

Abstract
Thimerosal is an organic mercurial compound used as a preservative in biomedical preparations. Little is known about the reactions of human neuronal and skin cells to its micro- and nanomo-lar concentrations, which can occur after using thimerosal-containing products. A useful combination of fluorescent techniques for the assessment of thimerosal toxicity is introduced. Short-term thimerosal toxicity was investigated in cultured human cerebral cortical neurons and in normal human fibroblasts. Cells were incubated with 125-nM to 250-μM concentrations of thimer-osal for 45 min to 24 h. A 4′, 6-diamidino-2-phenylindole dihy-drochloride (DAPI) dye exclusion test was used to identify non-viable cells and terminal transferase-based nick-end labeling (TUNEL) to label DNA damage. Detection of active caspase-3 was performed in live cell cultures using a cell-permeable fluorescent caspase inhibitor. The morphology of fluorescently labeled nuclei was analyzed. After 6 h of incubation, the thimerosal toxicity was observed at 2 μM based on the manual detection of the fluorescent attached cells and at a 1-μM level with the more sensitive GENios Plus Multi-Detection Microplate Reader with Enhanced Fluorescence. The lower limit did not change after 24 h of incubation. Cortical neurons demonstrated higher sensitivity to thimerosal compared to fibroblasts. The first sign of toxicity was an increase in membrane permeability to DAPI after 2 h of incubation with 250 μM thimerosal. A 6-h incubation resulted in failure to exclude DAPI, generation of DNA breaks, caspase-3 activation, and development of morphological signs of apoptosis. We demonstrate that thimerosal in micromolar concentrations rapidly induce membrane and DNA damage and initiate caspase-3– dependent apoptosis in human neurons and fibroblasts. We conclude that a proposed combination of fluorescent techniques can be useful in analyzing the toxicity of thimerosal.
Keywords: thimerosal, active caspase-3, apoptosis, toxicity, neurons, fibroblasts, DNA breaks, membrane damage, DAPI
Thimerosal (sodium ethylmercury-thiosalicylate) is an antibacterial and antifungal mercurial compound used as a preservative in biological products and vaccines, in concentrations ranging from 0.003 to 0.01% (30 –100 μg/ml) (Ball et al., 2001). Thimerosal contains 49.6 % mercury by weight and releases ethylmercury as a metabolite. In the body, ethylmer-cury can be converted to inorganic mercury, which then preferentially accumulates in the kidneys and brain (Blair et al., 1975). Inorganic mercury is known to induce membrane and DNA damage (Ferrat et al., 2002; Ben-Ozer et al., 2000), and in cell culture conditions it was shown to be mutagenic and generate DNA breaks in concentrations below 500 nM (Schurz et al., 2000). Ethylmercury can significantly increase the concentration of inorganic mercury in many organs (Magos et al., 1985). After in vivo administration, ethylmercury passes through cellular membranes and concentrates in cells in vital organs, including the brain, where it releases inorganic mercury, raising its concentrations higher than equimolar doses of its close and highly toxic relative methylmercury (Magos et al., 1985).
However, little is known about acute reactions of various types of human cells following short-time exposure to thimer-osal in micro- and nanomolar concentrations.
In this paper we used a convenient and easily reproducible combination of fluorescent techniques analyzing various markers of DNA and membrane damage, and investigated the toxicity of micromolar and nanomolar concentrations of thimerosal (125 nM–250 μM) occurring in the first 24 h of exposure in cultures of human cortical neuronal cells and in human fibroblasts.
We found that thimerosal in micromolar concentrations rapidly decreased cellular viability. Within several h after thimer-osal administration, cells lost their capability to exclude the fluorescent dye 4′,6-diamidino-2-phenylindole dihydrochlo-ride (DAPI) and developed multiple DNA breaks accompanied by caspase-3 activation and apoptotic morphology. Neuronal cell cultures demonstrated a higher sensitivity to thimerosal compared with fibroblasts.
MATERIALS AND METHODS
Cell cultures
HCN-1A Human cerebral cortical neurons (CRL-10442) were purchased from American Type Culture Collection (ATCC, Manassas, VA) and were cultured according to ATCC recommendations. The line was derived from cortical tissue removed from a patient undergoing hemispherec-tomy for intractable seizures. As recommended by ATCC, the cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4 mM L-glutamine, modified to contain 4.5g/l glucose and 1.5g/l sodium bicarbonate, supplemented with 10% fetal bovine serum, and the pH adjusted to 7.35 prior to filtration.
Normal neonatal human foreskin HCA 2 fibroblasts (PD32) were obtained from the laboratory of Dr. Olivia Smith-Pereira, Ph.D. The cells were grown in DMEM supplemented with 10% fetal bovine serum medium, and the pH was adjusted to 7.4 prior to filtration. For the experiments, all cells were subcultured in 24-well cell culture plates (Fisher, Pittsburgh, PA). All experiments were reproduced in triplicates. Each of the parallel series yielded identical results.
Thimerosal
Thimerosal (minimum 97% HPLC), SigmaUltra (Sigma, St. Louis, MO) was added to cell cultures in 30 μl of double-distilled water to final concentrations of 250 μM, 50 μM, 10 μM, 2 μM, 1 μM, 500 nM, 250 nM, and 125 nM. Concentrations of 1 μM–125 nM were used with neuronal cells only. Control cell cultures received 30 μl of water without thimerosal.
