

Abstract
Two novel ligands with a 4′-substitution on the phenyl ring B of biphenylthiol: 5-chloro-2-(2′-((dimethylamino)methyl)-4′-iodophenylthio)benzenamine, 7, and 2-(2′-((dimethylamino)methyl)-4′-methoxyphenylthio)-5-iodobenzenamine, 8, were prepared and tested as potential serotonin transporter (SERT) imaging agents. The new ligands displayed extremely high binding affinities to SERT (Ki = 0.22 ± 0.09 and 0.11 ± 0.04 nM, respectively), with very low binding affinities to dopamine and norepinephrine transporters (Ki > 1, 000 nM). The corresponding [125I]7 and [125I]8 were successfully prepared from the tri-n-butyltin derivatives. They showed good brain uptakes and prolonged retention after iv injection in rats (brain uptake was 1.77 and 0.98 %dose/g for [125I]7 and 0.92 and 0.29 %dose/g for [125I]8 at 2 and 120 minutes, respectively). Significantly, [125I]7 showed excellent uptake and prolonged retention in the hypothalamus, where the SERT concentration is the highest. The hypothalamus/cerebellum ratios (target to background ratios) were 4.24, 7.10, 8.24 and 12.6 at 2, 4, 6 and 12 hours, respectively. The hypothalamus/cerebellum ratios for [125I]8 were 3.97, 5.57 and 5.06 at 1, 2 and 4 hours, respectively. Adding the 4′-iodo- group to the phenyl ring B of 7 appeared to reduce the rate of clearance from the brain, and the kinetics favored uptake and retention in the hypothalamus. The localization of [125I]7 in the hypothalamus region in the brain of rats could be blocked by pretreatment with (+)McN5652, escitalopram and ADAM (2), all selective serotonin transporter ligands (at 2 mg/Kg dose, iv, 5 min pretreatment). Ex vivo autoradiograms of rat brain sections (at 4 hr after iv injection of [125I]7) showed intense labeling in regions of the brain known to have high SERT density. The excellent selective uptake and retention in the hypothalamus region suggests that [123I]7 is a potential lead compound for developing new imaging agents targeting SERT binding sites with single photon emission computed tomography (SPECT).
Introduction
The serotonin transporter (SERT) is a transmembrane protein located in the synapse which is responsible for removing serotonin from the synaptic cleft. The associations between serotonergic function and depression have been well documented [4–6]. It is commonly held that patients with major depression experience changes in the serotonergic neuronal transmission function in the brain [1–3]. A series of newer antidepressants preferentially increase 5-HT (serotonin) transmission by inhibiting serotonin reuptake. Selective serotonin reuptake inhibitors (a.k.a. SSRIs, i.e. citalopram, fluoxetine, fluvoxamine, paroxetine and sertraline) preferably inhibit serotonin uptake compared with dopamine or norepinephrine uptakes. SSRIs have revolutionized the management of depression for millions of patients. They are also widely prescribed for treating various other mental disorders such as obsessive compulsive and social phobic disorders [7, 8].
Recently, in vivo imaging of SERT binding sites in the brain by position emission tomography (PET) has been significantly improved. Encouraging results from human studies using the leading PET imaging agent, [11C]DASB (3) (Table 1) have demonstrated the potential of studying drug occupancy in living human brains [9–14]. Progress has also been made in imaging SERT binding sites in the brain by single photon emission computed tomography (SPECT) [15–17]. SPECT imaging studies using [123I]ADAM (2) were performed to investigate SERT occupancies after a single dose of escitalopram or citalopram. A test-retest study showed that the drug occupancy in the brain could be correlated with the level of antidepressant dose with good reproducibility [13, 15, 16, 18–20].
Table 1.
In vitro inhibition constants (Ki, nM) of SERT ligands
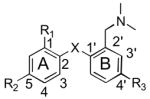
|
Ki (nM)
|
|||||||
|---|---|---|---|---|---|---|---|
| Compd | X | R1 | R2 | R3 | SERT | NET | DAT |
| 1 IDAM a | S | CH2OH | Cl | H | 0.1 ± 0.01 | 234 ± 26 | >1000 |
| 2 ADAM b | S | NH2 | I | H | 0.01 ± 0.00 | 699 ± 80 | 840 ± 100 |
| 3 OADAM c | O | NH2 | I | H | 0.12 ± 0.02 | 20 ± 2 | >1,000 |
| 4 c | CH2 | NH2 | I | H | 48.6 ± 4.10 | 17 ± 4 | >1,000 |
| 5 d | S | NH2 | Br | O(CH2)2F | 0.09 ± 0.07 | >1,000 | >1,000 |
| 6 DASB e | S | NH2 | CN | H | 1.1 ± 0.04 | >1,000 | >1,000 |
| 7 | S | NH2 | Cl | I | 0.22 ± 0.09 | >1,000 | >1,000 |
| 8 | S | NH2 | I | OMe | 0.11 ± 0.04 | >1,000 | >1,000 |
| 19 | S | NH2 | Cl | Br | 0.12 ± 0.09 | >1,000 | >1,000 |
| 21 | S | NH2 | Br | Br | 0.09 ± 0.01 | >1,000 | >1,000 |
| 20 | S | NH2 | Br | OMe | 0.03 ± 0.01 | >1,000 | >1,000 |
| 25 | S | NH2 | Br | OH | 0.14 ± 0.06 | >1,000 | >1,000 |
Up to this point, the development of SPECT or PET imaging agents targeting SERT binding sites has relied on four different core structures, 5-iodo-nitroquipazine [21–23], tropanes [24–26], (+)-McN5652 [27–31] and biphenylthiols [32–40]. Biphenylthiol derivatives, [123I]IDAM (1) [41] and [123I]ADAM (2) [39, 42], have been evaluated as potential agents for SPECT because there are several unique benefits associated with using this core structure (Table 1). Biphenylthiol contains no optical center and the synthesis is relatively simple. Most importantly, this group of compounds is highly selective, showing a high binding affinity to SERT (Ki in nM or sub nM) and relatively low binding affinity to the other monoamine transporters (dopamine transporter, DAT and norepinephrine transporter, NET) and other CNS receptors or binding sites. The biphenylthiol core structure is neutral and relatively small, properties essential for penetrating the intact blood-brain barrier and delivering imaging agents to SERT binding sites in the brain. These distinctive features have attracted many researchers to work on this series of compounds [32–37, 43].
Most of the biphenylthiols reported are derivatives containing substitutions on the phenyl ring A [32, 34]. Our laboratory has previously reported IDAM (1) and ADAM (2). In the ADAM (2) series of compounds, we have recently prepared two derivatives, the O-bridged, OADAM (3) and C-bridged, 4, and measured the kinetics of the brain uptake of the 125I labeled compounds in rats [44] (Table 1). The specific hypothalamus uptake of C-bridged, [125I]4, was inferior to that of OADAM (3) and ADAM (2). It was found that OADAM (3) consistently displayed a higher uptake in the hypothalamus (between 60–240 minutes post iv injection) than that of ADAM (2). The specific uptake of OADAM (3) in the hypothalamus exhibited the highest target to non-target ratio (hypothalamus/cerebellum, HY/CB, was 5.3 at 240 minutes post iv injection). But, one major drawback of OADAM (3) was that it appeared to show less selectivity. It had a higher binding affinity for norepinephrine transporter (NET) (Ki = 20 ± 2 nM) (Table 1), making it the least selective SERT ligand among the biphenylthiols reported so far.
In order to further explore the potential of compounds with a biphenylthiol core, we developed several derivatives, 7 and 8, with 4′-substitutions on the phenyl ring B. To our surprise, these 4′-substituted derivatives showed a remarkable in vivo biodistribution. In particular, the 4′-iodo-biphenylthiol derivative, 7, displayed extraordinary uptake and retention in the hypothalamus, a target region for SERT. In this paper, we report the first examples of iodinated biphenylthiol derivatives with a 4′-substitution on the phenyl ring B. When labeled with 123I (T1/2 = 13 hr and 159 KeV), they were promising agents for imaging SERT with SPECT.
Chemistry
The biphenylthiol derivatives described in this paper were prepared as outlined in Scheme 1. Commercially available 2-amino-5-bromobenzoic acid (9) and 2-amino-5-methoxybenzoic acid (10) were converted to corresponding thiols 11 and 12, which were coupled with either 2,5-dichloronitrobenzene or 2,5-dibromonitrobenzene. The resulting carboxylic acids 13–15 were treated with thionyl chloride and reacted with dimethylamine to yield amides 16–18. Reductions of nitro and amide groups simultaneously by excess borane-THF provided compounds 19, 20 and 21. The bromide groups of 19 and 20 were converted to the corresponding tri-n-butyltin derivatives. The compounds 19 and 20 were used as radio-iodination precursors. Non-radioactive iodinated compounds 7 and 8 were also prepared from 19 and 20 respectively by treatments with iodine in chloroform.
Figure 1.
