

Summary
A central question about the brain is how information is processed by large populations of neurons embedded in intricate local networks. Answering this question requires not only monitoring functional dynamics of many neurons simultaneously, but also interpreting such activity patterns in the context of neuronal circuitry. Here we introduce a versatile approach for loading Ca2+ indicators in vivo by local electroporation. The unique feature of this method is that Ca2+ imaging can be performed both at neuron population level and with exquisite subcellular resolution down to dendritic spines and axon boutons. This enabled mitral cell odor-evoked ensemble activity to be analyzed simultaneously with revealing their specific connectivity to different glomeruli. Co-labeling of Purkinje cell dendrites and intersecting parallel fibers allowed Ca2+ imaging of both presynaptic boutons and postsynaptic dendrites. This approach thus provides an unprecedented capability for in vivo visualizing active cell ensembles and tracing their underlying local neuronal circuits.
Introduction
Neural coding and processing takes form of complex spatiotemporal activity patterns in a large number of neurons that are interconnected into sophisticated circuits. To understand such a complicated process, monitoring activity of a single neuron or neuronal population is essential but not sufficient. It is equally important to interpret the recorded activity pattern within the context of specific local neural circuits. One example in this regard is odor processing in the mammalian olfactory bulb. Odors are initially represented as spatial patterns of activated olfactory glomeruli (Rubin and Katz, 1999; Uchida et al., 2000), which subsequently break down into distributed mitral cell ensemble codes. Understanding such an exquisite coding-pattern transformation requires the mitral cell activity pattern to be analyzed with reference to specific projections of their apical primary dendrites into different glomeruli (Shepherd et al., 2004). Similarly, analyzing the functional organization of orientation columns in the visual cortex also necessitates correlating the sharp shift of orientation tuning property with dendrite arborization and synaptic connectivity of pyramidal cells at inter-columnar borders (Ohki et al., 2005; Ohki et al., 2006).
Currently, such a concerted analysis of both neuronal ensemble dynamics and underlying functional connectivity remains technically difficult. One major approach to monitor the activity of many neurons simultaneously is optical imaging with voltage- or Ca2+-sensitive dyes. So far, most in vivo Ca2+ imaging studies utilize a glass pipette to inject Ca2+ dye directly into a recorded neuron. The effective dye diffusion from the pipette into a neuron allows imaging functional signals in small subcellular structures such as dendrites (Charpak et al., 2001; Helmchen et al., 1999; Svoboda et al., 1997). However, because of the technical difficulty, this loading procedure is impractical for analyzing neural networks composed of many cells. To facilitate in vivo imaging of neuron ensembles, an effective method has recently been developed to load large numbers of cells with membrane-permeant Ca2+ dyes in acetoxymethyl (AM) ester form (Stosiek et al., 2003; Garaschuk et al., 2006). With this method, thousands of neurons in the visual cortex can be labeled for imaging the functional organization of neuronal populations distributed across orientation borders and pinwheels (Ohki et al., 2005; Ohki et al., 2006). Compared with the intracellular staining, one drawback for the AM-ester dye bulk loading is that the universal and non-specific labeling of both neurons and glial cells creates a fluorescence background which generally limits the identification of individual small neuronal processes such as dendritic spines and axon terminals, the critical components of local circuits (Stosiek et al., 2003; Nimmerjahn et al., 2004; Heim et al. 2007; but see Kerr et al., 2005; Sullivan et al., 2005).
Dextran-conjugated cell markers such as BDA are routinely used in tracing long-range neural projections in the brain (Vercelli et al., 2000). They have a remarkable capability of labeling the full length of axons from cell body to presynaptic terminals. Dextran-bound Ca2+ indicators have been used for functional imaging through either retrograde or anterograde labeling of distally located neuronal somas or axonal terminals (Friedrich and Korsching, 1997; Kreitzer et al., 2000; Mulligan et al., 2001; O’Donovan et al., 1993; Wachowiak and Cohen, 2001). The applicability of this method in studying local circuits is, however, limited by its only labeling the neurons with direct axonal connection to a remote dye-injection site. On the other hand, electroporation is a widely used technique for delivering genes and dyes into neurons (Haas et al., 2001; Lodovichi et al., 2003; Pinault, 1996; Tabata and Nakajima, 2001). Recently, using an in vitro preparation, the O’Donovan’s group has shown elegantly that calcium dyes including dextran conjugates can be effectively loaded into neurons via en bloc electroporation of an isolated neonatal spinal cord placed between a pair of metal electrode plates (Bonnot et al., 2005).
To achieve simultaneous tracing and functional imaging of local neuronal circuits, we here introduce a simple but versatile method for loading synthetic Ca2+ dyes in vivo through local electroporation in various brain regions. This method features the combined advantages of intracellular single-cell staining and loading cell populations with an AM-ester dye. It enables not only imaging Ca2+ activity at the cell population level, but also visualizing local neuronal circuits with a pattern and resolution similar to those in Golgi silver staining.
Results
Local dextran-dye electroporation and its impact on circuit function
Dextran-conjugated Ca2+ indicators are impermeable to cell membrane (Haugland, 2002). Previous attempts to label neurons simply by pressure injection of dextran Ca2+ dyes into the brain only result in dye accumulation in the extracellular space (Bonnot et al., 2005; Stosiek et al., 2003). To overcome this problem, we carried out local electroporation of cell membrane with a glass pipette filled with a Ringer solution containing dextran-conjugated Ca2+ dyes. A small patch-like pipette with a tip inner diameter of 2–4 μm was used to minimize mechanical damages to local neuronal circuits. As illustrated schematically in Figure 1A, the pipette was inserted into a desired brain region. Electrical pulses were delivered from a stimulator to drive a regular electric isolator to output weak positive current (1–5 μA). The current flowed through the animal from the tip of the pipette to the grounded mouse tail. The waveform and amplitude of injected current were monitored using an oscilloscope to measure the voltage drop across a series resistor. Dextran-conjugated Ca2+ indicators electrophoresed from the pipette were found to be able to label not only cell bodies but also dendrites and axons passing through the electroporation site (Figures 1B, 3C1 and 4A). Instead of several days required for dextran anterograde or retrograde loading from a remote injection site, in vivo local electroporation took only two to three hours for elaborate neuronal structures to be clearly visualized down to individual dendritic spines. Ca2+ imaging was thus carried out routinely on the same day of dye loading, which obviated the complication of post-surgical animal care, brain infection and inflammation, as well as incidence of animal death before Ca2+ imaging is performed.
Figure 1.