Dye exclusion test using DAPI
DAPI is a nonintercalating DNA-specific dye with an emission maximum in the blue spectrum (Shapiro, 1985). It is widely used for counterstaining cellular nuclei in fixed sections and has been demonstrated to be useful for the detection of nonviable cells with compromised membranes in live cell cultures (Boutonnat et al., 1999; McCarthy and Hale, 1988).
The DAPI exclusion test was performed as described (Boutonnat et al., 1999). Briefly, cells were incubated with DAPI (Sigma, St. Louis, MO) diluted in a cell culture medium at a final concentration of 100 ng/ml for 30 min at 20°C (Boutonnat et al., 1999). A fluorescent signal was monitored and representative images were taken at 45 min and 2, 4, 6, and 24 h after the addition of thimerosal, The DAPI incubation started 30 min before each observation was made (at 15 min, 90 min, etc). Images were acquired using an Olympus IX-70 fluorescent microscope equipped with a MicroMax digital camera system (Princeton Instruments, Inc., Trenton, NJ) containing an RTE/CCD-1300-Y/HS array cooled by a Peltier device. Image acquisition was performed using the MetaMorph 4.1 program (Advanced Scientific, Inc., Meraux, LA). The micrographs were taken at central parts of the wells, where cellular density was most uniform.
Terminal transferase-based nick-end labeling (TUNEL)
Cells were fixed in ice cold methanol, and TUNEL staining for detection of DNA breaks was performed using the ApoTaq Fluorescein and ApoTaq Rhodamine kits for indirect immunofluorescence (Serologicals, Gaithersburg, MD), employing the standard technique recommended by the manufacturer. Following washing, the cells were counterstained with the DNA binding dye DAPI (1 μg/ml) for visualization of all cellular nuclei and were mounted in Vectashield (Vector Laboratories, Burlingame, CA) for observation by fluorescence microscopy.
Caspase-3 detection
Detection of active caspase-3 in live cell cultures was performed using an APO LOGIX™ carboxyfluorescein (FAM) caspase detection kit (Cell Technology, Minneapolis, MN). The kit detects active caspases in living cells through the use of a FAM-labeled DEVD fluoromethyl ketone (FMK) caspase inhibitor, which irreversibly binds to active caspase-3 (Amstad et al., 2000; Bedner et al., 2000; Smolewski et al., 2001). The inhibitor is cell permeable and noncytotoxic. With lesser affinity, FAM-DEVD-FMK binds to the other caspases participating in apoptosis: caspase-8 >caspase-7 >caspase-10 >caspase-6 in the order of decreasing binding affinity (Carcia-Calvo et al., 1998).
The kit was used as recommended by the manufacturer. Briefly, 10 μl of 30X Working Dilution FAM-Peptide-FMK was added to 300 μl of cell culture medium/per well, directly in 24-well cell culture plates after 5 h or 23 h of incubation with thimerosal. Cells were incubated for 1 h at 37°C under 5% CO2, protected from light. Then the medium was carefully removed, and the cells were washed twice with 2 ml/per well of 1X Working Dilution Wash Buffer. The fluorescent signal was observed under an Olympus IX-70 fluores-cent microscope equipped with a MicroMax digital camera system (Princeton Instruments, Inc.) containing an RTE/CCD-1300-Y/HS array cooled by a Peltier device. Caspase-positive cells appeared fluorescing green. Representative images were taken at 6 h and 24 h after the addition of thimerosal. Image acquisition was performed using the MetaMorph 4.1 program (Advanced Scientific, Inc.). Positive controls included cultures of cortical neurons treated with 0.5 μM staurosporin to induce caspase-3 activation. In several series of experiments, we added DAPI to cell cultures to the concentration of 100 ng/ml for 30 min immediately after 1 h of incubation with the FAM-Peptide-FMK solution. This made the co-localization of active caspase-3 and DAPI signals possible.
Fluorescence measurements using a microplate reader
In a separate set of experiments, we measured both active caspase-3 and DAPI signals in co-localization experiments using a GENios Plus Multi-Detection Microplate Reader with Enhanced Fluorescence (Tecan Inc., Research Triangle Park, NC). Neuronal cells were incubated with 1–250 μM concentrations of thimerosal for 6 h and processed as described for simultaneous DAPI and active caspase-3 detection. Both FITC and DAPI fluorescence were measured directly in 24-well plates using a Chroma Technology bandpass filter set: FITC excitation D490/40, emission 520/10; DAPI excitation D360/40, emission 460/20. The reactions were repeated twice and yielded the same dose-dependent increase in thimerosal toxicity. Background fluorescence was subtracted from the experimental series, and the results were represented as graphs of average values using Microsoft Excel.
RESULTS
Thimerosal-Induced Changes in Membrane Permeability and Cell Viability
Changes in cell viability rapidly occurred after administration of thimerosal in all cell cultures and were detected by the loss of ability to exclude the fluorescent dye DAPI. DAPI is classified as a semipermeant dye, which requires a relatively short (30-min) exposure time of cell cultures to the dye prior to the signal observation in a DAPI exclusion test (Boutonnat et al., 1999). Under these conditions, the dye has been shown to be useful for the detection of nonviable cells and can be utilized as a selective marker of membrane integrity. Indeed, it is a less toxic alternative to propidium iodide (PI) (Boutonnat et al., 1999).
