLs to accumulate within the liver and perform pathogenetic and/or antiviral effector functions. Future work intended to address the molecular basis of platelet/CTL interactions will not only expand our current knowledge of the host-virus relations that determine the pathogenesis of infection, but they may also guide us to the discovery of new approaches for the treatment of chronic HBV infection and its life-threatening complications.
Acknowledgments
This work was supported by grants HL31950, HL42846, HL78784 (ZMR) and AI40696 (LGG) from the National Institutes of Health, USA and from the VIRGIL European Network of Excellence on Antiviral Drug Resistance (LSHM-CT-2004-503359) (LGG).
Footnotes
Competing interests statement: The authors declare that they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. 2006;1(1):23–61. doi: 10.1146/annurev.pathol.1.110304.100230. [DOI] [PubMed] [Google Scholar]
- 2.Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med. 2004 Mar 11;350(11):1118–29. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 3.Brechot C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology. 2004 Nov;127(5 Suppl 1):S56–61. doi: 10.1053/j.gastro.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Chisari FV, Filippi P, McLachlan A, Milich DR, Riggs M, Lee S, et al. Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice. JVirol. 1986;60:880–7. doi: 10.1128/jvi.60.3.880-887.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilles PN, Fey G, Chisari FV. Tumor necrosis factor-alpha negatively regulates hepatitis B virus gene expression in transgenic mice. JVirol. 1992;66:3955–60. doi: 10.1128/jvi.66.6.3955-3960.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guidotti LG, Matzke B, Pasquinelli C, Shoenberger JM, Rogler C, Chisari FV. The hepatitis B virus (HBV) precore protein inhibits HBV replication in transgenic mice. JVirol. 1996;70:7056–61. doi: 10.1128/jvi.70.10.7056-7061.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guidotti LG, Martinez V, Loh YT, Rogler CE, Chisari FV. Hepatitis B virus nucleocapsid particles do not cross the hepatocyte nuclear membrane in transgenic mice. JVirol. 1994;68:5469–75. doi: 10.1128/jvi.68.9.5469-5475.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guidotti LG, Matzke B, Schaller H, Chisari FV. High level hepatitis B virus replication in transgenic mice. JVirol. 1995;69:6158–69. doi: 10.1128/jvi.69.10.6158-6169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koyama AH, Irie H, Fukumori T, Hata S, Iida S, Akari H, et al. Role of virus-induced apoptosis in a host defense mechanism against virus infection. J Med Invest. 1998;45(1–4):37–45. [PubMed] [Google Scholar]
- 10.Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001 Oct;14(4):778–809. doi: 10.1128/CMR.14.4.778-809.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 12.Guidotti LG, Rochford R, Chung L, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–9. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 13.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, et al. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003 Jan;77(1):68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wieland SF, Vega RG, Muller R, Evans CF, Hilbush B, Guidotti LG, et al. Searching for interferon-induced genes that inhibit hepatitis B virus replication in transgenic mouse hepatocytes. J Virol. 2003 Jan;77(2):1227–36. doi: 10.1128/JVI.77.2.1227-1236.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol. 2005 Jun;79(11):7269–72. doi: 10.1128/JVI.79.11.7269-7272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura K, Kakimi K, Wieland S, Guidotti LG, Chisari FV. Interleukin-18 inhibits hepatitis B virus replication in the livers of transgenic mice. J Virol. 2002 Nov;76(21):10702–7. doi: 10.1128/JVI.76.21.10702-10707.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimura K, Kakimi K, Wieland S, Guidotti LG, Chisari FV. Activated intrahepatic antigen-presenting cells inhibit hepatitis B virus replication in the liver of transgenic mice. J Immunol. 2002 Nov 1;169(9):5188–95. doi: 10.4049/jimmunol.169.9.5188. [DOI] [PubMed] [Google Scholar]