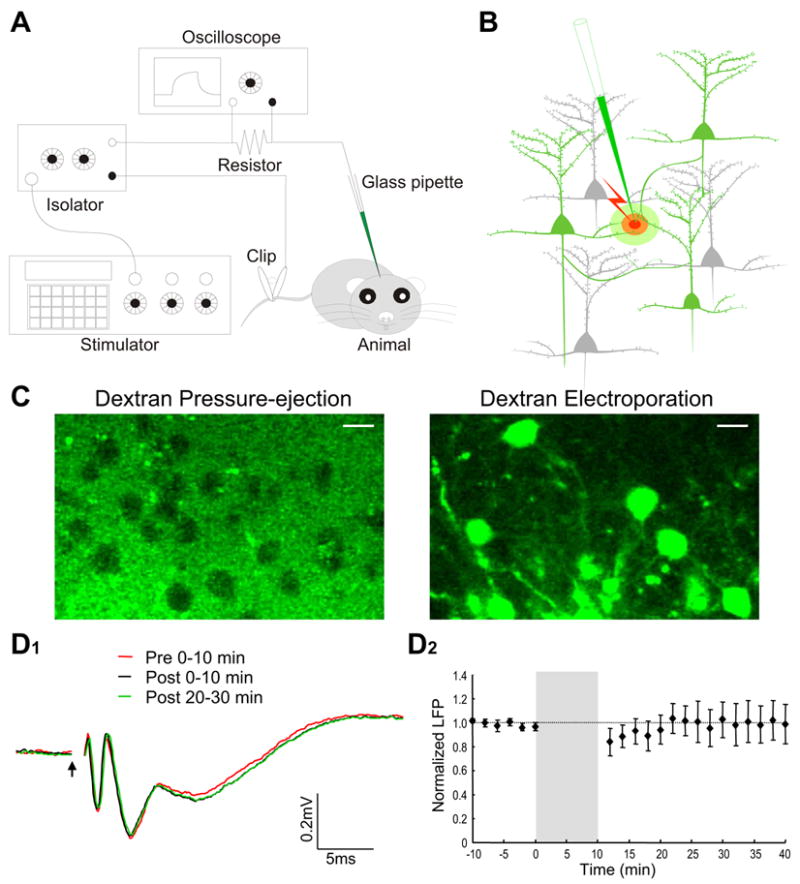
Equipment setup, dye loading mechanism and evaluating disruptive effect of electroporation on network function. A, Schematic diagram showing the experimental setup. An electric circuit, consisting of a pulse generator, a current-output isolator, a series resistor, a Ca2+ dye-filled glass pipette, an anesthetized animal and a metal clip attached to the mouse tail, was used for local electroporation. The waveform and amplitude of current pulses were monitored on an oscilloscope by measuring the voltage drop across a 100-KΩ resistor. B, Schematic illustration of how Ca2+ indicators are loaded to visualize local neuronal circuits. Cell membrane in close vicinity of the pipette tip was transiently ruptured by electric current pulses, and Ca2+ dyes were electrophoresed into the axons and dendrites that passed through a small effective dye-loading area. The loaded dyes were then transported or simply diffused along axons and dendrites to visualize the entire-neuron morphology. C, Evidence for the critical involvement of membrane electroporation in dextran dye labeling. Left panel, loading dextran dye into the barrel cortex simply by pressure ejection (6 psi, 5 sec) did not lead to successful neuronal labeling. Right panel, loading the same dextran-dye solution by electroporation led to the labeling of many neurons with both soma and processes visualized. The two images were taken at three hours after loading with 10% dextran-conjugated Calcium Green-1 (CG-1). Scale bar, 10 μm. D1, Local field potentials recorded in the olfactory bulb external plexiform layer before and after electroporation. Field potentials were evoked by electric stimulation of the lateral olfactory tract. The first negative peak at 1–3 ms after the stimulus onset (arrow) indicates the antidromic activation of mitral cells, while the second and third peaks reflect the functional activation of local synaptic circuits. Three different field-potential traces are superimposed for comparison, which are average of the recordings made, respectively, at 0–10 min before and 0–10 or 20–30 min after electroporation. D2, Plotting over time the normalized amplitude of the second negative peak before and after electroporation. The shaded area indicates 10-min electroporation. Field potentials were first normalized as percentage of the mean baseline amplitude before electroporation, and were then averaged across different experiments. Each data point is presented as mean±SEM.
Figure 3.
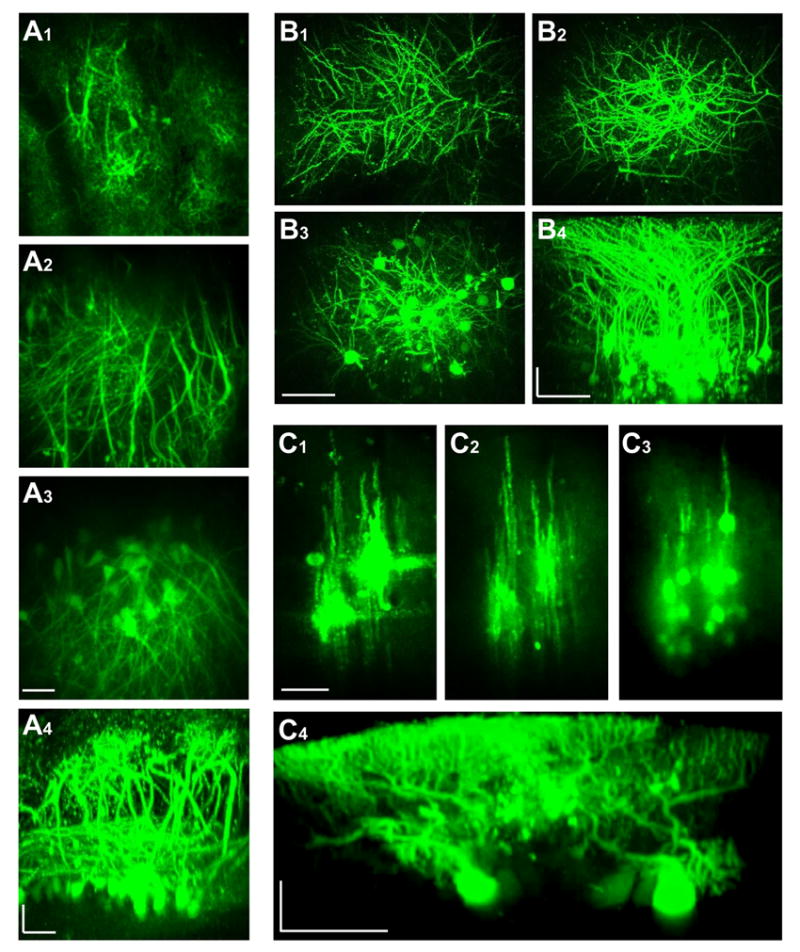
In vivo two-photon imaging of neuronal labeling with dextran-conjugated Ca2+ indicators in three representative brain regions. A, olfactory bulb; B, barrel cortex; C, cerebellum. A1, A2 and A3 are fluorescence images obtained by projecting in z-axis the images captured consecutively from three different ranges of depth (0–125, 125–250 and 250–400 μm). A4 is a side projection of the entire image stack from A1, A2 and A3. B1, B2 and B3 are three images obtained by z-projection for a depth range of 0–100, 100–200 and 200–300 μm, respectively. B4 is a side projection of the entire image stack from B1, B2 and B3. C1, C2 and C3 are compressed from images taken at the depth of 0–100, 100–150 and 150–250 μm. C4 is a side view reconstructed from a different image stack. Three video files (Movies A, B, C) are provided in Supplemental Materials to give a better 3-D illustration of labeling in these different brain regions. Scale bars, 50 μm. A4, B4 and C4 are horizontally stretched and thus carry two different scale bars.