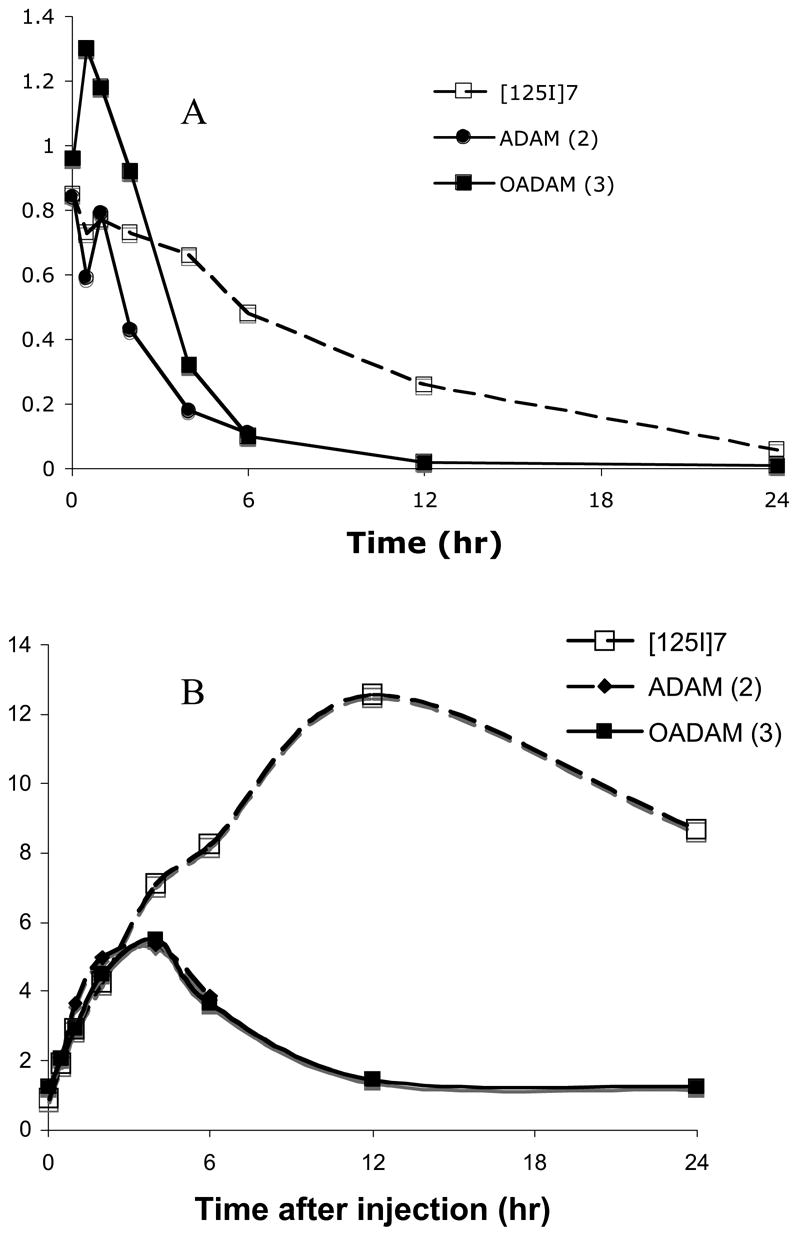
Comparison of total hypothalamus uptakes (A) and hypothalamus/cerebellum (HY/CB) ratios (B) between ADAM (2) [42], OADAM (3) [44] and [125I]7 in rats. The kinetics of the brain uptake and retention for [125I]7 was slower than that of ADAM (2) or OADAM (3). The hypothalamus/cerebellum ratios of ADAM (2) and OADAM (3) reached the highest value at 4 hr post iv injection (HY/CB = 5), while [125I]7 peaked at 12 hr after the tracer injection (HY/CB = 12.6).
Similar to that reported for this series of ligands [41, 42, 44], the preparation of [125I]7 and [125I]8 was successfully performed by using an oxidative iodination of the corresponding tri-n-butyltin derivatives (Scheme 2). The radiolabeling yields for [125I]7 and [125I]8 were 87% and 70%, respectively and the radiochemical purities were 99% and 99%, respectively.
Biological studies and discussion
We used an in vitro binding assay to determine the inhibition constants (Ki, nM) with membrane preparations of three different groups of LLC-PK1 cells, each expressing one specific monoamine transporter, either SERT, DAT or NET [45]. As expected, the binding affinities of 7 and 8 to SERT were excellent (Ki = 0.22 ± 0.07 and 0.11 ± 0.04 nM, respectively), while the affinities to DAT and NET were very low (Ki > 1, 000 nM). The results are consistent with the binding profiles reported for other biphenylthiol derivatives [35–37, 39, 40, 42]. Other 4′-substituted derivatives of this series, 19, 20, 21 and 25 similarly showed sub-nM binding affinities and excellent selectivity (low binding affinities to other monoamine transporters) (Table 1).
The most surprising and unexpected observation for this series was their relatively good brain uptake and the prolonged retention of [125I]7 and [125I]8. Biodistribution studies of [125I]7 and [125I]8 in normal rats (Table 2 and 3) showed that they penetrated the intact blood-brain barrier with initial brain uptakes of 1.77 and 0.92 %dose/organ at 2 min post iv injection, respectively. The brain uptake values showed a low, but steady, decline for both compounds. [125I]7 showed a slower decline, and at 2 and 4 hours there was 0.98 and 0.75 %dose/organ remaining in the brain. The brain uptake stayed relatively high at 6 hours (0.57 %dose/organ) (Table 2 and Fig. 1). In a similar biodistribution study, the brain uptake of [125I]8 declined faster and at 2 hours only 0.29 %dose/organ remained in the brain. There was a slightly slower washout rate from the lungs for [125I]7. We also observed a prolonged retention of [125I]8 in skin tissue. Since the other major organs (lungs, liver, kidneys and muscle) showed comparable uptake and washout, it is not apparent that the unique brain uptake and retention simply reflect the equilibrium of distribution in the major organs (Table 2 and 3). The partition coefficients were comparable (2,061 and 2,206 for [125I]7 and [125I]8, respectively). Therefore, it may not be a significant factor for consideration. It is likely that the unique brain kinetics of [125I]7 may be related to adding the 4′-substitution at the phenyl ring B. We decided to focus on [125I]7 for further characterization because 7 exhibited a slower decline in brain uptake and a higher retention than 8. We compared biodistribution data of [125I]7 with that of two other previously reported leading SERT imaging agents, ADAM (2) and OADAM (3) [42, 44].
Table 2.
Organ distribution (% dose/organ, n = 3) and regional brain uptake (% dose/g) of [125I]7 in rats (iv injection)
| Organ | 2 min | 30 min | 60 min | 2 hr | 4 hr | 6 hr | 12 hr | 24 hr |
|---|---|---|---|---|---|---|---|---|
| Blood | 3.45 ± 0.47 | 3.32 ± 0.13 | 4.06 ± 0.31 | 3.67 ± 0.28 | 2.43 ± 0.27 | 2.25 ± 0.59 | 2.34 ± 0.20 | 1.09 ± 0.11 |
| Heart | 1.71 ± 0.20 | 0.18 ± 0.05 | 0.11 ± 0.02 | 0.09 ± 0.01 | 0.06 ± 0.01 | 0.05 ± 0.01 | 0.04 ± 0.01 | 0.02 ± 0.00 |
| Muscle | 9.90 ± 1.41 | 11.7 ± 2.47 | 7.30 ± 0.28 | 4.93 ± 0.25 | 3.75 ± 0.13 | 3.02 ± 0.26 | 2.09 ± 0.38 | 1.24 ± 0.36 |
| Lung | 7.08 ± 1.21 | 2.34 ± 0.19 | 1.66 ± 0.17 | 0.79 ± 0.05 | 0.45 ± 0.03 | 0.34 ± 0.06 | 0.16 ± 0.03 | 0.06 ± 0.01 |
| Kidney | 5.28 ± 0.48 | 1.33 ± 0.04 | 1.03 ± 0.07 | 0.73 ± 0.03 | 0.68 ± 0.09 | 0.53 ± 0.15 | 0.40 ± 0.07 | 0.23 ± 0.05 |
| Spleen | 1.10 ± 0.30 | 0.47 ± 0.12 | 0.40 ± 0.06 | 0.21 ± 0.04 | 0.12 ± 0.02 | 0.09 ± 0.02 | 0.03 ± 0.00 | 0.02 ± 0.00 |
| Liver | 22.7 ± 5.03 | 5.82 ± 0.51 | 4.67± 0.35 | 3.55 ± 0.24 | 2.85 ± 0.21 | 2.74 ± 0.43 | 2.13 ± 0.11 | 1.30 ± 0.13 |
| Skin | 5.14 ± 0. 71 | 5.28 ± 0.74 | 5.97± 0.44 | 4.49 ± 0.61 | 4.66 ± 0.16 | 4.65 ± 1.41 | 2.18 ± 0.50 | 1.16 ± 0.24 |
| Brain | 1.77 ± 0.37 | 1.17 ± 0.09 | 1.21 ± 0.12 | 0.98 ± 0.10 | 0.75 ± 0.05 | 0.57 ± 0.11 | 0.26 ± 0.04 | 0.05 ± 0.01 |
| Thyroid | 0.07 ± 0.01 | 0.04 ± 0.01 | 0.03 ± 0.00 | 0.04 ± 0.01 | 0.08 ± 0.01 | 0.14 ± 0.02 | 0.23 ± 0.05 | 0.15 ± 0.02 |
| Regional brain distribution (% dose/g) | ||||||||
|
| ||||||||
| Region | 2 min | 30 min | 60 min | 2 hr | 4 hr | 6 hr | 12 hr | 24 hr |
|
| ||||||||
| Cerebellum | 0.95 ± 0.22 | 0.38 ± 0.04 | 0.27 ± 0.04 | 0.17 ± 0.01 | 0.09 ± 0.01 | 0.06 ± 0.01 | 0.02 ± 0.01 | 0.01 ± 0.00 |
| Striatum | 0.81 ± 0.14 | 0.69 ± 0.04 | 0.68 ± 0.05 | 0.63 ± 0.02 | 0.51 ± 0.02 | 0.38 ± 0.12 | 0.16 ± 0.02 | 0.04 ± 0.01 |