The results of the experiments show a dose- and time-dependent increase of membrane permeability to DAPI, first detected after 2 h of incubation with thimerosal and resulting in the penetration of the dye into the nuclei and DNA staining (Figs. 1 and 2). Figure 1 presents experiments performed on human cultured cortical neurons (HCN-1A) and shows that, after 2 h of incubation with thimerosal at a concentration of 250 μM, the DAPI penetrated through cellular membranes and stained the cellular nuclei. The inability to exclude the dye indicates the loss of cellular membrane integrity and cell death (Boutonnat et al., 1999; McCarthy and Hale, 1988). After 4 h of incubation, thimerosal-induced membrane permeability and DNA staining were observed at a concentration of 10 μM. After 6 h of incubation with thimerosal, changes in membrane permeability were detected at concentrations as low as 2 μM, based on the appearance of DAPI-stained cells attached to the bottom of the wells. In control cell cultures, which were treated with DAPI alone, only sporadic dead cells were detected, and their numbers stayed the same 2, 4, and 6 h after the addition of DAPI (Fig. 1). There was no change in cell membrane permeability for DAPI for up to 24 h if no thimerosal was added.
FIG. 1.
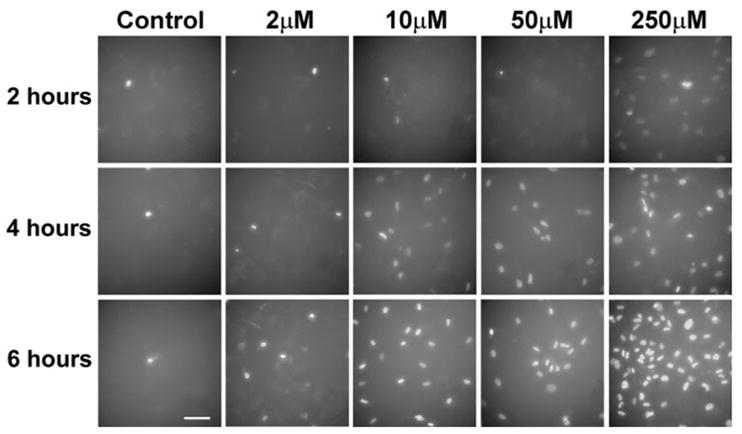
Nonviable human cortical neurons in cell culture detected by a DAPI exclusion test after incubation with various concentrations of thimerosal. All panels have the same density of cells plated on a dish. Only nonviable cells are visualized by nuclear staining with DAPI, when cellular membranes are compromised and cells are either dead or dying. Solitary dying cells can be seen in controls, whereas the majority of cells incubated with thimerosal have compromised membranes. (Bar = 100 μm).
FIG. 2.
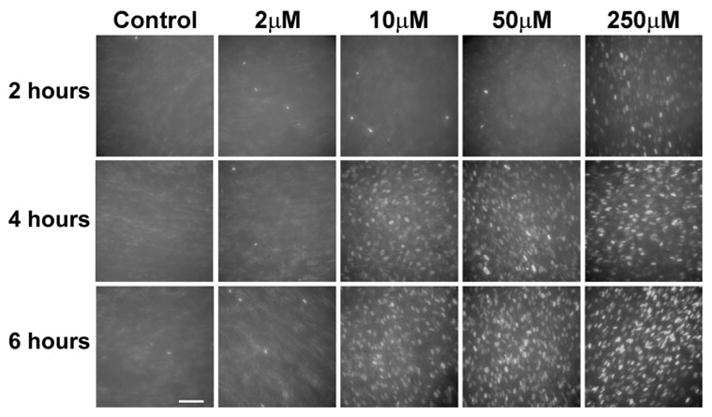
Nonviable human skin fibro-blasts (PD 32) in cell culture detected by a DAPI exclusion test after incubation with various concentrations of thimero-sal. All panels have the same density of cells plated on a dish and represent young fibroblasts that underwent only 32 population doublings in cell culture conditions. Exclusively nonviable cells are visualized by nuclear staining with DAPI, when cellular membranes are compromised and cells are either dead or dying. (Bar = 100 μm).
We performed direct counts of DAPI-positive cells for the initial quantitative assessment of our results. We counted all DAPI-positive cells in two ×40 fields of view for each of the thimerosal concentrations after 6 h of incubation. All counts were taken in central parts of the wells, where the cellular density was most uniform. The comparison with the average density of cells in these areas revealed that, at 2-μM thimer-osal, 11% were DAPI-positive; at 10-μM thimerosal, 58% were DAPI-positive; at 50-μM thimerosal, 61% of the cells were DAPI-positive; and at 250-μM thimerosal, 100% of the neurons had compromised cellular membranes. In controls, less than 1% of the cells were DAPI positive, due to cell death naturally occurring in the cell cultures.
No changes in membrane permeability and DAPI staining were observed with thimerosal concentrations lower than 2 μM at times of incubation up to 24 h.
Since dying cells disattach from the bottom shortly after death and float in the media, they cannot be counted. This explains the similar numbers of DAPI-positive cells counted after 10- and 50-μM thimerosal treatments, and it could have some affect on the sensitivity of the lower limit of toxicity measurements. To address this issue and to take into consideration all DAPI-stained cells, we used a fluorescent microplate reader, which detects the fluorescence of both attached and floating dead cells (see Fig. 3). Using a GENios Plus Microplate Reader, we detected the lower limit of thimerosal toxicity for neuronal cells after 6 h of incubation to be at 1-μM concentration of thimerosal.
FIG. 3.