Figure 4.
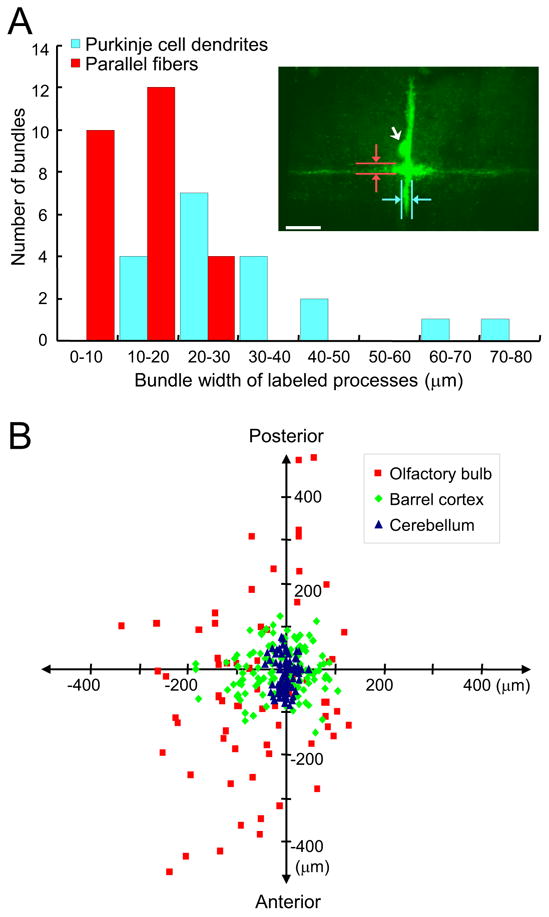
Small restricted dye-loading area and broad distribution of labeled neurons. A, Quantifying the labeled bundle widths of both parallel fibers and Purkinje cell dendrites in the cerebellum. Red columns show the width distribution of labeled parallel fibers, and cyan columns are distribution of labeled Purkinje cell dendrites. The inset shows an example of labeling obtained by z-projection of an image stack. The loading conditions were 5% dextran-bound OGB-1 (10,000 m.w.), 2 μA current, 100 ms pulse width and 900 pulses delivered at 5 Hz. Please note that in this case only one Purkinje cell was labeled, with its soma indicated by a white arrow. The vertical fluorescent bundle is Purkinje cell dendrites, and the horizontal bundle is labeled parallel fibers. Also indicated is how the widths of labeling were measured. Scale bar, 50 μm. B, Summary of the distribution patterns of labeled neuronal somas in three brain regions. The dye injection sites are aligned at the center of the diagram. Blue triangles indicate the distribution of labeled cells in the cerebellum, green diamonds reflect labeling in the barrel cortex, and red squares are labeling in the olfactory bulb. The vertical and horizontal axes correspond, respectively, to the rostro-caudal and medio-lateral directions.
Because of high water solubility, the unloaded dextran dye remaining in extracellular space was easily washed out, leaving sometimes no background at all, while at other times only a small fluorescence spot confined in close proximity to the loading pipette. The exact reason for such a variation in fluorescence background was unclear. It could be due to changes in local blood circulation, which appeared to be critically involved in removing excessive dyes from the electroporation site.
To confirm further that dextran dye loading was indeed due to cell-membrane electroporation rather than other mechanisms such as active dye uptake, we injected the same dextran dye solution directly into the barrel cortex by pressure ejection through the same pipette. Over three hours after the pressure ejection, we did not observe any neuronal labeling. Under two-photon microscope, neuronal somas appeared as many dark holes in fluorescence background (Figure 1C). This suggests that the pressure-ejected dextran dye was not taken up by neurons, but instead remained in extracellular space until being washed. Therefore, the action of electric current pulses is not just limited to dextran dye iontophoresis, but also electroporates the membrane for dye penetration.
Given such a membrane-eletroporating action, one major concern regarding this method is the potential damages incurred to neurons, which may compromise normal local-circuit functions. However, the current amplitude that we used was much lower than those routinely applied for stimulating brain tissues. To assess the actual effect of electroporation on local network function, field potentials were recorded in the olfactory bulb external plexiform layer, and mitral cell populations were antidromically activated by stimulating the lateral olfactory tract (LOT). Such an LOT-evoked field potential has been a traditional measure to assess the functionality of olfactory bulb local dendritic circuits (Rall and Shepherd, 1968). By comparing the field potentials recorded before and after electroporation, neither the waveform nor amplitude of the LOT-evoked potentials was affected, except a slight depression sometimes observed in the first ten minutes following electroporation (Figure 1D1, D2). A statistical analysis of the field-potential amplitudes using ANOVA also confirmed that there was no significant difference between the potentials recorded at any time points. Similar experiments were carried out in the barrel cortex, and no significant changes were noted in the whisker deflection-evoked local field potentials (data not shown). These results suggest that the functioning of local circuits is not compromised by the electroporation protocol used in this study.
Exploring for optimal dye-loading conditions
To optimize the electroporation dye loading conditions, we systematically varied three major parameters: the current pulse amplitude (from 0.1 to 10 μA), current pulse duration (from 5 to 200 ms) and dextran dye concentration (from 0.1% to 10%), while fixing other conditions such as the pipette tip size (~2.5 μm inner diameter), the frequency (2 Hz) and total number of pulses delivered (1200 pulses). Calcium Green-1 (CG-1) dextran conjugate in 10,000 molecular weight (m.w.) was used in this set of experiments in the barrel cortex. The first three panels in Figure 2 illustrate the relations of the three parameters with the total number of neurons labeled per electroporation.
Figure 2.