- 18.Kakimi K, Lane TE, Chisari FV, Guidotti LG. Cutting edge: Inhibition of hepatitis B virus replication by activated NK T cells does not require inflammatory cell recruitment to the liver. J Immunol. 2001;167(12):6701–5. doi: 10.4049/jimmunol.167.12.6701. [DOI] [PubMed] [Google Scholar]
- 19.Kakimi K, Guidotti LG, Koezuka Y, Chisari FV. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J Exp Med. 2000;192(7):921–30. doi: 10.1084/jem.192.7.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 21.Chisari FV, Ferrari C. Hepatitis B virus immunopathogenesis. Annu Rev Immunol. 1995;13:29–60. doi: 10.1146/annurev.iy.13.040195.000333. [DOI] [PubMed] [Google Scholar]
- 22.Tsuji M, Romero P, Nussenzweig RS, Zavala F. CD4+ cytolytic T cell clone confers protection against murine malaria. J Exp Med. 1990 Nov 1;172(5):1353–7. doi: 10.1084/jem.172.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shoukry NH, Cawthon AG, Walker CM. Cell-mediated immunity and the outcome of hepatitis C virus infection. Annu Rev Microbiol. 2004;58:391–424. doi: 10.1146/annurev.micro.58.030603.123836. [DOI] [PubMed] [Google Scholar]
- 24.Steinman RM, Inaba K, Turley S, Pierre P, Mellman I. Antigen capture, processing, and presentation by dendritic cells: recent cell biological studies. Hum Immunol. 1999;60(7):562–7. doi: 10.1016/s0198-8859(99)00030-0. [DOI] [PubMed] [Google Scholar]
- 25.Sallusto F, Lanzavecchia A. Mobilizing dendritic cells for tolerance, priming, and chronic inflammation. J Exp Med. 1999;189(4):611–4. doi: 10.1084/jem.189.4.611. [comment] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994 Jan 28;76(2):301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 27.van Wely CA, Blanchard AD, Britten CJ. Differential expression of alpha3 fucosyltransferases in Th1 and Th2 cells correlates with their ability to bind P-selectin. Biochem Biophys Res Commun. 1998 Jun 18;247(2):307–11. doi: 10.1006/bbrc.1998.8786. [DOI] [PubMed] [Google Scholar]
- 28.Lalor PF, Shields P, Grant A, Adams DH. Recruitment of lymphocytes to the human liver. Immunol Cell Biol. 2002 Feb;80(1):52–64. doi: 10.1046/j.1440-1711.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- 29.Austrup F, Vestweber D, Borges E, Lohning M, Brauer R, Herz U, et al. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature. 1997 Jan 2;385(6611):81–3. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- 30.Borges E, Tietz W, Steegmaier M, Moll T, Hallmann R, Hamann A, et al. P-selectin glycoprotein ligand-1 (PSGL-1) on T helper 1 but not on T helper 2 cells binds to P-selectin and supports migration into inflamed skin. J Exp Med. 1997 Feb 3;185(3):573–8. doi: 10.1084/jem.185.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferrari C, Missale G, Boni C, Urbani S. Immunopathogenesis of hepatitis B. J Hepatol. 2003;39 (Suppl 1):S36–42. doi: 10.1016/s0168-8278(03)00137-5. [DOI] [PubMed] [Google Scholar]
- 32.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996 Jan;4(1):25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 33.Guidotti LG, McClary H, Loudis JM, Chisari FV. Nitric oxide inhibits hepatitis B virus replication in the livers of transgenic mice. J Exp Med. 2000 Apr 3;191(7):1247–52. doi: 10.1084/jem.191.7.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wieland SF, Guidotti LG, Chisari FV. Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J Virol. 2000 May;74(9):4165–73. doi: 10.1128/jvi.74.9.4165-4173.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robek MD, Wieland SF, Chisari FV. Inhibition of hepatitis B virus replication by interferon requires proteasome activity. J Virol. 2002 Apr;76(7):3570–4. doi: 10.1128/JVI.76.7.3570-3574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robek MD, Boyd BS, Wieland SF, Chisari FV. Signal transduction pathways that inhibit hepatitis B virus replication. Proc Natl Acad Sci U S A. 2004 Feb 10;101(6):1743–7. doi: 10.1073/pnas.0308340100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wieland SF, Eustaquio A, Whitten-Bauer C, Boyd B, Chisari FV. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc Natl Acad Sci U S A. 2005 Jul 12;102(28):9913–7. doi: 10.1073/pnas.0504273102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heise T, Guidotti LG, Chisari FV. Characterization of nuclear RNases that cleave hepatitis B virus RNA near the La protein binding site. J Virol. 2001 Aug;75(15):6874–83. doi: 10.1128/JVI.75.15.6874-6883.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heise T, Guidotti LG, Chisari FV. La autoantigen specifically recognizes a predicted stem-loop in hepatitis B virus RNA. J Virol. 1999;73(7):5767–76. doi: 10.1128/jvi.73.7.5767-5776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heise T, Guidotti LG, Cavanaugh VJ, Chisari FV. Hepatitis B virus RNA-binding proteins associated with cytokine-induced clearance of viral RNA from the liver of transgenic mice. J Virol. 1999;73(1):474–81. doi: 10.1128/jvi.73.1.474-481.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Isogawa M, Furuichi Y, Chisari FV. Oscillating CD8(+) T cell effector functions after antigen recognition in the liver. Immunity. 2005 Jul;23(1):53–63. doi: 10.1016/j.immuni.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Ando K, Guidotti LG, Wirth S, Ishikawa T, Missale G, Moriyama T, et al. Class I restricted cytotoxic T lymphocytes are directly cytopathic for their target cells in vivo. JImmunol. 1994;152:3245–53. [PubMed] [Google Scholar]
- 43.Ando K, Moriyama T, Guidotti LG, Wirth S, Schreiber RD, Schlicht HJ, et al. Mechanisms of class I restricted immunopathology. A transgenic mouse model of fulminant hepatitis. JExpMed. 1993;178:1541–54. doi: 10.1084/jem.178.5.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kakimi K, Lane TE, Wieland S, Asensio VC, Campbell IL, Chisari FV, et al. Blocking chemokine responsive to gamma-2/interferon (IFN)-gamma inducible protein and monokine induced by IFN-gamma activity in vivo reduces the pathogenetic but not the antiviral potential of hepatitis B virus-specific cytotoxic T lymphocytes. J Exp Med. 2001;194(12):1755–66. doi: 10.1084/jem.194.12.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bertoletti A, Maini MK. Protection or damage: a dual role for the virus-specific cytotoxic T lymphocyte response in hepatitis B and C infection? Curr Opin Immunol. 2000;12(4):403–8. doi: 10.1016/s0952-7915(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 46.Sitia G, Isogawa M, Kakimi K, Wieland SF, Chisari FV, Guidotti LG. Depletion of neutrophils blocks the recruitment of antigen-nonspecific cells into the liver without affecting the antiviral activity of hepatitis B virus-specific cytotoxic T lymphocytes. Proc Natl Acad Sci U S A. 2002 Oct 15;99(21):13717–22. doi: 10.1073/pnas.172521999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sitia G, Isogawa M, Iannacone M, Campbell IL, Chisari FV, Guidotti LG. MMPs are required for recruitment of antigen-nonspecific mononuclear cells into the liver by CTLs. J Clin Invest. 2004 Apr;113(8):1158–67. doi: 10.1172/JCI21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999 Jul 9;274(28):19919–24. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 49.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995 Oct 27;270(43):25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 50.Sitia G, Iannacone M, Muller S, Bianchi ME, Guidotti LG. Treatment with HMGB1 inhibitors diminishes CTL-induced liver disease in HBV transgenic mice. J Leukoc Biol. 2006 Aug 25; doi: 10.1189/jlb.0306173. [DOI] [PubMed] [Google Scholar]
- 51.Jin RC, Voetsch B, Loscalzo J. Endogenous mechanisms of inhibition of platelet function. Microcirculation. 2005 Apr-May;12(3):247–58. doi: 10.1080/10739680590925493. [DOI] [PubMed] [Google Scholar]
- 52.Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol. 2004 Sep;25(9):489–95. doi: 10.1016/j.it.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol. 2003 Dec;23(12):2131–7. doi: 10.1161/01.ATV.0000095974.95122.EC. [DOI] [PubMed] [Google Scholar]