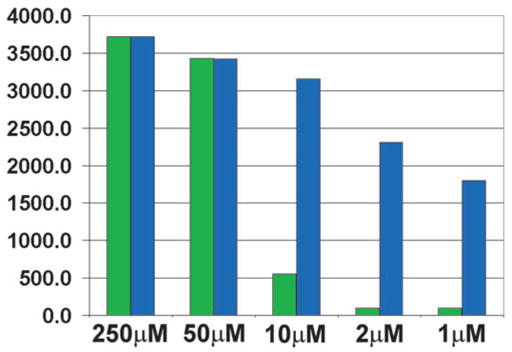
Simultaneous detection of caspase-3 activation and nonviable cells (DAPI test) in cultured human cortical neurons after 6-h incubation with various concentrations of thimerosal. A GENios Plus Multi-Detection Micro-plate Reader with Enhanced Fluorescence was used for the detection. Labeling of active caspase-3 was performed in live cell cultures using a FAM-labeled DEVD fluoromethyl ketone caspase inhibitor (green) with the simultaneously performed DAPI exclusion test (blue), showing dead or dying cells. Fluorescence from both floating and attached nonviable cells was recorded. A Chroma Technology bandpass filter set was used to acquire single-color images: FITC excitation D490/40, emission 520/10; DAPI excitation D360/40, emission 460/20. The reactions were repeated twice, and averages are shown. For the easier comparisons, the caspase-3 signal at 250 μM was equalized (multiplied 6.71 times) with DAPI signal. Y-axis – arbitrary GENios Plus readings of fluorescence. Note that the DAPI exclusion test reveals the early appearance of cells with compromised membranes and a stronger DAPI signal compared to the caspase-3 signal.
Experiments with cultured human fibroblasts produced similar results, although, when compared with neuronal cells, the fibroblasts demonstrated a slightly lower sensitivity to thimer-osal toxicity by the DAPI exclusion test in terms of the number of DAPI-stained cells (Fig. 2).
Similar to neuronal cells, significant numbers of DAPI-stained nuclei were first observed after 2 h of incubation with thimerosal at 250 μM concentration in the fibroblast culture experiments (Fig. 2). After 4 h of incubation, nuclear staining was detected at 10-μM concentration of thimerosal. However, unlike the neuronal cells, the human fibroblasts did not show toxicity at 2-μM concentration of thimerosal after 6 h of incubation.
Detection of Thimerosal-Induced DNA Damage
We used TUNEL to detect DNA breaks generated in neurons and fibroblasts after 6 h of incubation with thimerosal. Following incubation, the cells were fixed, labeled by TUNEL, and counterstained by DAPI, which in these experiments was employed as a fluorescent DNA marker to visualize all cell nuclei in fixed cell cultures.
The results of these experiments are presented in Figure 4. The figure demonstrates that TUNEL-positive cells were detected in all cell cultures after 6 h of incubation, up to the concentration of 2 μM of thimerosal.
FIG. 4.
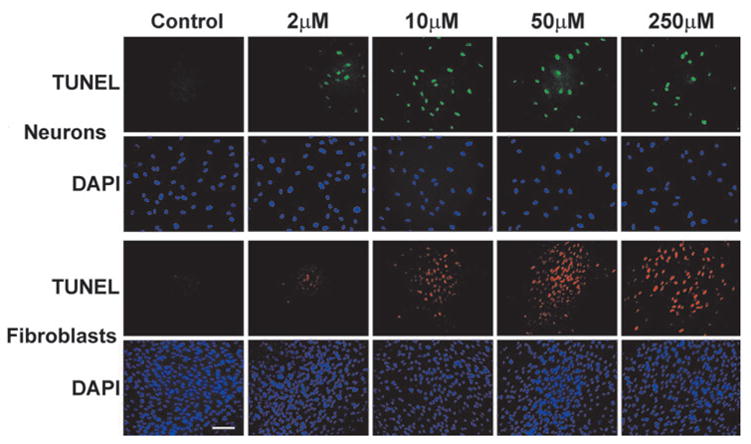
Detection of DNA breaks by TUNEL in cultures of human cortical neurons (green fluorescence) and human fibroblasts (PD32) (red fluorescence) after 6 h of incubation with various concentrations of thimerosal. Blue fluores-cence – DAPI counterstaining was performed on fixed cells to visualize all cellular nuclei. (Bar = 100 μm).
To determine if extending the time of incubation with thimerosal at concentrations below 2μM would result in the generation of DNA breaks, we extended the time of incubation to 24 h in a separate series of experiments. After 24 h, a TUNEL signal was detected in neuronal cells at 1-μM concentration of thimerosal (versus 2 μM at 6 h) (not shown). Incubation of neuronal cells for 24 h with concentrations of thimerosal below 1 μM (125, 250, and 500 nM) did not produce a TUNEL signal.
Detection of Apoptotic Morphology in Thimerosal-Treated Cells
We performed a morphological evaluation of the fixed and fluorescently stained cell cultures after thimerosal treatment for the purpose of identifying apoptotic cells. To identify apoptotic morphology, the cells were fixed and then stained by DAPI. In this experiment, DAPI was employed not as a vital dye, as in our previous study, but rather as a fluorescent histological nuclear stain. Although DAPI is an important marker used in live cell cultures to selectively label nonviable cells (Boutonnat et al., 1999; McCarthy and Hale, 1988), it is also frequently used in fixed cells to visualize nuclear morphology and apo-ptotic bodies. We used it for this purpose in these tests.