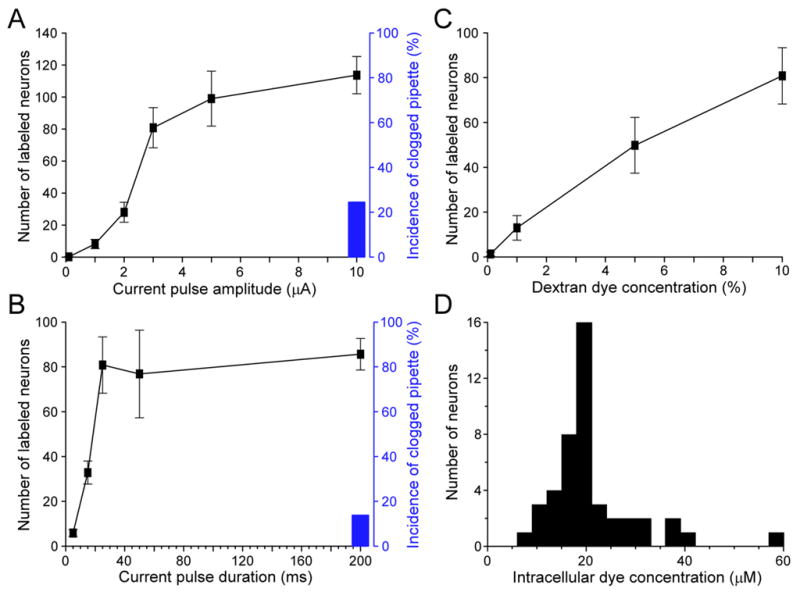
Effects of different parameters on dextran-dye loading efficiency and an estimate of intracellular Ca2+ dye concentration. A, B and C, Plots of the total number of labeled neurons per electroporation in the barrel cortex with the current pulse amplitude (A), current pulse duration (B) and dextran-conjugated CG-1 (10,000 m.w.) concentration (C). For A, the other two loading parameters were 25 ms current pulse width and 10% dextran CG-1; for B, the parameters were 3 μA current and 10% dextran; and for C, 3 μA current amplitude and 25 ms pulse duration. All the loading pipettes had a tip inner diameter of ~2.5 μm, and a total of 1200 pulses were delivered at 2 Hz. In A and B, black bars indicate the percentage of incidence in which the loading pipette got clogged during electroporation. The clogging of pipette usually happened at a large current amplitude (10 μA for 25 ms pulses) or with a long pulse duration (200 ms for 3 μA current). D, Histogram showing the distribution of estimated CG-1 concentration in 45 neurons labeled by local electroporation with parameters of 3 μA current, 25 ms pulse duration, 1200 pulses at 2 Hz and 10% CG-1 dextran conjugate (10,000 m.w.).
The number of labeled neurons, as counted by stained cell bodies, increased sigmoidally with the amplitude of current pulses (Figure 2A). The most dramatic increase of labeling occurred in a current range of 2 to 3 μA; beyond 5 μA, there was much less further increase. Figure 2B shows that increasing the pulse duration also increased the number of labeled neurons. Such an increase occurred abruptly at the short pulse range, reaching a plateau level at ~25 ms. Finally, the labeling of neurons increased linearly with the dextran-dye concentration (Figure 2C). Besides the number of labeled neurons, another factor of consideration was the clogging of dye-loading pipette during electroporation. This usually happened with 10% dextran concentration either at a large current amplitude or a long pulse duration, as shown by black bars in Figure 2A and B. These results thus provide a set of optimal parameters for dye electroporation, which are 3–5 μA electric current, 25 ms pulse duration and using 5–10% dextran-conjugated Ca2+ dyes.
Intracellular Ca2+ dye concentration loaded with dextran electroporation
To estimate the intracellular Ca2+ dye concentration loaded by local electroporation, we first carried out in vivo loading of 10% dextran-conjugated CG-1 into the mouse barrel cortex, and then sacrificed the animal to make slice preparations. In the slices containing dextran-labeled neurons, whole-cell patch recording was made on an unlabeled neuron with a pipette containing 25 μM CG-1 hexapotassium salt. After intracellular dialysis reached equilibrium, fluorescence intensity of the patched cell was compared to that of the neighboring neurons labeled with CG-1 dextran electroporation; and the intracellular dye concentration was estimated by assuming a linear correlation between dye concentration and fluorescence intensity. Figure 2D shows a distribution histogram of the estimated CG-1 concentration in 45 dextran-labeled neurons. On average, the loaded dye concentration was 21.0 ± 9.1 μM (mean ± SD). The detailed loading conditions for estimating intracellular dye concentration is presented in the legend of Figure 2D.
Visualizing local circuits in various brain regions
To test the versatility of our loading procedure, three major brain regions were studied with either CG-1 dextran conjugate or Oregon Green 488 BAPTA-1 (OGB-1) conjugate. In the olfactory bulb, dextran-bound Ca2+ indicators were electroporated into the deep half of the external plexiform layer (EPL). Cell labeling was found to be very extensive, with many mitral cells and their entire dendritic structures clearly visualized. In the superficial glomerular layer, in vivo two-photon imaging revealed the fine structures of mitral cell distal dendritic tufts (Figure 3A1). With the focus of microscope moving downward, many thin dendritic branches bifurcated at different levels of glomerular tufts gradually converged into thicker primary dendrite trunks, which left glomeruli and extended across the EPL (Figure 3A2, for better visualization see Movie A in Supplemental Materials). In the deep part of EPL, the mitral cell lateral secondary dendrites were also revealed extending horizontally, forming complicated mesh-like networks. Further below, many mitral cell somas were distributed widely in the mitral cell soma layer (Figure 3A3). A computer-reconstructed side view in Figure 3A4 illustrates how well the detailed layout of mitral cell dendritic network was visualized in vivo for Ca2+ imaging.
In the mouse barrel cortex, many thin spiny dendrites were found below the pia, extending laterally at an oblique angle (Figure 3B1). These branches gradually converged into thicker apical dendrites that extended vertically into deep layers (Figure 3B2) to join different cell bodies (Figure 3B3). The pyramidal cell basal dendrites were also clearly visible (Figure 3B3). A side projection of the image stack reveals that labeled neurons could be easily identified, and that their morphology could be traced from a cell body all the way to dendritic tips (Figure 3B4, also see 3D rotation of labeled barrel cortex in Supplemental Movie B).
In the 4th and 5th lobules of the cerebellum, CG-1 dextran conjugate was injected into the molecular layer. Unlike those observed in the olfactory bulb and barrel cortex, staining in the cerebellum was much more restricted to the electroporation site. The labeled Purkinje cell somas and dendrites were arranged in longitudinal stripes, as shown in Figure 3C by three images taken at different depths. A better visualization is provided as Supplemental Movie C. The parallel fibers were also labeled as horizontal fluorescent bundles crossing perpendicularly the labeled Purkinje cell dendrites at the point of dye electroporation (Figure 3C1, two crosses for two injection sites). Such a cross-like pattern is best illustrated in the inset of Figure 4A, in which electroporation was performed at only one single site and fluorescence background due to unloaded dye at the site of injection was completely washed out. This indicates that both dendrites and axons can be loaded with dextran-dye electroporation. A side projection in Figure 3C4 shows the extensive labeling of Purkinje cell dendrite.
The unique organization of cerebellar local circuits presents a valuable opportunity to quantify the actual tissue volume in which dextran-conjugated Ca2+ dye can be effectively loaded by local electroporation. Figure 4A shows the width distribution of labeled Purkinje-cell dendrite stripes and parallel-fiber axon bundles. The average width of labeling was 13.4 ± 5.3 μm for parallel fibers and 32.6 ± 15.3 μm for Purkinje cell dendrites (mean ± SD). These numbers suggest that the effective dye-loading area of electroporation, using a 2–4 μm pipette tip and less than 5 μA current, was quite constricted, within a diameter of a few tens of microns. The labeled cell bodies were, however, distributed much more broadly than this range, suggesting that most neurons were labeled via their dendrites or axons passing through the small effective dye-loading area.