- 54.Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, et al. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat Med. 2005 Nov;11(11):1167–9. doi: 10.1038/nm1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002 Nov;8(11):1227–34. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 56.Diacovo TG, Puri KD, Warnock RA, Springer TA, von Andrian UH. Platelet-mediated lymphocyte delivery to high endothelial venules. Science. 1996 Jul 12;273(5272):252–5. doi: 10.1126/science.273.5272.252. [DOI] [PubMed] [Google Scholar]
- 57.Ludwig RJ, Schultz JE, Boehncke WH, Podda M, Tandi C, Krombach F, et al. Activated, not resting, platelets increase leukocyte rolling in murine skin utilizing a distinct set of adhesion molecules. J Invest Dermatol. 2004 Mar;122(3):830–6. doi: 10.1111/j.0022-202X.2004.22318.x. [DOI] [PubMed] [Google Scholar]
- 58.Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. 2005 Oct 1;106(7):2334–9. doi: 10.1182/blood-2005-04-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yadav SS, Howell DN, Steeber DA, Harland RC, Tedder TF, Clavien PA. P-Selectin mediates reperfusion injury through neutrophil and platelet sequestration in the warm ischemic mouse liver. Hepatology. 1999 May;29(5):1494–502. doi: 10.1002/hep.510290505. [DOI] [PubMed] [Google Scholar]
- 60.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005 Mar;5(3):215–29. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 61.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998 Feb 5;391(6667):591–4. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 62.Guidotti LG, Ando K, Hobbs MV, Ishikawa T, Runkel L, Schreiber RD, et al. Cytotoxic T lymphocytes inhibit hepatitis B virus gene expression by a noncytolytic mechanism in transgenic mice. Proc Natl Acad Sci U S A. 1994 Apr 26;91(9):3764–8. doi: 10.1073/pnas.91.9.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. 2002 Sep 17;106(12):1523–9. doi: 10.1161/01.cir.0000028590.02477.6f. [DOI] [PubMed] [Google Scholar]
- 64.Cattaneo M. Aspirin and clopidogrel: efficacy, safety, and the issue of drug resistance. Arterioscler Thromb Vasc Biol. 2004 Nov;24(11):1980–7. doi: 10.1161/01.ATV.0000145980.39477.a9. [DOI] [PubMed] [Google Scholar]
- 65.Gachet C. Regulation of platelet functions by P2 receptors. Annu Rev Pharmacol Toxicol. 2006;46:277–300. doi: 10.1146/annurev.pharmtox.46.120604.141207. [DOI] [PubMed] [Google Scholar]
- 66.Storey RF. Biology and pharmacology of the platelet P2Y12 receptor. Curr Pharm Des. 2006;12(10):1255–9. doi: 10.2174/138161206776361318. [DOI] [PubMed] [Google Scholar]
- 67.Malinin AI, Atar D, Callahan KP, McKenzie ME, Serebruany VL. Effect of a single dose aspirin on platelets in humans with multiple risk factors for coronary artery disease. Eur J Pharmacol. 2003 Feb 21;462(1–3):139–43. doi: 10.1016/s0014-2999(02)02956-4. [DOI] [PubMed] [Google Scholar]
- 68.McKenzie ME, Malinin AI, Bell CR, Dzhanashvili A, Horowitz ED, Oshrine BR, et al. Aspirin inhibits surface glycoprotein IIb/IIIa, P-selectin, CD63, and CD107a receptor expression on human platelets. Blood Coagul Fibrinolysis. 2003 Apr;14(3):249–53. doi: 10.1097/01.mbc.0000046182.72384.ab. [DOI] [PubMed] [Google Scholar]
- 69.Storey RF, Judge HM, Wilcox RG, Heptinstall S. Inhibition of ADP-induced P-selectin expression and platelet-leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thromb Haemost. 2002 Sep;88(3):488–94. [PubMed] [Google Scholar]
- 70.Nakamoto Y, Suda T, Momoi T, Kaneko S. Different procarcinogenic potentials of lymphocyte subsets in a transgenic mouse model of chronic hepatitis B. Cancer Res. 2004 May 1;64(9):3326–33. doi: 10.1158/0008-5472.can-03-3817. [DOI] [PubMed] [Google Scholar]
- 71.Nakamoto Y, Guidotti LG, Pasquetto V, Schreiber RD, Chisari FV. Differential target cell sensitivity to CTL-activated death pathways in hepatitis B virus transgenic mice. J Immunol. 1997 Jun 15;158(12):5692–7. [PubMed] [Google Scholar]