Apoptotic morphology was detected in thimerosal-treated cells. Figure 5 demonstrates that, after 6 h of incubation, both fibroblasts and neurons showed morphological signs of apo-ptosis, which included chromatin condensation on the nuclear membrane, the appearance of characteristic doughnut-shaped nuclei, different stages of apoptotic body formation, and freely positioned apoptotic bodies. After 6 h of incubation, apoptotic morphology was observed at concentrations as low as 2 μM of thimerosal (Fig. 5), whereas, at 24 h after incubation, similar apoptotic morphology was observed at concentrations as low as 1 μM.
FIG. 5.
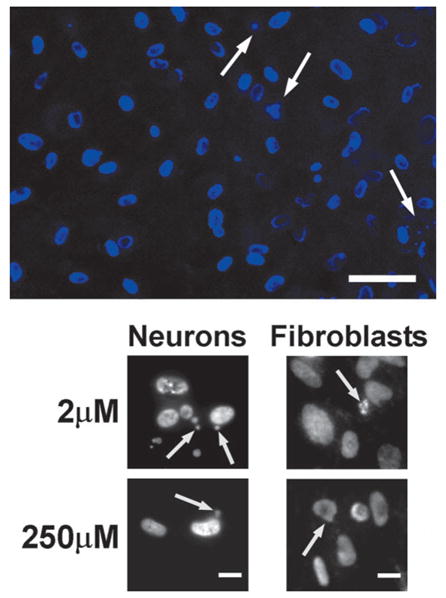
Apoptotic morphology in human neurons and fibroblasts after 6 h of incubation with 2- and 250-μM concentrations of thimerosal. Cell were fixed and stained by DAPI (1 μg/ml) to visualize nuclear morphology and apoptotic bodies. Upper image – DAPI-stained neuronal nuclei (blue) after 6 h of incubation with 2-μM thimerosal. Apoptotic doughnut-shaped nuclei with chromatin condensation on the nuclear membrane and apoptotic bodies are seen. Arrows indicate forming and free located apoptotic bodies (Bar = 50 μm). Lower panel – black/white image of DAPI-stained neuronal and fibro-blast cells after 6 h of incubation with 2- or 250-μM thimerosal. Arrows indicate condensation of chromatin on nuclear membrane or formation of apoptotic bodies. (Bar = 10 μm).
To further confirm the apoptotic nature of cell death induced by thimerosal, we performed detection of active caspase-3, which is a sensitive and specific indicator of apoptosis.
Active Caspase-3 in Thimerosal-Treated Cells
Caspase-3 activation serves as a sensitive marker of ap-optosis, developing through caspase-3– dependent mechanisms, which constitutes one of the most frequent apoptotic pathways. We employed visualization of active caspase-3 directly in living cells through the use of a FAM-labeled peptide caspase inhibitor (FAM-Peptide-FMK) (see Materials and Methods).
We detected caspase-3–positive neuronal cells after 6 h of incubation with thimerosal at concentrations ranging from 250 to 2 μM. The intensity of the signal was dose-dependent and much lower at the 2-μM concentration, compared to higher concentrations, probably due to an earlier stage of caspase-3 activation (Fig. 6).
FIG. 6.
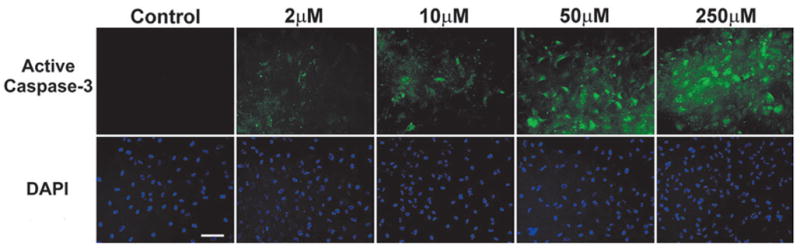
Caspase-3 activation in cultured human cortical neurons after 6-h incubation with various concentrations of thimerosal. Detection of active caspase-3 was performed in live cell cultures using APO LOGIX™ carboxyfluorescein (FAM) caspase detection kit, which employs a FAM-labeled DEVD fluoromethyl ketone (FMK) caspase inhibitor (green fluorescence). The inhibitor irreversibly binds to active caspase-3. Note the primarily cytoplasmic localization of active caspase-3 at lower concentrations of thimerosal, whereas the higher concentrations demonstrate the predominantly nuclear localization of active caspase-3, indicating a later stage in progression towards cell death. Blue fluorescence – DAPI counterstaining performed on fixed cells to visualize all cellular nuclei. (Bar = 100 μm).
Assessment of 200 cells per well randomly, using the fluo-rescent microscope, revealed that active caspase-3 was expressed in 20% of the cells at 2-μM thimerosal, 26% at 10-μM thimerosal, 83% at 50-μM thimerosal, and 97% of the neurons at 250-μM thimerosal concentration. In the controls, less than 1% of the cells was caspase-3–positive, due to cell death naturally occurring in the cell cultures.
At 2-μM thimerosal, the active caspase-3 signal was predominantly observed in the cytoplasm, which represents the early stage of its activation, whereas, at higher concentrations of thimer-osal, the signal was detected in both the cytoplasm and the nuclei (Fig. 6). (Nuclear localization of active caspase-3 is characteristic for later stages of the apoptotic process.)
When we used a fluorescent microplate reader, which detects signals from the detached cells, we detected active caspase-3 activation at 1-μM concentration of thimerosal after 6-h incubation, probably due to the added contribution from floating dead cells (Fig. 3).