Figure 4B compares the spatial distributions of labeled cell bodies in the mouse cerebellum, barrel cortex, and olfactory bulb. The X and Y coordinates indicate the relative distance of each labeled neuron to the dye-loading pipette in the medio-lateral and rostro-caudal direction, respectively. The Purkinje cell labeling was relatively close to the electroporation site, with an elongated distribution of cell bodies along Y axis. This is consistent with the fan-shape orientation of Purkinje cell dendrites. Compared with the cerebellum, labeled neurons in the barrel cortex exhibited a much wider and more symmetrical distribution. The most extensive labeling was observed in the olfactory bulb, which covered approximately 1,000 μm of antero-posterior range. Due to the limited visual accessibility of the olfactory bulb circumference, this distribution appeared to be under-estimated, especially for the medio-lateral dimension. Such a wide staining pattern is presumably due to the long extension of mitral cell lateral dendrites (Mori et al., 1983; Xiong and Chen, 2002). These results suggest that the range of dye labeling achieved by a single electroporation depends on regional differences in the architecture of local neuronal circuits. For brain regions like the cerebellum, electroporation at multiple sites is needed to achieve more extensive labeling for network functional imaging.
Electroporation with regular salt-form Ca2+ indicators
We also applied our optimized electroporation protocol to testing the use of regular salt-form Ca2+-sensitive dyes, which so far can only be loaded into a neuron by direct intracellular injection. We used three different dyes in potassium salt: CG-1 hexapotassium, OGB-1 hexapotassium and X-rhod-1 tripotassium. Interestingly, in vivo local electroporation was able to load all these three cell-impermeant dyes into neurons, just as with dextran conjugates. Figure 5A illustrates such an example in which two separate electroporations were made in the barrel cortex. One was with 5% CG-1 hexapotassium (green color) and the other with 5% X-rhod-1 tripotassium (red color). This led to dual-color labeling of two neuron populations, with their dendrites (Figure 5A1) and somas (Figure 5A2) visualized at a resolution down to dendritic spines. In some experiments, a portion of neurons was labeled with both OGB-1 and X-rhod-1, yielding a yellowish color (see Supplemental Movie D). Electrical stimulation of the superficial layer evoked reliable Ca2+ responses in labeled pyramidal cells, confirming that both neurons and Ca2+ dyes are functional (Figure 5B). The applicability of electroporation to the salt-form Ca2+ indicators makes a large variety of synthetic Ca2+ dyes (with different Kd values) now all usable for in vivo functional imaging. Especially, this extends the color spectrum range for multi-color Ca2+ imaging, which is highly desired for in vivo functional study of many GFP-targeted mouse models.
Figure 5.
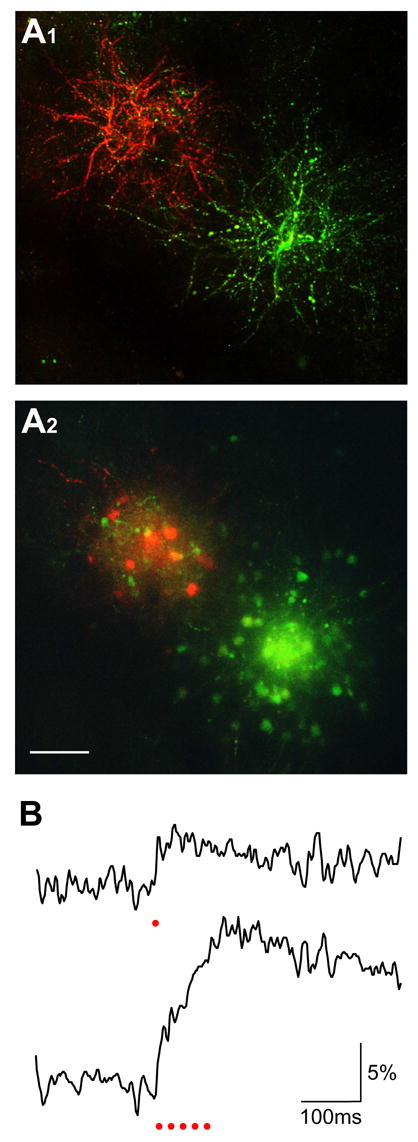
Electroporation using regular salt-form Ca2+ dyes. A, In vivo two-photon imaging of the barrel-cortex neurons labeled by two separate electroporations using 5% CG-1 hexapotasssium and 5% X-rhod-1 tripotassium, respectively. The loading parameters were both -3 μA current amplitude, 25 ms pulse duration and 1200 pulses delivered at 2 Hz. A1 shows the z-projection of labeled dendrites imaged at a depth range of 50 to 115 μm below the pia. A2 is the z-projection of images between 185 and 250 μm, showing the labeled cell bodies. Scale bar, 50 μm. B, Ca2+ responses of a neuron labeled with CG-1 hexapotassium salt. The two traces show the responses to one (upper) and five (lower) electric stimuli as indicated by red dots. For each trace, five imaging trials were averaged.
In vivo Ca2+ imaging of dendrites, spines and presynaptic boutons
Subcellular compartments such as dendrites, spines and axon terminals play crucial roles in signal processing by individual neurons. To date, loading Ca2+ dye with a sharp electrode or patch pipette is the only effective way to image dendritic functions in vivo at a high resolution (Debarbieux et al., 2003; Helmchen et al., 1999; Svoboda et al., 1997). However, the difficulties in obtaining and holding intracellular recording long enough for adequate dye diffusion have limited the use of this method. In this regard, Ca2+ dye electroporation presents a much easier and more efficient way to study the functions of these small local-circuit components.
To illustrate this point, we first carried out in vivo Ca2+ imaging in the cerebellum. Figure 6A1 shows a top view of dextran-labeled Purkinje cell dendrites, which was composed of several vertical rows of fluorescence spots. A side view in Figure 6A2 reveals that each row of spots was actually the cross section of an array of distal dendritic arbors that originated from a Purkinje cell soma. With a train of 50-Hz electric pulses delivered laterally to the parallel fibers, line-scan two-photon imaging revealed that Ca2+ responses in these dendritic arbors followed closely the consecutive stimuli in a step-wise fashion (Figure 6A3, a). Superimposing Ca2+ responses to 2, 4, 6 and 10 stimuli indicated a linear correlation of Ca2+ concentration with the number of delivered pulses (Figure 6A3, b). However, as the stimulus number further increased, the Ca2+ fluorescence signal started to become non-linear and eventually reached saturation (Supplemental Figure 1).
Figure 6.