| Hippocampus | 0.76 ± 0.17 | 0.56 ± 0.04 | 0.58 ± 0.05 | 0.50 ± 0.06 | 0.39 ± 0.07 | 0.28 ± 0.06 | 0.12 ± 0.03 | 0.02 ± 0.00 |
| Cortex | 1.16 ± 0.25 | 0.68 ± 0.03 | 0.68 ± 0.07 | 0.54 ± 0.04 | 0.40 ± 0.08 | 0.25 ± 0.07 | 0.08 ± 0.03 | 0.02 ± 0.00 |
| Hypothalamus | 0.85 ± 0.17 | 0.73 ± 0.05 | 0.77 ± 0.02 | 0.73 ± 0.04 | 0.66 ± 0.06 | 0.48 ± 0.08 | 0.26 ± 0.05 | 0.06 ± 0.01 |
| Ratio to Cerebellum | ||||||||
|
| ||||||||
| Region | 2 min | 30 min | 60 min | 2 hr | 4 hr | 6 hr | 12 hr | 24 hr |
| Striatum | 0.85 ± 0.10 | 1.81 ± 0.11 | 2.52 ± 0.19 | 3.64 ± 0.15 | 5.51 ± 0.22 | 6.24 ± 0.97 | 7.84 ± 2.18 | 5.36 ± 1.80 |
| Hippocampus | 0.80 ± 0.05 | 1.47 ± 0.04 | 2.17 ± 0.17 | 2.90 ± 0.26 | 4.15 ± 0.47 | 4.77 ± 0.14 | 5.61 ± 0.40 | 3.43 ± 0.62 |
| Cortex | 1.22 ± 0.09 | 1.79 ± 0.10 | 2.51 ± 0.13 | 3.10 ± 0.16 | 4.23 ± 0.65 | 4.27 ± 0.69 | 3.72 ± 0.62 | 2.68 ± 0.49 |
| Hypothalamus | 0.89 ± 0.03 | 1.91 ± 0.07 | 2.90 ± 0.40 | 4.24 ± 0.22 | 7.10 ± 0.47 | 8.24 ± 0.40 | 12.6 ± 1.11 | 8.67 ± 1.23 |
Table 3.
Organ distribution (% dose/organ, n = 3) and regional brain uptake (% dose/g) of [125I]8 in rats (iv injection)
| Organ | 2 min | 30 min | 60 min | 120 min | 240 min |
|---|---|---|---|---|---|
| Blood | 11.22 ± 0.26 | 5.19 ± 0.63 | 4.48 ± 0.07 | 4.11 ± 0.24 | 3.76 ± 0.20 |
| Heart | 0.57 ± 0.05 | 0.14 ± 0.03 | 0.09 ± 0.01 | 0.10 ± 0.01 | 0.06 ± 0.01 |
| Muscle | 15.37 ± 1.42 | 9.23 ± 1.27 | 6.64 ± 0.14 | 6.03 ± 0.60 | 4.95 ± 0.67 |
| Lung | 3.90 ± 0.44 | 1.41 ± 0.20 | 0.63 ± 0.00 | 0.36 ± 0.07 | 0.22 ± 0.01 |
| Kidney | 2.79 ± 0.11 | 1.66 ± 0.23 | 1.32 ± 0.14 | 0.74 ± 0.10 | 0.45 ± 0.05 |
| Spleen | 0.64 ± 0.19 | 0.37 ± 0.11 | 0.19 ± 0.00 | 0.13 ± 0.03 | 0.08 ± 0.00 |
| Liver | 21.2 ± 1.80 | 5.70 ± 0.65 | 5.32 ± 1.10 | 4.81 ± 0.47 | 3.81 ± 0.30 |
| Skin | 8.44 ± 0.91 | 13.2 ± 0.17 | 11.76 ± 0.48 | 11.0 ± 1.54 | 11.5 ± 4.49 |
| Brain | 0.92 ± 0.12 | 0.74 ± 0.16 | 0.55 ± 0.01 | 0.29 ± 0.07 | 0.08 ± 0.01 |
| Thyroid | 0.07 ± 0.00 | 0.16 ± 0.04 | 0.44 ± 0.07 | 1.24 ± 0.47 | 3.75 ± 1.00 |
| Regional brain distribution (% dose/g) | |||||
|
| |||||
| Region | 2 min | 30 min | 60 min | 120 min | 240 min |
|
| |||||
| Cerebellum | 0.48 ± 0.06 | 0.22 ± 0.06 | 0.11 ± 0.01 | 0.04 ± 0.01 | 0.02 ± 0.00 |
| Striatum | 0.46 ± 0.05 | 0.43 ± 0.09 | 0.35 ± 0.01 | 0.19 ± 0.03 | 0.05 ± 0.00 |
| Hippocampus | 0.41 ± 0.06 | 0.38 ± 0.08 | 0.30 ± 0.02 | 0.16 ± 0.04 | 0.05 ± 0.00 |
| Cortex | 0.59 ± 0.08 | 0.45 ± 0.09 | 0.30 ± 0.02 | 0.13 ± 0.03 | 0.03 ± 0.00 |
| Hypothalamus | 0.46 ± 0.05 | 0.50 ± 0.09 | 0.43 ± 0.04 | 0.24 ± 0.05 | 0.08 ± 0.01 |
| Ratio to Cerebellum | |||||
|
| |||||
| Region | 2 min | 30 min | 60 min | 120 min | 240 min |
|
| |||||
| Cerebellum | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 |
| Striatum | 0.95 ± 0.03 | 2.01 ± 0.11 | 3.28 ± 0.31 | 4.39 ± 0.16 | 3.09 ± 0.01 |
| Hippocampus | 0.86 ± 0.03 | 1.76 ± 0.07 | 2.77 ± 0.23 | 3.57 ± 0.16 | 2.80 ± 0.13 |
| Cortex | 1.24 ± 0.02 | 2.11 ± 0.11 | 2.76 ± 0.05 | 2.87 ± 0.07 | 2.00 ± 0.11 |
| Hypothalamus | 0.97 ± 0.01 | 2.32 ± 0.17 | 3.97 ± 0.08 | 5.57 ± 0.12 | 5.06 ± 0.60 |
It is apparent that [125I]7 showed a prolonged retention in the hypothalamus. The hypothalamus retention at 4 hours (0.75 %dose/organ) was higher than that of ADAM, 4 or OADAM, 3 [42, 44] (Fig. 1A). Regional brain distribution of [125I]7 showed the highest uptake in the hypothalamus, where SERT binding sites are densely packed. The hypothalamus to cerebellum ratios (HY/CB) displayed a steady increase between 2 and 720 minutes, and the ratios were very high between 2 and 24 hours (HY/CB ratio = 4.24, 7.10, 8.24, 12.56 and 8.67 for 2, 4, 6, 12 and 24 hr, respectively, see Table 2 and Fig. 1B). The absolute values of HY/CB ratios for [125I]7 were higher than those of [125I]ADAM (2) [42] and OADAM (3) [44] (HY/CB ratios peaked between 5–6 in 4 hours) (Fig. 1B). It is important to note that the absolute peak hypothalamus uptake value for [125I]7 was higher than that for OADAM (3) (0.66 and 0.32 %dose/g at 4 hr, for [125I]7 and OADAM (3), respectively) (Table 2 and Fig. 1) [44]. The high brain uptake and HY/CB ratio may be an advantage, because it translates to a higher specific binding for SPECT imaging. HY/CB ratios for [125I]8 were comparable to those of [125I]ADAM(2) [42], but the hypothalamus uptake and retention were lower than those of OADAM (3) and [125I]7 [44]. These critical differences in brain uptake and hypothalamus retention are clearly associated with the substitutions on the biphenylthiols and by the particular positions from which substituents are attached. The biodistribution studies of [125I]7 in rat brains demonstrated the unique effects of the 4′-iodo substitution on the hypothalamus uptake and retention. We believe that this is the first example of an iodinated SPECT agent based on a biphenylthiol core structure with a 4′-iodo substitution. The improved hypothalamus uptake may be useful. However, the uptake kinetics, peaking at 6–12 hours may be too slow for SPECT imaging. It may be worth testing an alternative substitution group, other than the chlorine atom at the 5-position of ring A, while retaining the 4′-iodo group on the phenyl ring B. We are currently investigating additional derivatives to further explore this novel series of compounds.