When we extended the incubation time with thimerosal from 6 to 24 h, detectable numbers of attached cells with active caspase-3 were observed at 1-μM concentration of thimerosal (Fig. 7). An active caspase-3 signal at 1-μM concentration was cytoplasmic, demonstrating an earlier stage of caspase-3 activation. Interestingly, after 24 h of incubation, the neurons treated with 2-μM thimerosal showed the migration of caspase-3 from the cytoplasm to the nuclei (Fig. 7). The majority of caspase-3–positive cells were also DAPI-positive, which indicates membrane damage occurring simultaneously with apoptotic response. However, at the higher 250-μM concentration of thimerosal, a number of cells were only DAPI-positive without caspase-3 activation, demonstrating necrotic death (Fig. 7). We did not detect active caspase-3 at 24 h of incubation in untreated neurons, or in neuronal cultures treated with lower concentrations of thimerosal (500, 250, and 125 nM).
FIG. 7.
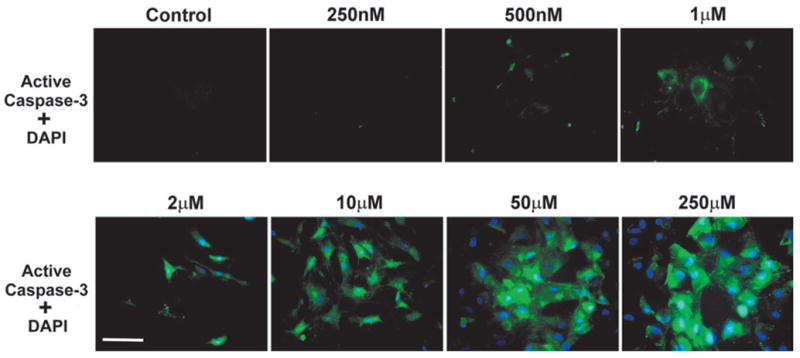
Simultaneous detection of caspase-3 activation and nonviable cells in cultured human cortical neurons after 24-h incubation with various concentrations of thimerosal. Detection of active caspase-3 was performed in live cell cultures using a FAM-labeled DEVD fluoromethyl ketone caspase inhibitor (green fluorescence), which irreversibly binds to active caspase-3. Only nonviable cells are visualized by the simultaneously performed DAPI exclusion test (blue fluorescence), showing dead or dying cells with compromised membranes. Note that, in this case, DAPI is not used as a counterstain for fixed cells, like in the previous figure, but is employed as an exclusion dye (see Materials and Methods). 6 h of incubation with 2-μM concentration of thimerosal resulted in cytoplasmic localization of active caspase-3, while after 24 h active caspase-3 is now localized in the nuclei, indicating a later stage in cell death progression. Compare the cytoplasmic localization of active caspase-3 at 1-μM concentration of thimerosal to its nuclear localization at 2-μM concentration. The majority of caspase-3–positive cells have compromised cellular membranes (Bar = 100 μm). The composite images were created in MetaMorph 4.1 (Advanced Scientific, Inc.) by overlaying single color images. A Chroma Technology bandpass filter set was used to acquire single-color images: FITC excitation D490/40, emission 520/10; DAPI excitation D360/40, emission 460/20.
DISCUSSION
Our data indicate that thimerosal is toxic to human neurons and fibroblasts if applied in micromolar concentrations (1–250 μM). An early sign of thimerosal toxicity is a change in cellular membrane permeability to the vital dye DAPI, which is associated with the loss of cell viability (Boutonnat et al., 1999; McCarthy and Hale, 1988). This can be detected as early as 2 h after incubation.
DAPI proved to be useful for analyzing thimerosal toxicity, because it is a sensitive marker of membrane integrity. It is employed as a propidium iodide substitute in cell viability assays and labels nuclei of dying cells, which lack an intact plasma membrane (Boutonnat et al., 1999; Castro-Hermida et al., 2000; McCarthy and Hale, 1988; Robertson et al., 1998). Dual staining experiments using propidium iodide and DAPI co-staining with FACS analysis demonstrated that DAPI stains only dead cells (McCarthy and Hale, 1988). Viable cells that are not stained by PI also exclude DAPI (McCarthy and Hale, 1988).
The nature of cell death labeled by DAPI in the case of thimerosal treatment deserves additional discussion. The DAPI exclusion method relies on the fact that this dye is largely impermeable to cells with an intact plasma membrane. However, when cell membrane integrity becomes compromised, DAPI gains access to the nucleus, where it complexes with DNA and renders the nucleus highly fluorescent. Early compromised integrity of plasma membranes is a characteristic feature of necrotic cell death, whereas, in apoptosis, cellular membranes are compromised at later times. This is why intra-cellular staining by DAPI (and also by its more toxic substitute propidium iodide) is regularly interpreted as a sign of necrosis (Boutonnat et al., 1999). However, in the case of thimerosal, the changes in membrane permeability coincided with the activation of apoptosis-specific caspase-3 (Fig. 3). In our opinion, this indicates a separate direct membrane damaging effect of thimerosal that developed simultaneously with apoptotic changes, such as caspase-3 activation.