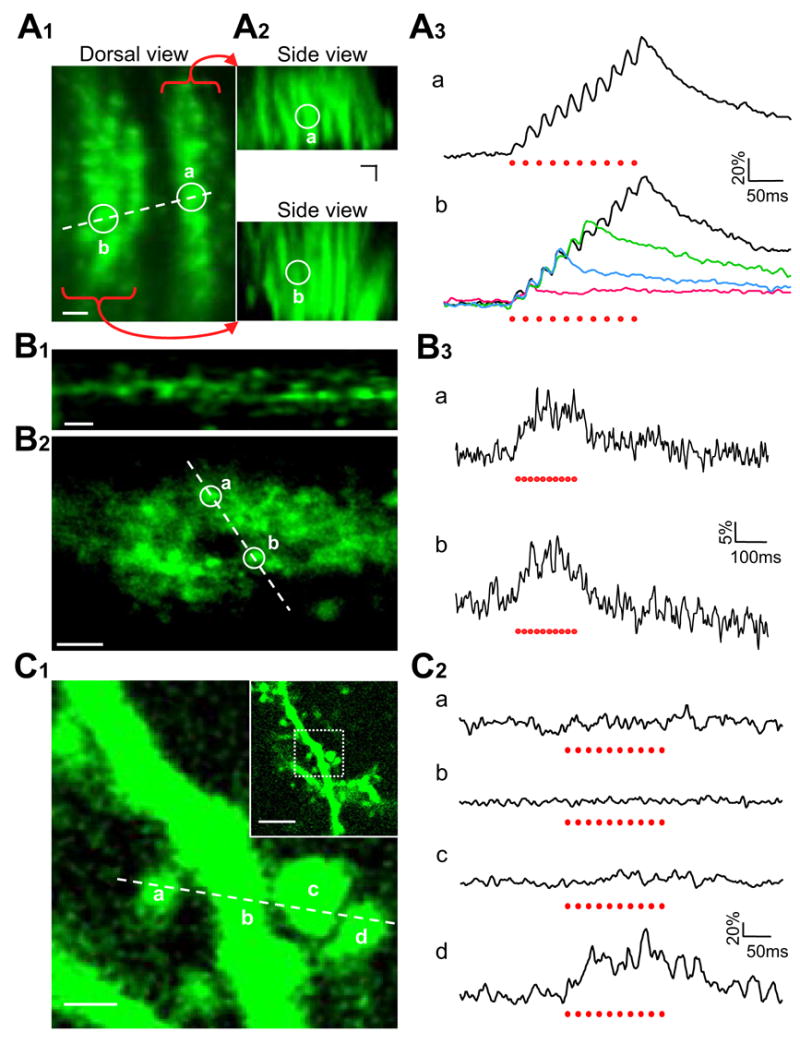
Local electric stimulation-evoked Ca2+ responses in different subcellular structures. A, Ca2+ response in Purkinje cell dendrites. A1, A top view of labeled distal dendritic arbors. The dashed line indicates the location where line-scan two-photon imaging was performed. A2, Upper and lower panels show the side views of two labeled Purkinje-cell distal dendritic arbor arrays. Their correspondence to the top-view image in A1 is indicated by two white circles (a, b), where Ca2+ responses shown in A3 were measured. A3, Upper trace is a dendritic Ca2+ response evoked at site a by ten electric pulses delivered laterally to unlabeled parallel fibers. Bottom traces are superimposed responses at site b to 2, 4, 6, and 10 stimuli. Ten trials were averaged for each trace. B, Imaging Ca2+ activity in vivo in parallel-fiber presynaptic boutons. B1, A representative fluorescence image showing presynaptic boutons along the parallel fibers. For better visualization see Supplemental Movie E. B2, Line-scan imaging was performed, with Ca2+ responses in boutons a and b shown in B3. B3, Responses of boutons a and b to ten electric pulses applied laterally to the labeled parallel fibers. Ten trials were averaged for each trace. C, In vivo Ca2+ imaging of pyramidal cell dendritic spines in the barrel cortex. C1, Two-photon images showing a pyramidal cell apical dendrite covered with many spines. The inset in the up-right corner is a low-magnification image, in which a white square box indicates the area that was zoomed in for Ca2+ imaging. Line-scan recording was made along a dashed line which covered three spines (a, c, d) and their parental dendrite (b). C2, Only one out of three spines showed a Ca2+ response to ten electric stimuli. No Ca2+ increase was observed in the parental dendritic shaft. Eight trials were averaged for each trace. Red dots below the traces in A3, B3 and C2 indicate individual electric stimuli. Scale bars represent 5 μm in A1, A2, B1, B2 and in the inset of C1, but 1 μm in the major image of C1.
As described above in Figure 3C1, dextran dye electroporation also labeled the cerebellar parallel fibers. High-magnification two-photon imaging was able to resolve individual presynaptic boutons along these parallel fibers. The boutons looked like chains of tiny fluorescence dots linked with much fainter thin threads (Figure 6B1, for better visualization see Supplemental Movie E). In vivo Ca2+ imaging of presynaptic boutons remains so far technically impossible due to brain pulsations released by craniotomy. The efficiency of Ca2+ dye loading achieved by local electroporation allowed us to drill a very tiny hole (~100 μm diameter) in the skull for inserting a single dye-loading pipette just for once. This ensured the level of stability required for imaging Ca2+ signals in individual axonal boutons. With line-scan two-photon imaging, presynaptic Ca2+ increase was recorded in labeled parallel fiber boutons in response to a train of ten electric stimuli delivered at 50 Hz (Figures 6B2 and 6B3). These results, together with Ca2+ imaging of Purkinje cell postsynaptic dendrite (Figure 6A), demonstrate the potential of imaging pre- and postsynaptic Ca2+ activity simultaneously for revealing functional connectivity in the brain.
Another important element in local neuronal circuits is dendritic spines, which are the major postsynaptic apparatus for receiving excitatory synaptic input as well as the key locus for synaptic plasticity associated with learning and memory. Though the function of spines has been extensively studied in vitro with brain slices and tissue cultures (Yuste and Denk, 1995), in vivo analysis has so far been restricted to imaging plastic changes in spine morphology or glutamate receptor insertion (Takahashi et al., 2003; Trachtenberg et al., 2002). With Ca2+ dye electroporation, it is now possible to study the physiological functions of spines in vivo. In the barrel cortex, numerous spines were readily identifiable on dextran-labeled pyramidal cell dendrites (Figure 6C1, inset). Using line-scan mode as indicated by a dashed line in Figure 6C1, three neighboring spines were imaged simultaneously with their parental dendritic shaft. In response to a train of electric shocks, only one spine displayed a Ca2+ increase while the other two spines and their dendritic shaft did not show any responses (Figure 6C2). These data may provide the first in vivo evidence supporting the well-established concept that dendritic spines function as independent biochemical compartments.
The experiments described so far involve imaging Ca2+ responses evoked by artificial electric stimulation. To elucidate how natural sensory signals are processed by a subcellular microcircuit, Ca2+ imaging was performed on dextran-dye labeled mitral cells in response to odor stimuli. We took advantage of the mitral cells located at the anterior edge of dorsal olfactory bulb, where their primary dendrites are horizontally oriented. Under such circumstances, the mitral cell soma, primary dendrite and major glomerular-tuft branches were located largely within the same imaging plane, which allowed simultaneous imaging of Ca2+ signals from these different subcellular structures (Figure 7A). A series of aliphatic aldehydes with different carbon-chain length were delivered to characterize the odor response profile in different mitral cell compartments. As shown in Figure 7B, pentanal (5CHO) activated the imaged mitral cell more strongly than other aldehydes. It evoked a Ca2+ increase not only in the cell body but also in the primary dendrite and glomerular tuft branches. Five out of six tested aldehydes, including butanal (4CHO), pentanal (5CHO), hexanal (6CHO), heptanal (7CHO) and octanal (8CHO), induced a clear Ca2+ response in the glomerular tuft. However, the mitral cell soma only showed a distinct response to pentanal. These in vivo imaging results very likely reflect the sharpening of odor-tuning curve from the glomerular layer down to the mitral cell soma layer. However, it remains to be resolved whether they could also be due to differences of dye concentration in the four subcellular compartments or to different amplitude of Ca2+ increase in relation with differences in the surface-to-volume ratio. Nevertheless, these experiments demonstrate the potential of using Ca2+ dye electroporation to study signal transmission and processing in vivo in subcellular neuronal microcircuits.