In a separate effort focused on developing 18F labeled SERT imaging agents for PET, we prepared 4′-(2-fluoroethoxy)- derivative, 5, (see Table 1). It also showed excellent uptake and retention in the hypothalamus region of the rat brain (Kung et al., un-published data). The results of in vivo biodistribution in rats for [18F]5 also displayed the same unique kinetics in the hypothalamus as those observed for [125I]7. It is likely that the 4′-substitution with the 2-fluoroethoxy- group led to an excellent in vitro binding affinity and a high uptake and retention in the hypothalamus, properties that would be useful in a SERT imaging agent for PET.
To demonstrate that the uptake in the hypothalamus region of the brain for [125I]7 was specifically linked to SERT binding, we performed a blocking study. The results (Fig. 2) clearly indicated that the binding of [125I]7 in the hypothalamus is due to specific SERT binding. Only the selective SERT ligands, (+)McN5652, escitalopram and ADAM (2), were able to block uptake in the hypothalamus region. Other drugs, nisoxetine-NET ligand; GBR-12909-DAT ligand, showed no inhibition. (+)McN5652 and escitalopram were able to block more efficiently than ADAM (2). It is likely that binding of the “cold” ADAM (2) (a dose of 2 mg/kg, injected at 5 min prior to [125I]7 injection) may be slower than the delivery of the tracer. The timing of the peak uptake and total retention of ADAM (2) may not be fast enough to compete with the binding of [125I]7 to SERT binding sites in the hypothalamus, resulting in less than complete blockage (Fig. 2).
Figure 2.
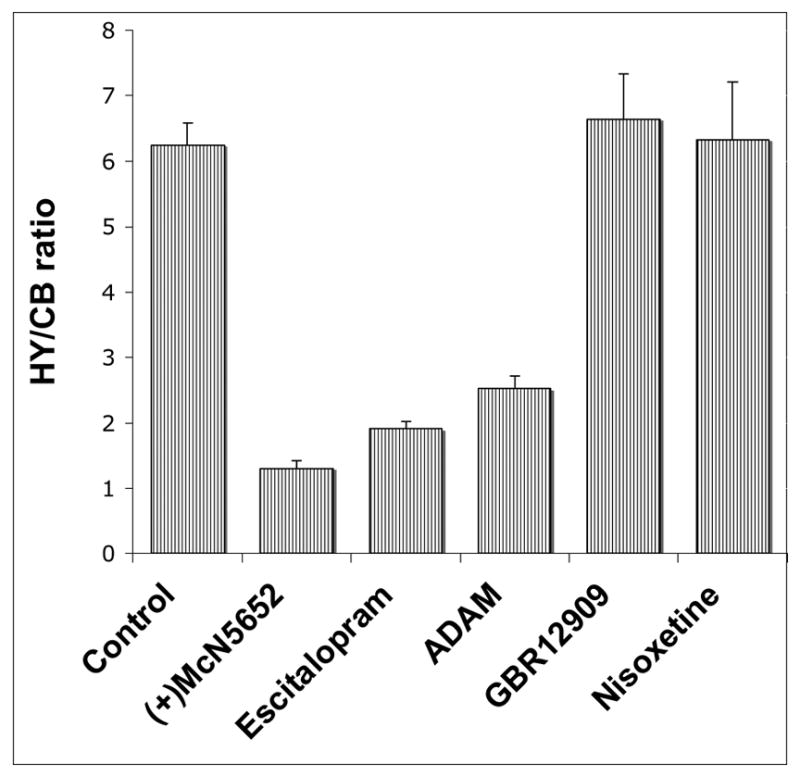
Competitive studies of [125I]7 in rat brains in vivo. Rats were pretreated with drugs with a dose of 2 mg/kg, iv. 5 min prior to the tracer administration. At 4 hr after the tracer injection, hypothalamus/cerebellum (HY/CB) ratios were compared between saline-pretreated (control) and drug-pretreated rats. The values are presented as the average ± SD of three rats in each point. (+)McN5652, Escitapram and ADAM (2) are serotonin transporter (SERT) ligands. Nisoxetine is a norepinephrine transporter (NET) ligand and GBR-12909 is a dopamine transporter (DAT) ligand.
Ex vivo autoradiography was performed 4 hours after an iv injection of [125I]7 in rat brains. The brain sections showed intense labeling in several regions [46], i.e. the lateral hypothalamic area (LH), dorsomedial hypothalamic nucleus (DM), interpeduncular nucleus (IP), thalamus nuclei (ThN), dorsal raphe (DR), medial raphe (MR), superior colliculus (SC), locus coeruleus (LC), sustantia nigra (SN), globus pallidus (GP), all areas known to have high densities of SERT sites (Fig. 3) [47]. Lower, but detectable, labeling was also found in the frontal cortex, caudate putamen, ventral pallidum and hippocampus, areas containing a lower number of SERT sites. The regional distribution observed with [125I]7 is consistent with those reported for other SERT ligands [48, 49]. The distribution is completely in agreement with those of [125I]ADAM (2) [42] and [125I]IDAM (1) [41]. But, due to its higher selectivity, (note the contrast of the autoradiography at 4 hr post iv injection) [125I]7 is likely to be superior to any other previously reported agent for mapping SERT binding sites in the brain.
Figure 3.
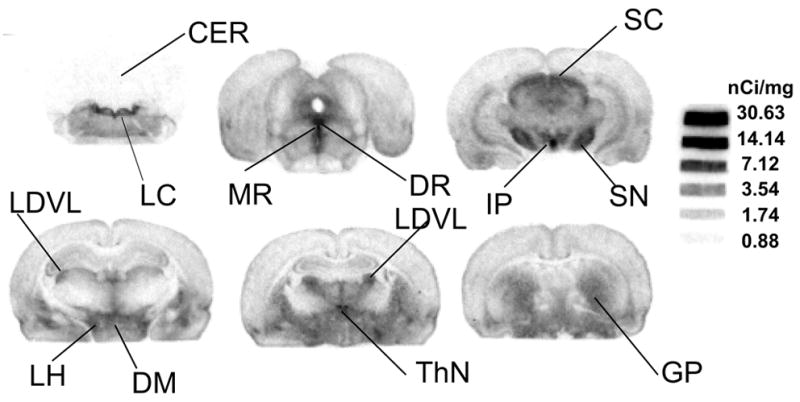
Ex vivo autoradiographic localization of [125I]7 binding sites in rats 4 hours post-injection in a normal rat – High levels of radioactivity were observed in areas containing high densities of serotonin transporter sites. Rat brain sections showed intense labeling in several regions [46], i.e. lateral hypothalamic area (LH), dorsomedial hypothalamic nucleus (DM), interpeduncular nucleus (IP), thalamus nuclei (ThN), dorsal raphe (DR), medial raphe (MR), superior colliculus (SC), locus coeruleus (LC), sustantia nigra (SN), globus pallidus (GP), areas known to have high densities of SERT sites [47]. Lower, but detectable, labeling was also found in the frontal cortex, caudate putamen, ventral pallidum and hippocampus, areas containing a significantly lower number of SERT sites. The coronal sections correspond to the stereotaxic atlas [46].
In conclusion, a novel biphenylthiol derivative, [125I]7, was prepared and tested as a potential lead compound for developing new SPECT imaging agents targeting SERT binding sites. This new ligand has a 4′-iodo- substitution group on the phenyl ring B, which preserves the binding affinity and selectivity. Yet, the 4′-iodo-biphenylthiol derivative dramatically changed the kinetics of in vivo biodistribution resulting in improved hypothalamus uptake and retention, a property desirable for a SPECT imaging agent. This novel ligand, or analogs with similar substitutions, may be potential lead candidates for studying serotonin transporters with in vivo SPECT imaging.
Experimental
1H and NMR spectra were performed on a Bruker DPX 200 spectrometer using tetramethylsilane as an internal standard. High resolution mass spectrometries were measured at McMaster Regional Centre for Mass Spectrometry. Microwave reactions were performed on a Biotage Initiator™ microwave reactor. Tetrahydrofuran (THF) was distilled immediately before use from sodium benzophenone ketyl. All other chemicals were purchased from Aldrich Chemical Co. and used without further purification. Two HPLC systems were used to determine the purities of target compounds. Two HPLC measurements were performed using an Agilent 1100 Series with an isocratic pump and a UV detector. Measurements were performed on a Phenomenex Gemini analytical C-18 column (250 × 4.6 mm, 5 micron) with acetonitrile/ammonium formate buffer (10 mM) 80/20, v/v flow rate 1.0 mL/min at 254 nm (System A), and on a Hamilton PRP-1 analytical column (250 × 4.1, 10 micron) with acetonitrile/dimethyl glutarate buffer (5 mM pH 7) 90/10, v/v flow rate 1.0 mL/min at 254 nm (System B).
Male CD1 rats weighing 200–225 g were used in all the studies. No carrier added [125I]NaI (0.1N NaOH solution) was purchased from NEN Research Products (PerkinElmer Life Sciences, Boston, MA). IDAM and ADAM, as well as the respective corresponding tri-n-butyltin precursors needed for the radioiodination of IDAM and ADAM, were prepared in our laboratory [38, 42]. (+)McN5652 was kindly provided by Research Biochemicals Int. (Natick, MA) through a drug synthesis program supported by the National Institute of Mental Health. The radioiodinated nisoxetine was prepared in our laboratory as described previously [50]. All other chemicals used were of reagent grade.