In many cases, the importance of caspase-3 activation is related to its connection to specific and extensive apoptotic DNA cleavage (Porter and Janicke, 1999). This DNA fragmentation can be labeled by the TUNEL technique and is widely used for the visualization of apoptotic cells. A caspase-activated deoxyribonuclease (CAD, or DFF 40) is implicated as a direct executioner of the cleavage (Liu et al., 1997; Mukae et al., 1998). Most of the time, the enzyme is kept inactive by the binding of an inhibitor (ICAD, or DFF 45). Activation of the nuclease occurs when the inhibitor is cleaved by activated caspase-3 (Enari et al., 1998; Sakahira et al., 1998). However, the exact sequence of events in case of a human brain is likely different from this scheme. In human CNS neurons, other caspase-3–related pathways and possibly the other DNA cleaving enzymes are more important, and the role of the CAD-mediated mechanism is likely limited, because no expression of CAD mRNA was detected in human brain cells (Mukae et al., 1998).
Similar to our results, high cellular toxicity of thimerosal in low micromolar concentrations was recently reported using another cell culture model (Makani et al., 2002). The effects of different concentrations of thimerosal were examined in Jurkat cells. The cells were incubated with 5- to 0.5-μM concentrations of thimerosal for 24 h. Concentration-dependent apopto-sis was detected and measured by TUNEL. Caspase-3 activation was also detected after 4 and 6 h of incubation with thimerosal. The study concluded that thimerosal induced caspase-3– dependent apoptosis in Jurkat cells. This apoptosis was associated with the depolarization of the mitochondrial membrane and release of cytochrome c. In this same study, a significantly enhanced generation of reactive oxygen species was also detected, as a result of incubation with thimerosal (Makani et al., 2002). We hypothesize that these elevated levels of free radicals and the subsequent oxidation may play role in apoptosis induction and might also be involved in the direct membrane-damaging effects of thimerosal identified in our study.
We showed that the concentrations of thimerosal that induced toxic effects in human cortical neurons ranged from 1 to 250 μM. However, comparisons of the nuclear morphology of dying cells after incubation with higher versus lower concentrations of thimerosal demonstrate important differences. Although caspase-3 activation was detected in both high and low concentrations of thimerosal, the morphology of dying cells was different in these two situations. The cell bodies of neurons treated with higher concentrations of thimerosal (50 –250 μM) were swollen, which is more characteristic of necrotic cell death, whereas cells treated with low concentrations (2–10 μM) were shrunken, as is typical for apoptosis (Fig. 7). Similarly, the nuclei of dying neurons treated with 250-μM thimer-osal were larger in size and swollen, in contrast to the shrunken nuclei of cells treated with 2-μM thimerosal (Fig. 7). Thus, cell death occurring after incubation of neuronal cells with higher concentrations of thimerosal has features of both apoptosis (caspase-3 activation) and necrosis (cell edema and nuclei swelling). This can be explained by a direct membrane-damaging effect of thimerosal, which rapidly leads to the loss of membrane integrity and cell swelling. This process likely occurs simultaneously with apoptosis induction, the initiation of the caspase cascade, and the activation of caspase-3. At lower concentrations of thimerosal, direct membrane-damaging effects are weaker, and no swelling is observed.
Investigation of thimerosal toxicity is especially important at the present time, because this compound is used in biological products and can be administered in toxic doses either accidentally or intentionally (Ball et al., 2001).
In our study, the concentrations of thimerosal that induced toxic effects ranged from 1 μM (405 μg/l) to 250 μM (101 mg/l), which is equivalent to the levels of inorganic mercury from 201 μg/l to 50 mg/l. In clinical cases of accidental or intentional usage in high concentrations, thimerosal was administered in doses from 3 mg/kg to several hundred mg/kg (Ball et al., 2001). Such doses resulted in local necrosis at the application site and severe central nervous system and kidney injury.
Much lower concentrations are reached during normal vaccination, when thimerosal-containing vaccines are used. In the case of a full series of vaccinations containing thimerosal, up to 403 μg of thimerosal (equivalent to 200 μg of mercury) are received by 6 months of age (calculated from Ball et al., 2001). This results in the administration of 200/3.81 = 52 μg/kg, 200/5.22 = 38 μg/kg, and 200/6.27 = 32 μg/kg of mercury. These calculations utilize averages of the 5th, 50th, and 95th% weight for females at birth (2.36 kg, 3.23 kg, 3.81 kg) and at 6 months (5.25 kg, 7.21 kg, 8.73 kg) = 3.81 kg, 5.22 kg, 6.27 kg, reported by (Ball et al., 2001) when used in calculating exposure limits for mercury in comparisons of various agencies guidelines.
The lowest toxic concentration of mercury contained in the thimerosal doses in our present study (201 μg/l) is less than four times higher than some of these estimated concentrations. The rapidly developing toxicity of thimerosal in low micro-molar concentrations over short time frames is of concern and suggests that additional research is necessary to estimate the effects of prolonged exposure to thimerosal in lower doses.
In this paper we demonstrated that extending the time of incubation with thimerosal from 2 to 6 h is associated with toxicity that was not seen after a shorter time of exposure. For this reason, further studies of lower concentrations and longer exposure times appear to be warranted. These results indicate that additional research is needed to more fully delineate the dose- and time-dependent toxicity of thimerosal in sub-micro-molar concentrations and suggests that toxicity may occur at even lower doses than those utilized in these experiments, with longer times of exposure. Because mercury can be retained in body organs for months to years, the study of longer incubation times is warranted. We also conclude that a proposed combination of fluorescent techniques combining the assessment of DNA, membrane damage, and active caspase-3 is useful in studying thimerosal toxicity.