Figure 7.
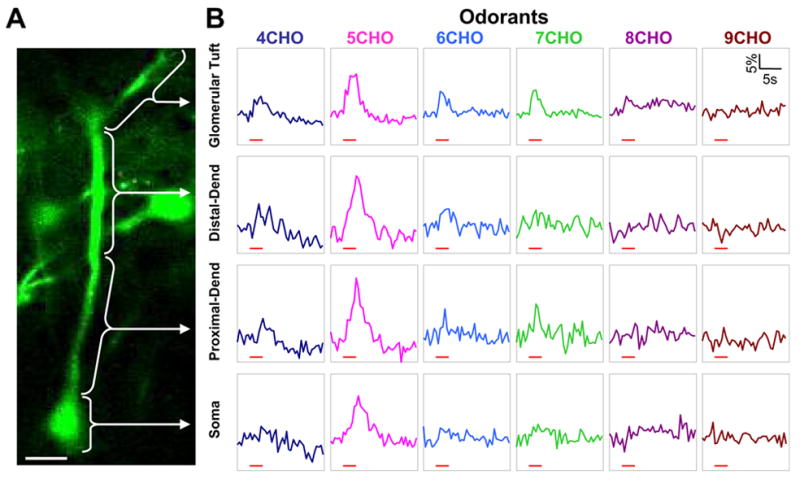
Imaging odor-evoked Ca2+ response along the mitral cell soma-primary dendrite axis. A, A labeled mitral cell with its soma, primary dendrite trunk and glomerular tuft branches visualized within the same imaging plane. Ca2+ responses were analyzed respectively in the glomerular tuft branches, the distal and proximal primary dendrite trunk, and the soma. Scale bar, 25 μm. B, Odor-evoked Ca2+ increase was imaged at 2 Hz in frame-scan mode and subsequently measured in the four subcellular compartments as indicated in A. Aliphatic aldehydes with different carbon-chain length were delivered to activate olfactory sensory input. In each vertical column are Ca2+ responses in the different compartments to the same aldehyde stimulus, and each row shows the responses to different aldehydes within a certain compartment. Red bars under each trace indicate the timing of odor delivery.
Imaging neuron ensemble response to natural sensory input
Our original goal in developing this method was to study neuronal ensemble activity in visualized local circuits. This goal was tested in the barrel cortex in response to whisker deflection as well as in the olfactory bulb to odor stimulation. In the barrel cortex, a single whisker deflection usually evokes a fast neuronal response that involves only one or two spike discharges (Brecht et al., 2003; Wilent and Contreras, 2004). To record such a fast response, two-photon Ca2+ imaging was carried out in line-scan mode. In Figure 8A1, two labeled pyramidal cell somas were imaged along a dashed line. Both neurons responded to an air puff-induced whisker deflection with a transient Ca2+ increase (Figure 8A2). The fast rising phase of Ca2+ signals may correspond to a single action potential as reported previously (Kerr et al., 2005; Svoboda et al., 1997).
Figure 8.
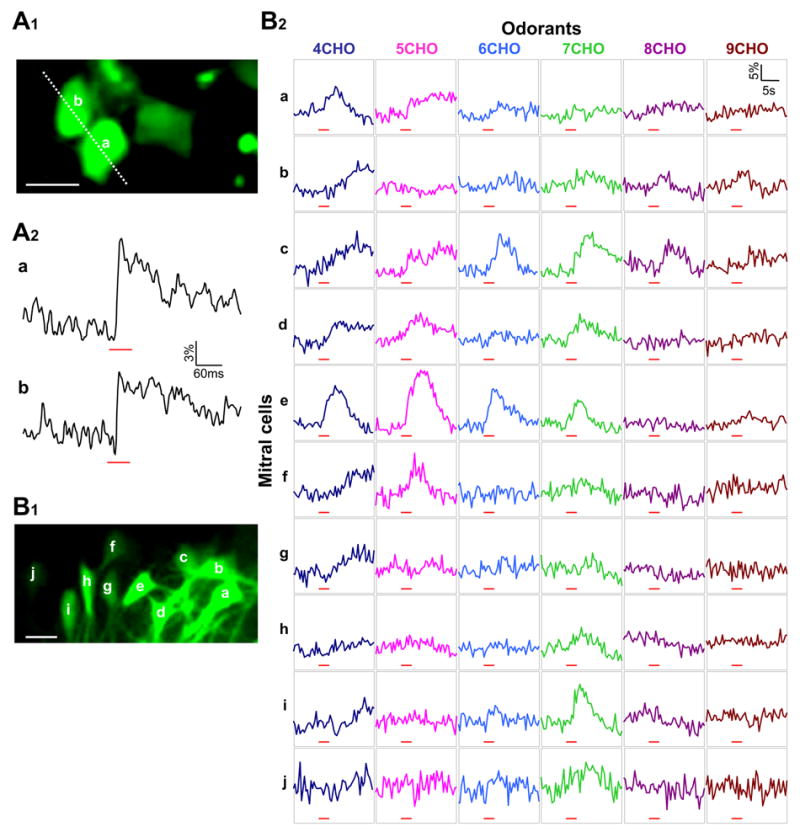
Ca2+ imaging of multiple neurons for monitoring cell ensemble activity in a natural sensory process. A, Two labeled neurons in the barrel cortex were line-scan imaged to reveal their Ca2+ activity in response to whisker stimulation. A1, An imaged area in the barrel cortex that was located 260 μm below the pia. Scale bar, 10 μm. A2, The two neurons indicated in A1 displayed a Ca2+ increase to whisker deflection. Red bars indicate the timing of an air puff. B, Imaging odor responses of 10 mitral cells in the olfactory bulb. B1, The relative locations of ten mitral cells within the same focus plane. The dendritic projection of these neurons is illustrated partially in a lower-magnification image shown in Figure 3A. Scale bar, 25 μm. B2, Simultaneous Ca2+ recording from the ten mitral cells designated as a to j in B1. Each column represents the responses of ten different mitral cells to a given aldehyde, and each row shows the responses of a certain mitral cell to the six different aldehydes. Red bars indicate the timing of odor delivery, and Ca2+ imaging was carried out in frame-scan mode at 2 Hz.