5′-Bromo-2′-(4-chloro-2-nitrophenylthio)benzoic acid (13)
To a solution of 2-amino-5-bromobenzoic acid (9) (3.57 g, 16.5 mmol) in 1.6 mL of 50% sodium hydroxide, water (22 mL) and NaNO2 (1.14 g, 16.5 mmol) were slowly added. The resulting mixture was poured into a mixture of concentrated HCl (5 mL) and 7 g of ice with external cooling with salt/ice. The mixture was stirred at 0°C for 1 h and neutralized by an addition of potassium acetate (~1 g). The mixture was slowly added with vigorous stirring to a solution of O-ethylxanthic acid, potassium salt (7.7 g, 48 mmol) in 28 mL water at 80 °C. The temperature was maintained at 80 °C during the addition and stirring was continued until the evolution of N2 gas had subsided. After cooling down to room temperature by external cooling in an ice bath, the mixture was acidified by the addition of concentrated HCl to pH 3 and extracted by dichloromethane (20 mL × 2) under argon atmosphere. The organic layer was dried with sodium sulfate under argon and the solvent was removed in vacuo. The crude product, 2-mercapto-5-bromobenzoic acid (11), was quickly dissolved in 15 ml of hot ethanol and added to a mixture of 2,5-dichloronitrobenzene (3.2 g, 16.7 mmol) and sodium ethoxide (0.76 g sodium in 33 mL ethanol). The resulting solution was heated to reflux for 2 hours. After cooling down, the solvent was removed in vacuo and water was added to the residue. The solution was acidified by an addition of concentrated HCl and extracted with ethyl acetate. Combined organic layers were washed with water and dried with sodium sulfate. The solvent was removed in vacuo. The residue was purified by silica gel column (ethyl acetate/methanol 10%) to provide an oily yellow solid. (2.6 g, 41 %): 1H NMR (CDCl3): δ 8.14 (d, J = 2.0 Hz, 1H), 8.12 (d, J = 2.0 Hz, 1H), 7.69 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 7.46 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H).
2′-(4-Bromo-2-nitrophenylthio)-5′-methoxybenzoic acid (14)
The same procedure described above for compound 13 was followed starting with 2-amino-5-methoxybenzoic acid (10) (2.5 g, 15 mmol). The crude intermediate, 2-mercapto-5-methoxybenzoic acid (12), was coupled with 2,5-dibromonitrobenzene (4.64 g, 16.5 mmol). Yellow solid. (3.2 g, 56 %): mp-80–82°C; 1H NMR (CDCl3): δ 13.4 (s, 1H), 8.39 (s, 1H), 7.81 (d, J = 8.8 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.44 (d, J = 2 Hz, 1H), 7.26 (dd, J = 8.6 Hz, J = 2 Hz, 1H), 6.86 (d, J = 8.8 Hz, 1H), 3.91 (s, 3H).
5′-Bromo-2′-(4-bromo-2-nitrophenylthio)benzoic acid (15)
5-Bromo-2-mercaptobenzoic acid (11), prepared as described above from 2-amino-5-bromobenzoic acid (3.57 g, 16.5 mmol) and was coupled with 2,5-dibromonitrobenzene (4.67 g, 16.7 mmol) to provide title compound, a yellow oil (2.9 g, 41%). 1 H NMR (CDCl3/CD3OD 10%): 8.23 (d, J = 2.1 Hz, 1H), 8.08 (d, J = 2.1 Hz, 1H), δ7.45–7.67 (m, 2H), 6.96 (d, J = 8.6 Hz, 1H), 7.21 (d, J = 8.4 Hz, 1H).
5′-Bromo-2′-(4-chloro-2-nitrophenylthio)-N,N-dimethylbenzamide (16)
A solution of 13 (0.8 g, 2.06 mmol) and thionyl chloride (1.5 mL) in chloroform (30 mL) was heated to reflux for 2 hours. The solvent and excess thionyl chloride were removed in vacuo. The residue was dissolved in chloroform (20 mL) and added to the 2M solution of dimethylamine in tetrahydrofuran (10 mL) at 0 °C. The mixture was stirred at room temperature for 3 hours. The solvent was removed and the residue was dissolved in ethyl acetate and washed with water and brine. The organic layer was passed through a short pad of silica gel and the solvent was removed in vacuo. The residue was spectroscopically pure without further purifications. Yellow oil (0.9 g, 69%); 1H NMR (CDCl3): δ 8.17 (d, J = 2.2 Hz, 1H), 7.56–7.77 (m, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.34 (dd, J = 8.6 Hz, 2,1 Hz, 1H), 6.88 (d, J = 8.6 Hz, 1H), 3.02 (s, 3H), 2.87 (s, 3H).
2′-(4-Bromo-2-nitrophenylthio)-5′-methoxy-N,N-dimethylbenzamide (17)
The procedure described above to prepare 16 was applied to 14 (2 g, 5.2 mmol) provided a title compound, a yellow oil (1.5 g, 68%); 1H NMR (CDCl3): δ 8.31 (d, J = 2.2 Hz, 1H), 7.39–7.50 (m, 2H), 6.92–7.02 (m, 2H), 6.80 (d, J = 8.8 Hz, 1H), 3.87 (s, 3H), 3.02 (s, 3H), 2.85 (s, 3H).
5′-Bromo-2′-(4-bromo-2-nitrophenylthio)-N,N-dimethylbenzamide (18)
The procedure described above to prepare 16 was applied to 15 (1 g, 2.31 mmol) provided a title compound, also a yellow oil (0.8 g, 75%); 1H NMR (CDCl3): δ 8.32 (d, J = 2.1 Hz, 1H), 7.56–7.63 (m, 2H), 7.40–7.50 (m, 2H), 6.81 (d, J = 8.6 Hz, 1H), 3.02 (s, 3H), 2.87 (s, 3H).
2-(4′-Bromo-2′-((dimethylamino)methyl)phenylthio)-5-chlorobenzenamine (19)
To a solution of 16 (200 mg, 0.48 mmol) in THF at 0°C, 1.0 M BH3-THF (5 mL) was added. The mixture was heated to reflux for 24 hours. After cooling down, 0.5 mL concentrated HCl was cautiously added and the solvent was removed in vacuo. Water (5 mL) was added to the residue and the mixture was heated to reflux for 30 min. The mixture was adjusted to pH 10 with 1N NaOH and then extracted with ethyl acetate (5 mL × 2). The organic layer was dried (Na2SO4) and purified by silica gel column (ethyl acetate). Pale yellow oil (150 mg, 83 %); 1 HNMR (CDCl3): δ 7.34–7.39 (m, 2H), 7.19 (dd, J = 8.4 Hz, 2.1 Hz, 1H), 6.66–6.76 (m, 3H), 4.63 (br s, 2H), 3.50 (s, 2H), 2.30 (s, 6H); HRMS calcd for C15H16BrClN2S [MH+] 370.9984 obsd 370.9971; HPLC: 99% tR = 11.7 min (System A), 97 % tR = 6.58 min, (System B).
5-Bromo-2′-(2-((dimethylamino)methyl)-4′ methoxyphenylthio)benzenamine (20)
The same procedure described above to prepare 19 was applied to 17 (200 mg, 0.48 mmol) to provide the title compound, a pale yellow oil (89 mg, 50 %); 1H NMR (CDCl3/CD3OD 10%): δ 7.19–7.25 (m, 2H), 6.82–6.91 (m, 3H), 6.75 (dd, J = 8.2 Hz, 2.0 Hz, 1H), 4.41 (s, 2H), 3.80 (s, 3H), 2.90 (s, 6H); HRMS calcd for C16H19BrN2OS [M+] 366.0401 obsd 366.0403; HPLC: 96% tR = 5.9 min (System A), 96% tR = 4.15 min, (System B).
5-Bromo-2-(4′-bromo-2′-((dimethylamino)methyl)phenylthio)benzenamine (21)
The same procedure described above to prepare 19 was applied to 18 (200 mg, 0.43 mmol) to provide the title compound, a pale yellow oil (120 mg, 66%); 1 H NMR (CDCl3): δ 7.38 (d, J = 2.2 Hz, 1H), 7.29 (d, J = 8.2 Hz, 1H), 7.19 (dd, J = 8.4 Hz, 2.2 Hz, 1H). 6.82–6.87 (m, 2H), 6.74 (d, J = 8.4 Hz, 1H), 4.62 (br s, 2H), 3.50 (s, 2H), 2.29 (s, 6H); HRMS calcd for C15H16Br2N2S [M+] 415.9380 obsd 415.9387; HPLC: 98% tR = 12.8 min (System A), 98% tR = 7.96 min, (System B).
5-Chloro-2-(2′-((dimethylamino)methyl)-4′ (tributylstannyl)phenylthio)benzenamine (22)
A mixture of 19 (48 mg, 0.129 mmol) and bistributyl tin (400 mg) and tetrakistriphenylphosphinepalladium(0) (25 mg) in triethyl amine (1 mL) was heated at 120°C in a sealed tube overnight. The crude mixture was purified by silica gel preparatory plate (ethyl acetate/dichlomethane 1/1) (35 mg, 47%) 1H NMR (CDCl3): δ 7.41 (d, J = 8.0 Hz, 1H), 7,18 (m, 1H), 6.80–6.86 (m, 2H), 6.64–6.76 (m, 2H), 4.75 (br s, 2H), 3.55 (s, 2H), 2.29 (s, 6H), 0.82–1.50 (m, 27H).