Acknowledgments
We are grateful to Katie Fewell and Dr. Benxiao Zhang for valuable technical assistance in experiments and in manuscript preparation. This research was supported by grant R01 CA78912-01 from the National Cancer Institute, National Institutes of Health (D.S.B.); by grant 004949-054 from the Texas Higher Education Coordinating Board (D.S.B. and V.V.D.); and by grants from DeBakey Medical Foundation (V.V.D.), Baylor College of Medicine (V.V.D.), the Taub Foundation (D.S.B.), the Henry J. N. Taub Fund for Neurosurgical Research (D.S.B.), the George A. Robinson IV Foundation (D.S.B.), the Blanche Greene Estate Fund of the Pauline Sterne Wolff Memorial Foundation (D.S.B.), and the Seigo Arai and Koppelman Funds of the Neurological Research Foundation (D.S.B.).
References
- Amstad P, Johnson G, Lee B, Dhawan S. An in situ marker for the detection of activated caspases. Biotechnol Lab. 2000;18:52–56. [Google Scholar]
- Ball L, Ball R, Pratt R. An assessment of thimerosal use in childhood vaccines. Pediatrics. 2001;107:1147–1154. doi: 10.1542/peds.107.5.1147. [DOI] [PubMed] [Google Scholar]
- Bedner E, Smolewski P, Amstad P, Darzynkiewicz Z. Activation of caspases measured in situ by binding of fluorochrome-labeled inhibitors of caspases (FLICA): Correlation with DNA fragmentation. Exp Cell Res. 2000;259:308 –313. doi: 10.1006/excr.2000.4955. [DOI] [PubMed] [Google Scholar]
- Ben-Ozer E, Rosenspire A, McCabe M, Worth R, Kindzelskii A, Warra N, Petty H. Mercuric chloride damages cellular DNA by a non-apoptotic mechanism. Mutat Res. 2000;470:19 –27. doi: 10.1016/s1383-5718(00)00083-8. [DOI] [PubMed] [Google Scholar]
- Blair A, Clark B, Clarke A, Wood P. Tissue concentrations of mercury after chronic dosing of squirrel monkeys with thimerosal. Toxicology. 1975;3:171–176. doi: 10.1016/0300-483x(75)90082-7. [DOI] [PubMed] [Google Scholar]
- Boutonnat J, Barbier M, Muirhead K, Mousseau M, Ronot X, Seigneurin D. Optimized fluorescent probe combinations for evaluation of proliferation and necrosis in anthracycline-treated leukaemic cell lines. Cell Prolif. 1999;32:203–213. doi: 10.1046/j.1365-2184.1999.3240203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcia-Calvo M, Peterson E, Leiting B, Ruel R, Nicholson D, Thornberry N. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem. 1998;273:32608 –32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Castro-Hermida J, Freire-Santos F, Oteiza-Lopez A, Ares-Mazas E. Unexpected activity of beta-cyclodextrin against experimental infection by Cryptosporidium parvum. J Parasitol. 2000;86:1118 –1120. doi: 10.1645/0022-3395(2000)086[1118:UAOCAE]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Ferrat L, Romeo M, Gnassia-Barelli M, Pergent-Martini C. Effects of mercury on antioxidant mechanisms in the marine phanerogam Posidonia oceanica. Dis Aquat Organ. 2002;50:157–160. doi: 10.3354/dao050157. [DOI] [PubMed] [Google Scholar]
- Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Magos L, Brown AW, Sparrow S, Bailey E, Snowden RT, Skipp WR. The comparative toxicology of ethyl- and methylmercury. Arch Toxicol. 1985;57:260 –267. doi: 10.1007/BF00324789. [DOI] [PubMed] [Google Scholar]
- Makani S, Gollapudi S, Yel L, Chiplunkar S, Gupta S. Biochemical and molecular basis of thimerosal-induced apoptosis in T-cells: Major role of mitochondrial pathway. Genes Immun. 2002;3:270 –278. doi: 10.1038/sj.gene.6363854. [DOI] [PubMed] [Google Scholar]
- McCarthy K, Hale M. Flow cytometry techniques in radiation biology. Toxicol Lett. 1988;43:219 –233. doi: 10.1016/0378-4274(88)90030-6. [DOI] [PubMed] [Google Scholar]
- Mukae N, Enari M, Sakahira H, Fukuda Y, Inazawa J, Toh H, Nagata S. Molecular cloning and characterization of human caspase-activated DNase. Proc Natl Acad Sci USA. 1998;95:9123–9128. doi: 10.1073/pnas.95.16.9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter A, Janicke R. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99 –104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- Robertson L, Campbell A, Smith H. Viability of Cryptospo-ridium parvum oocysts: Assessment by the dye permeability assay. Appl Environ Microbiol. 1998;64:3544 –3545. doi: 10.1128/aem.64.9.3544-3545.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96 –99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- Schurz F, Sabater-Vilar M, Fink-Gremmels J. Mutagenicity of mercury chloride and mechanisms of cellular defense: The role of metal-binding proteins. Mutagenesis. 2000;15:525–530. doi: 10.1093/mutage/15.6.525. [DOI] [PubMed] [Google Scholar]
- Shapiro H. Practical Flow Cytometry. Liss; New York: 1985. pp. 84–154. [Google Scholar]
- Smolewski P, Bedner E, Du L, Hsieh T, Wu J, Phelps J, Darzyn-kiewicz Z. Detection of caspase activation by fluorochrome-labeled inhibitors: Multiparameter analysis by laser scanning cytometry. Cytometry. 2001;44:73– 82. doi: 10.1002/1097-0320(20010501)44:1<73::aid-cyto1084>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]