2-(2′-((Dimethylamino)methyl)-4′-methoxyphenylthio)-5-(tributylstannyl)benzenamine (23)
The procedure above was applied to compound 20 (44 mg, 0.12 mmol) to yield the title compound (40 mg, 58%); 1H NMR (CDCl3): δ 7.23 (m, 1H), 6.89–7.02 (m, 2H), 6.66–6.98 (m, 3H), 3.77 (s, 3H), 3.64 (s, 2H), 2.25 (s, 6H), 0.88–1.57 (m, 27H).
5-Chloro-2-(2′-((dimethylamino)methyl)-4′ iodophenylthio)benzenamine (7)
To a solution of 22 (10 mg, 0.0172 mmol) in chloroform, 1N iodine in chloroform was added dropwise until the red color of the iodine persisted. The mixture was stirred 10 more minutes at room temperature and washed with 1N solution of sodium thiosulfate and purified by prep plate. Colorless oil (4.6 mg, 64%); 1 H NMR (CDCl3): δ 7.56 (d, J = 2.0 Hz, 1H), 7.34–7.40 (m, 2H), 6.66–6.71 (m, 2H), 6.58 (d, J = 8.2 Hz, 1H), 4.62 (br s, 2H), 3.48 (s, 2H), 2.29 (s, 6H); C15H16ClIN2S [M+] 417.9768 obsd 417.9753; HPLC: 96%, tR = 12.9 min (System A), 98% tR = 8.06 min, (System B).
2-(2′-((Dimethylamino)methyl)-4′-methoxyphenylthio)-5-iodobenzenamine (8)
The above procedure was applied to compound 23 (10 mg, 0.0173 mmol) and yielded the title compound (6 mg, 83 %); 1 H NMR (CDCl3): δ 6.88–7.08 (m, 4H), 6.88 (d, J = 2.8 Hz, 1H), 6.70 (dd, J = 8.4 Hz, 2.8 Hz, 1H), 4.67 (br s, 2H), 3.77 (s, 3H), 3.58 (s, 2H), 2.31 (s, 6H); C16H19IN2OS [M+] 414.0263 obsd 414.0208; HPLC: 96% tR = 6.5 min (System A), 97% tR = 5.3 min, (System B).
2′-(4-Bromo-2-nitrophenylthio)-5′-hydroxy-N,N-dimethylbenzamide (24)
A solution of 17 (320 mg, 0.78 mmol) in dichloromethane (3 mL) was added to a 1.0 M solution of BBr3 in dichloromethane (1.1 mL). The mixture was irradiated with microwave at 100°C for 20 minutes and washed with water (2 mL). The organic layer was dried (Na2SO4) and purified by silica gel column (ethyl acetate/dichloromethane 1/4) to yield a yellow oil (300 mg, 97 %). 1H NMR (CDCl3): δ 8.92 (br s, 1H), 8.34 (d, J = 2.1 Hz, 1H), 7.53 (dd, J = 8.8 Hz, 2.1 Hz, 1H), 7.37 (d, J = 8.2 Hz, 1H), 6.70–6.90 (m, 3H), 3.08 (s, 3H), 2.92 (s, 3H).
4′-(2-Amino-4-bromophenylthio)-3′-((dimethylamino)methyl)phenol (25)
To a solution of 24 (20 mg, 0.05 mmol) in THF at 0°C, 1.0 M BH3-THF (3 mL) was added. The mixture was heated to reflux for 24 hours. After cooling down, 0.5 mL concentrated HCl was cautiously added and the solvent was removed in vacuo. Water (5 mL) was added to the residue and the mixture was heated to reflux for 30 minutes. The mixture was adjusted to pH 10 with 1N NaOH and extracted with ethyl acetate (5 mL × 2). The organic layer was dried (Na2SO4) and purified by silica gel column (ethyl acetate). Pale yellow oil (10 mg, 56%); 1 H NMR (CDCl3): δ 7.19 (d, J = 8.0 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 6.75–6.83 (m, 3H), 6.60 (dd, J = 8.4 Hz, 2.8 Hz, 1H), 3.52 (s, 2H), 2.29 (s, 6H); HRMS calcd for C15H17BrN2OS [M+] 352.0245 obsd 352.0225; HPLC: 96% tR 3.7 min (System A), 97 % tR = 3.6 min (1 mL/min) (System B).
In vitro binding assays
Membrane homogenates from LLC-PK1 cells expressing three separate monoamine transporters (DAT, NET, and SERT) [45] were used for the binding assays. Competitive binding assays were performed in a final volume of 0.25 mL. Aliquots of membrane suspensions (100 μL corresponding to 30–40 μg protein) were mixed with 50 μM Tris-HCl, pH 7.4, 120 mM NaCl and 0.1% bovine serum albumin, 0.4 nM of either [125I]IPT [51], 0.2 nM [125I]IDAM [41] or 0.1nM [125I]2-iodo-nisoxetine [50] and 8–10 concentrations (10−10 to 10−5M) of competing drugs. Nonspecific binding was defined by using 1μM of GBR12909 for DAT, 5μM nisoxetine for NET and 1 μM IDAM for SERT assays. Incubation was carried out for 1 hour at room temperature and the bound ligand was collected on glass fiber filters presoaked with 1% polyethylenimine (Sigma, St. Louis, MO) and counted in a gamma counter (Packard 5000, Downers Grove, IL). The results of competition experiments were subjected to nonlinear regression analysis using Graphpad Prism 4 software (Graphpad Software, San Diego, CA).
Biodistribution in rats
Three rats per group were used for each biodistribution study. While under isoflurane anesthesia, 0.2 ml of a saline solution containing 10–100 mCi of radioactive tracer was injected into the femoral vein. The rats were sacrificed at the time indicated by cardiac excision while under anesthesia. Organs of interest were removed and weighed, and the radioactivity was counted. The percent dose per organ was calculated by comparing the tissue counts to counts of 1% of the initial dose (aliquots of the injected material diluted 100 times) measured at the same time. Regional brain distribution was measured after an iv injection of the radioactive tracer. Samples from different brain regions [cortex (CX), striatum (ST), hippocampus (HP), cerebellum (CB) and hypothalamus (HY)] were dissected, weighed and counted. The percentage dose/g of each sample was calculated by comparing sample counts with the counts of the diluted initial dose, as described above. The ratio of specific binding in each region was obtained by dividing the percentage dose/g of each region by that of the cerebellum [region/CB]. The cerebellum was used as a background region because it contains very low concentration of SERT.
In vivo competitive binding of various compounds in the regional uptake of [125I]7 was investigated by pretreating animals with various competing drugs (2 mg/kg, each, injected iv 5 min prior to injection of [125I]7). The competing drugs included: (+)McN5652, Escitalopram and ADAM (2), which are serotonin transporter (SERT) ligands, nisoxetine, a selective norepinephrine transporter (NET) ligand and GBR-12909, a dopamine transporter (DAT) ligand. Regional brain distributions were then determined at 4 hr after injection of [125I]7, as described above. The reduction of regional specific binding in the drug-pretreated rats was compared to binding in control animals with saline pretreatment.
Scheme 1.
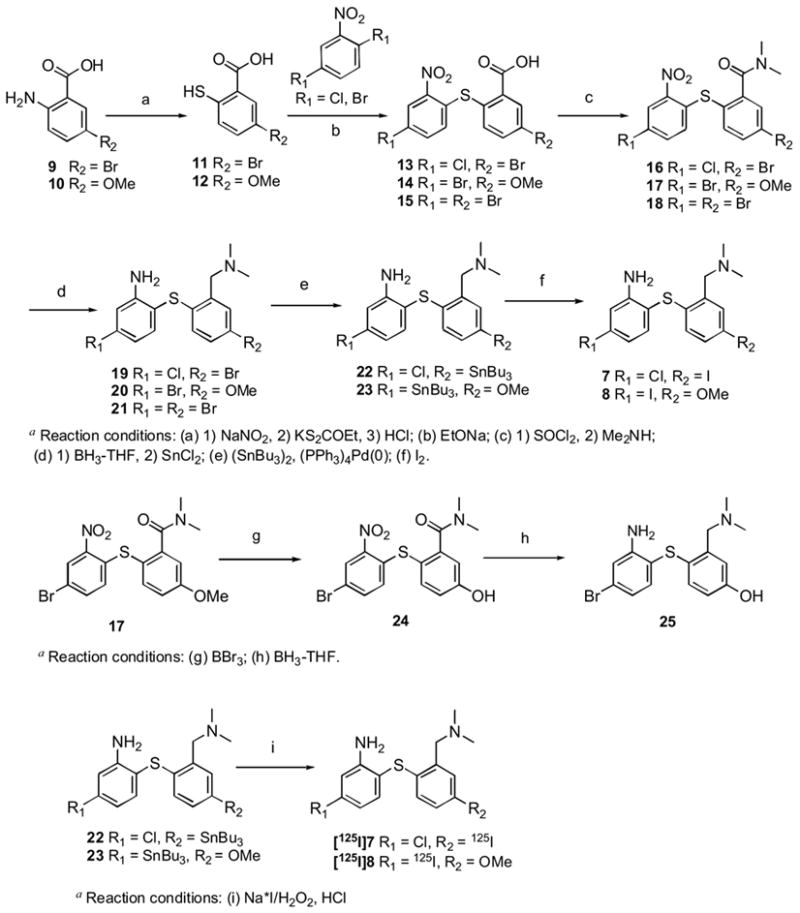
Synthesis of SERT ligands 7,8,19–21,25a
Acknowledgments
This work was supported by a grant awarded from the National Institutes of Health (R01-MH068782, H.F.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Owens MJ. Selectivity of antidepressants: from the monoamine hypothesis of depression to the SSRI revolution and beyond. J Clin Psychiatry. 2004;4:5–10. [PubMed] [Google Scholar]
- 2.Holtzheimer PE, 3rd, Nemeroff CB. Advances in the treatment of depression. NeuroRx. 2006;3:42–56. doi: 10.1016/j.nurx.2005.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nemeroff CB. The burden of severe depression: A review of diagnostic challenges and treatment alternatives. J Psychiatr Res. 2006 doi: 10.1016/j.jpsychires.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Owens MJ, Nemeroff CB. Role of serotonin in the pathophysiology of depression: focus on the serotonin transporter. Clin Chem. 1994;40:288–95. [PubMed] [Google Scholar]
- 5.Frazer A. Serotonergic and noradrenergic reuptake inhibitors: prediction of clinical effects from in vitro potencies. J Clin Psychiatry. 2001;62:16–23. [PubMed] [Google Scholar]
- 6.Frazer A. Antidepressants. J Clin Psychiatry. 1997;6:9–25. [PubMed] [Google Scholar]
- 7.White KJ, Walline CC, Barker EL. Serotonin transporters: implications for antidepressant drug development. AAPS J. 2005;7:E421–33. doi: 10.1208/aapsj070242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavanagh J, Patterson J, Pimlott S, Dewar D, Eersels J, Dempsey MF, Wyper D. Serotonin Transporter Residual Availability During Long-Term Antidepressant Therapy Does Not Differentiate Responder and Nonresponder Unipolar Patients. Biol Psychiatry. 2005 doi: 10.1016/j.biopsych.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 9.Parsey RV, Kent JM, Oquendo MA, Richards MC, Pratap M, Cooper TB, Arango V, Mann JJ. Acute Occupancy of Brain Serotonin Transporter by Sertraline as Measured by [(11)C]DASB and Positron Emission Tomography. Biol Psychiatry. 2005 doi: 10.1016/j.biopsych.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Giovacchini G, Lang L, Ma Y, Herscovitch P, Eckelman WC, Carson RE. Differential effects of paroxetine on raphe and cortical 5-HT1A binding: a PET study in monkeys. Neuroimage. 2005;28:238–48. doi: 10.1016/j.neuroimage.2005.05.042. [DOI] [PubMed] [Google Scholar]
- 11.Buchert R, Schulze O, Wilke F, Berding G, Thomasius R, Petersen K, Brenner W, Clausen M. Is Correction for Age Necessary in SPECT or PET of the Central Serotonin Transporter in Young, Healthy Adults? J Nucl Med. 2006;47:38–42. [PubMed] [Google Scholar]
- 12.Buchert R, Thomasius R, Petersen K, Wilke F, Obrocki J, Nebeling B, Wartberg L, Zapletalova P, Clausen M. Reversibility of ecstasy-induced reduction in serotonin transporter availability in polydrug ecstasy users. Eur J Nucl Med Mol Imaging. 2006;33:188–99. doi: 10.1007/s00259-005-1850-8. [DOI] [PubMed] [Google Scholar]
- 13.Brust P, Hesse S, Mueller U, Szabo Z. Neuroimaging of the serotonin transporter: possibilities and pitfalls. Current Psychiatry Reviews. 2006;2:111–49. [Google Scholar]
- 14.Parsey RV, Kent JM, Oquendo MA, Richards MC, Pratap M, Cooper TB, Arango V, Mann JJ. Acute occupancy of brain serotonin transporter by sertraline as measured by [11C]DASB and positron emission tomography. Biol Psychiatry. 2006;59:821–8. doi: 10.1016/j.biopsych.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Sacher J, Asenbaum S, Klein N, Geiss-Granadia T, Mossaheb N, Poetzi C, Attarbaschi T, Lanzenberger R, Spindelegger C, Rabas A, Heinze G, Dudczak R, Kasper S, Tauscher J. Binding kinetics of 123 I[ADAM] in healthy controls: a selective SERT radioligand. Int J Neuropsychopharmacol. 2006:1–8. doi: 10.1017/S1461145706006511. [DOI] [PubMed] [Google Scholar]
- 16.Klein N, Sacher J, Geiss-Granadia T, Attarbaschi T, Mossaheb N, Lanzenberger R, Potzi C, Holik A, Spindelegger C, Asenbaum S, Dudczak R, Tauscher J, Kasper S. In vivo imaging of serotonin transporter occupancy by means of SPECT and [(123)I]ADAM in healthy subjects administered different doses of escitalopram or citalopram. Psychopharmacology (Berl) 2006;188:263–72. doi: 10.1007/s00213-006-0486-0. [DOI] [PubMed] [Google Scholar]
- 17.Booij J, de Win MM. Brain kinetics of the new selective serotonin transporter tracer [(123)I]ADAM in healthy young adults. Nucl Med Biol. 2006;33:185–91. doi: 10.1016/j.nucmedbio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Newberg AB, Amsterdam JD, Wintering N, Ploessl K, Swanson RL, Shults J, Alavi A. 123I-ADAM binding to serotonin transporters in patients with major depression and healthy controls: a preliminary study. J Nucl Med. 2005;46:973–7. [PubMed] [Google Scholar]
- 19.Erlandsson K, Sivananthan T, Lui D, Spezzi A, Townsend CE, Mu S, Lucas R, Warrington S, Ell PJ. Measuring SSRI occupancy of SERT using the novel tracer [123I]ADAM: a SPECT validation study. Eur J Nucl Med Mol Imaging. 2005;32:1329–36. doi: 10.1007/s00259-005-1912-y. [DOI] [PubMed] [Google Scholar]
- 20.Guilloteau D, Chalon S. PET and SPECT exploration of central monoaminergic transporters for the development of new drugs and treatments in brain disorders. Curr Pharm Des. 2005;11:3237–45. doi: 10.2174/138161205774424744. [DOI] [PubMed] [Google Scholar]
- 21.Mathis CA, Biegon A, Taylor SE, Enas JD, Hanrahan SM. [I-125]5-Iodo-6-nitro-2-piperazinylquinoline: a potent and selective ligand for the serotonin uptake complex. Eur J Pharmacol. 1992;210:103–04. doi: 10.1016/0014-2999(92)90658-q. [DOI] [PubMed] [Google Scholar]
- 22.Jagust WJ, Eberling JL, Biegon A, Taylor SE, VanBrocklin HF, Jordan S, Hanrahan SM, Roberts JA, Brennan KM, Mathis CA. Iodine-123-5-iodo-6-nitroquipazine: SPECT radiotracer to image the serotonin transporter. J Nucl Med. 1996;37:1207–14. [PubMed] [Google Scholar]
- 23.Mathis CA, Taylor SE, Biegon A, Enas JD. [125I]5-Iodo-6-nitroquipazine: a potent and selective ligand for the 5-hydroxytryptamine uptake complex I in vitro studies. Brain Res. 1993;619:229–35. doi: 10.1016/0006-8993(93)91616-z. [DOI] [PubMed] [Google Scholar]
- 24.Stehouwer JS, Plisson C, Jarkas N, Zeng F, Voll RJ, Williams L, Martarello L, Votaw JR, Tamagnan G, Goodman MM. Synthesis, radiosynthesis, and biological evaluation of carbon-11 and fluorine-18 (N-fluoroalkyl) labeled 2beta-carbomethoxy-3beta-(4′-(3-furyl)phenyl)tropanes and -nortropanes: candidate radioligands for in vivo imaging of the serotonin transporter with positron emission tomography. J Med Chem. 2005;48:7080–3. doi: 10.1021/jm0504095. [DOI] [PubMed] [Google Scholar]
- 25.Willeit M, Praschak-Rieder N, Neumeister A, Pirker W, Asenbaum S, Vitouch O, Tauscher J, Hilger E, Stastny J, Brucke T, Kasper S. [123I]-β-CIT SPECT imaging shows reduced brain serotonin transporter availability in drug-free depressed patients with seasonal affective disorder. Biol Psychiatry. 2000;47:482–89. doi: 10.1016/s0006-3223(99)00293-0. [DOI] [PubMed] [Google Scholar]
- 26.Bergstrom KA, Halldin C, Hall H, Lundkvist C, Ginovart N, Swahn CG, Farde L. In vitro and in vivo characterisation of nor-beta-CIT: a potential radioligand for visualisation of the serotonin transporter in the brain. Eur J Nucl Med. 1997;24:596–601. doi: 10.1007/BF00841395. [DOI] [PubMed] [Google Scholar]
- 27.Marjamaki P, Zessin J, Eskola O, Gronroos T, Haaparanta M, Bergman J, Lehikoinen P, Forsback S, Brust P, Steinbach J, Solin O. S-[18F]fluoromethyl-(+)-McN5652, a PET tracer for the serotonin transporter: evaluation in rats. Synapse. 2003;47:45–53. doi: 10.1002/syn.10150. [DOI] [PubMed] [Google Scholar]
- 28.Zessin J, Eskola O, Steinbach J, Marjamaki P, Bergman J, Gronroos T, Haaparanta-Solin M, Brust P, Lehikoinen P, Solin O, Johannsen B. Imaging of the serotonin transporter with the [18F]fluoromethyl analogue of (+)-McN5652. Synthesis and Applications of Isotopically Labelled Compounds, Proceedings of the International Symposium, 7th; Dresden, Germany. June 18–22, 2000; 2001. pp. 377–79. [Google Scholar]
- 29.Suehiro M, Musachio JL, Dannals RF, Mathews WB, Ravert HT, Scheffel U, Wagner HN. An improved method for the synthesis of radiolabeled McN5652 via thioester precursors. Nucl Med Biol. 1995;22:543–45. doi: 10.1016/0969-8051(94)00114-y. [DOI] [PubMed] [Google Scholar]
- 30.Maryanoff EM, Vaught JL, Shank RP, McComsey DF, Costanzo MJ, Nortey SO. Pyrroloisoquinoleines antidepressants. 3. A focus on serotonin. J Med Chem. 1990;33:2793–97. doi: 10.1021/jm00172a018. [DOI] [PubMed] [Google Scholar]
- 31.Maryanoff BE, McComsey DF, Gardocki JF, Shank RP, Costanzo MJ, Nortey SO, Schneider CR, Setler PE. Pyrroloisoquinoline antidepressants. In-depth exploration of structure-activity relationships. J Med Chem. 1987;30:1433–54. doi: 10.1021/jm00391a028. [published erratum appears in J Med Chem 1988 Jan;31(1):276] [DOI] [PubMed] [Google Scholar]
- 32.Vercouillie J, Mavel S, Galineau L, Ragusa T, Innis R, Kassiou M, Chalon S, Dolle F, Besnard JC, Guilloteau D, Emond P. Synthesis and in vitro evaluation of novel derivatives of diphenylsulfide as serotonin transporter ligands. Bioorg Med Chem Lett. 2006;16:1297–300. doi: 10.1016/j.bmcl.2005.11.066. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S, Zhen J, Reith ME, Dutta AK. Design, synthesis, and preliminary SAR study of 3- and 6-side-chain-extended tetrahydro-pyran analogues of cis- and trans-(6-benzhydryl-tetrahydropyran-3-yl)-benzylamine. Bioorg Med Chem. 2006 doi: 10.1016/j.bmc.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 34.Zessin J, Deuther-Conrad W, Kretzschmar M, Wust F, Pawelke B, Brust P, Steinbach J, Bergmann R. [11C]SMe-ADAM, an imaging agent for the brain serotonin transporter: synthesis, pharmacological characterization and microPET studies in rats. Nucl Med Biol. 2006;33:53–63. doi: 10.1016/j.nucmedbio.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Jarkas N, Votaw JR, Voll RJ, Williams L, Camp VM, Owens MJ, Purselle DC, Bremner JD, Kilts CD, Nemeroff CB, Goodman MM. Carbon-11 HOMADAM: a novel PET radiotracer for imaging serotonin transporters. Nucl Med Biol. 2005;32:211–24. doi: 10.1016/j.nucmedbio.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 36.Jarkas N, McConathy J, Votaw JR, Voll RJ, Malveaux E, Camp VM, Williams L, Goodman RR, Kilts CD, Goodman MM. Synthesis and characterization of EADAM: a selective radioligand for mapping the brain serotonin transporters by positron emission tomography. Nucl Med Biol. 2005;32:75–86. doi: 10.1016/j.nucmedbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 37.Huang Y, Bae SA, Zhu Z, Guo N, Roth BL, Laruelle M. Fluorinated diaryl sulfides as serotonin transporter ligands: synthesis, structure-activity relationship study, and in vivo evaluation of fluorine-18-labeled compounds as PET imaging agents. J Med Chem. 2005;48:2559–70. doi: 10.1021/jm0400808. [DOI] [PubMed] [Google Scholar]
- 38.Oya S, Kung MP, Acton PD, Mu M, Hou C, Kung H. A new single-photon emission computed tomography imaging agent for serotonin transporters: [123I] IDAM, 5-Iodo-2-((2-((dimethylamino)methyl)phenyl)thio)benzyl alcohol. J Med Chem. 1999;42:333–35. doi: 10.1021/jm9806751. [DOI] [PubMed] [Google Scholar]
- 39.Oya S, Choi SR, Hou C, Mu M, Kung MP, Acton PD, Siciliano M, Kung HF. 2-((2-((Dimethylamino)methyl)phenyl)thio)-5-iodophenylamine (ADAM): An Improved Serotonin Transporter Ligand. Nucl Med Biol. 2000;27:249–54. doi: 10.1016/s0969-8051(00)00084-6. [DOI] [PubMed] [Google Scholar]
- 40.Oya S, Choi SR, Coenen H, Kung HF. New PET Imaging Agent for the Serotonin Transporter: [18F]ACF (2-[(2-Amino-4-chloro-5-fluorophenyl)thio]-N,N-dimethyl-benzenmethanamine) J Med Chem. 2002;45:4716–23. doi: 10.1021/jm020167y. [DOI] [PubMed] [Google Scholar]
- 41.Kung MP, Hou C, Oya S, Mu M, Acton PD, Kung HF. Characterization of [123I]IDAM as a novel single-photon emission tomography tracer for serotonin transporters. Eur J Nucl Med. 1999;26:844–53. doi: 10.1007/s002590050458. [DOI] [PubMed] [Google Scholar]
- 42.Choi SR, Hou C, Oya S, Mu M, Kung MP, Siciliano M, Acton PD, Kung HF. Selective in vitro and in vivo binding of [(125)I]ADAM to serotonin transporters in rat brain. Synapse. 2000;38:403–12. doi: 10.1002/1098-2396(20001215)38:4<403::AID-SYN5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 43.Guilloteau D, Chalon S. PET and SPECT exploration of central monoaminergic transporters for the development of new drugs and treatments in brain disorders. Curr Pharm Des. 2005;11:3237–45. doi: 10.2174/138161205774424744. [DOI] [PubMed] [Google Scholar]
- 44.Kung HF, Newman S, Choi SR, Oya S, Hou C, Zhuang ZP, Acton PD, Plossl K, Winkler J, Kung MP. 2-(2-(dimethylaminomethyl)phenoxy)-5-iodophenylamine: an improved serotonin transporter imaging agent. J Med Chem. 2004;47:5258–64. doi: 10.1021/jm049917p. [DOI] [PubMed] [Google Scholar]
- 45.Gu H, Wall SC, Rudnick G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J Biol Chem. 1994;269:7124–30. [PubMed] [Google Scholar]
- 46.Paxinos G, Watson C. The Rat Brain In Stereotaxic Coordinates. New York: Academic Press; 1986. [Google Scholar]
- 47.Cortes R, Soriano E, Pazos A, Probst A, Palacios JM. Autoradiography of antidepressant binding sites in the human brain: localization using [3H]paroxetine. Nueroscience. 1988;27:473–96. doi: 10.1016/0306-4522(88)90282-5. [DOI] [PubMed] [Google Scholar]
- 48.Biegon A, Mathis CA, Hanrahan SM, Jagust WJ. [125I]5-Iodo-6-nitroquipazine: a potent and selective ligand for the 5-hydroxytryptamine uptake complex II in vivo studies in rats. Brain Res. 1993;619:236–46. doi: 10.1016/0006-8993(93)91617-2. [DOI] [PubMed] [Google Scholar]
- 49.De Souza EB, Kuyatt BL. Autoradiographic localization of 3H-paroxetine-labeled serotonin uptake sites in rat brain. Synapse. 1987;1:488–96. doi: 10.1002/syn.890010513. [DOI] [PubMed] [Google Scholar]
- 50.Kung MP, Choi SR, Hou C, Zhuang ZP, Foulon C, Kung HF. Selective binding of 2-[125I]iodo-nisoxetine to norepinephrine transporters in the brain. Nucl Med Biol. 2004;31:533–41. doi: 10.1016/j.nucmedbio.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 51.Kung M-P, Essman WD, Frederick D, Meegalla S, Goodman M, Mu M, Lucki I, Kung HF. IPT: a novel iodinated ligand for the CNS dopamine transporter. Synapse. 1995;20:316–24. doi: 10.1002/syn.890200405. [DOI] [PubMed] [Google Scholar]
- 52.Wilson AA, Ginovart N, Schmidt M, Meyer JH, Threlkeld PG, Houle S. Novel radiotracers for imaging the serotonin transporter by positron emission tomography: synthesis, radiosynthesis, and in vitro and ex vivo evaluation of 11C-labeled 2-(phenylthio)araalkylamines. J Med Chem. 2000;43:3103–10. doi: 10.1021/jm000079i. [DOI] [PubMed] [Google Scholar]