In the olfactory bulb, mitral cells usually respond to odor stimulation with a train of action potentials (Nagayama et al., 2004). Such a relatively long-lasting response has allowed us to use the frame-scan mode to carry out two-photon Ca2+ imaging. Figure 8B1 shows ten labeled mitral cells located close to each other, within a small area of ~130 × 80 μm. Given that the average diameter of mouse olfactory glomeruli is ~100 μm (Bozza et al., 2004), we speculated that these mitral cells should receive odor signals from one or very few common glomeruli and accordingly display similar odor tuning properties. To our surprise, these mitral cells exhibited quite diverse odor response profiles to a series of tested aldehydes (Figure 8B2). A careful tracing of their labeled primary dendrites revealed that despite close neighborhood all these cells (except h and i) had their primary dendrites projected into different glomeruli. Cells h and i shared a common glomerulus and displayed a similar odor response profile, although the specific response to heptanal (7CHO) was weaker in Cell h than in Cell i. These results highlight the importance of analyzing mitral cell ensemble codes in the light of their local network connectivity. In this perspective, one major benefit of this method is to present a practical and effective means to carry out Ca2+ imaging of neuron populations simultaneously with revealing their local functional circuits.
Discussion
Electroporation conditions and intracellular dye concentration
To optimize the electroporation parameters for effective dye loading, we have systematically varied the electric current amplitude, current pulse duration and dextran dye concentration (Figure 2A–C). Our results revealed a combination of 5–10% dye concentration, 3–5 μA current and 25 ms pulse duration as a good parameter set. However, it should be noted that our parameter search was carried out while fixing other conditions such as the tip size of loading pipette, current-pulse shape and delivery pattern, as well as the total number of pulses delivered. It is likely that changing these other conditions may lead to a different set of optimal parameters. For example, a loading pipette with a larger tip may require higher current amplitude in order to reach enough local current density for electroporating the membrane.
Using an in vitro neonatal spinal cord preparation, the O’Donovan’s group has previously reported the loading of Ca2+ dyes into neurons by electroporating the entire isolated spinal cord sandwiched with two metal electrode plates (Bonnot et al., 2005). Such an en bloc electroporation appears to be very effective, as only five to seven electric pulses of 50–75 ms duration were enough, in contrast to over hundreds of pulses delivered in our local electroporation procedure. Another major difference is that, for electroporation to take effect, Ca2+ dye was first distributed through blood vasculature and central canal to reach deep tissues in the spinal cord. It appears that such global electroporation is more suited to in vitro and in utero preparations. As for in vivo adult brain, it is difficult to position a pair of metal electrode plates in an electrically effective way. Besides, perfusing Ca2+ dye into deep and large volume of brain tissue, especially in vivo through blood stream, would consume a large amount of dye and significantly raise the experiment cost. To minimize the electroporation-induced depression of neural function, this in vitro study used a low Ca2+/high Mg2+ solution to perfuse the spinal cord for blocking electroporation-induced transmitter release and excitotoxicity. Similarly, for in vivo local electroporation, even though our results showed no sign of disrupting circuit function, a cocktail of glutamate receptor antagonists can be introduced before electroporation, either by bath-perfusing the exposed brain or being included in the dye-loading pipette.
It is also reported that dextran-bound Ca2+ dye can be successfully loaded into neurons of the frog olfactory bulb, just by inserting several glass pipettes that are dry-coated with Ca2+ dye dextran conjugate (Mulligan et al., 2001). It is not clear whether this is due to active dye uptake or to membrane damage caused mechanically by pipette penetration. As cell membrane in cold-blooded animals may be more fragile to mechanic disruption, it remains to be tested whether this method works only in frogs or has a wide applicability to mammalian species.
The concentration of electroporation-loaded Ca2+ dye is an important parameter that can affect both intracellular Ca2+ buffering capacity and time course of Ca2+ fluorescence signal. The optimized loading conditions in this study have yielded an estimated dye concentration of 21.0 ± 9.1 μM, which is surprisingly close to that reported for the AM-ester bulk loading method (Stosiek et al., 2003). Due to some complicating factors, this value is only a rough estimate. For example, photo-bleaching of the dextran-loaded neurons when identifying the electroporation area for patch recording can cause an under-estimate. In spite of this, the dye concentration achieved with local electroporation appears to be in a range that does not significantly perturb neuronal functions. Another issue of consideration is the uneven labeling of neuron populations or subcellular compartments. Such a variation in dye concentration poses a potential problem for interpreting the imaged data. To correct for differences in dye-loading concentration in different neurons or compartments, dextran-conjugated Ca2+ dye can be co-loaded with a Ca2+ non-responsive Alexa dextran conjugate, which has the same molecular weight but a separable fluorescence spectrum. In this way, the functional Ca2+ signal can be first normalized to the Alexa fluorescence intensity in each labeled cell, and then compared across different neurons.
Sparse vs. universal labeling of neuronal circuits
One major difference between Ca2+-dye local electroporation and AM-ester bulk loading is the number of neurons labeled. The membrane-permeant AM ester dye labels almost all cells within an area of 200–400 μm diameter (Stosiek et al., 2003; Ohki et al. 2005). On the other hand, the effective loading area for electroporation is restricted to less than a few tens of microns (Figure 4A); but the labeled neurons can be much more widely distributed (Figure 4B), owing to dye transport or diffusion along dendrites and axons passing through the small effective loading area. Compared with AM-ester dye labeling, such a different loading mechanism renders the distribution pattern of electroporation-labeled neurons dependent on distinct local-circuit architecture in different brain regions (Figure 4B). In the cerebellum, in order to compensate for the limited range of labeling, multi-site electroporation can be applied, and each site usually takes only 10–15 minutes.
The relatively sparse labeling of neurons actually contributes to a most important advantage of this method, i.e. to ensure clear visibility of fine neuronal processes. A recent work from Helmchen’s group demonstrates that although axon terminals cannot be resolved individually with AM-dye labeling, these thin neural processes are indeed loaded with Ca2+ dyes. The neuropil composed of axon terminals can generate a “field potential-like” optical signal, which they term optical encephalogram (Kerr et al., 2005). Accordingly, labeling too many cellular components can obscure the thin and densely packed neuronal processes. In addition, sparsely labeling a neural network also makes it practical to trace individual axons and dendrites for studying functional connectivity. Actually, since this study has demonstrated the suitability of a large variety of Ca2+ dyes for electroporation loading, our method can be combined with the AM-ester dye labeling for dual-color imaging to take the full advantages of both methods.
Neuron vs. glial cell labeling
Glial cells are another major cell population in the brain. Co-labeling neurons with glial cells may complicate the interpretation of imaged Ca2+ signals (Nimmerjahn et al., 2004). It is previously reported that astrocytes and blood-vessel pericytes are labeled a few days after pressure injection of dextran-conjugated Ca2+ dye into the brain (Hirase et al., 2004). The labeling of pericytes is also noted in a study using en bloc electroporation of an isolated neonatal spinal cord (Bonnot et al., 2005). To our surprise, glial cells were rarely labeled by local dextran dye electroporation. Only on one occasion did we notice a labeled astrocyte with its characteristic radiating processes. The exact reason for such a difference between our results and the other two studies is currently unclear. The paucity of glia labeling in local electroporation could be due to the fact that most glial cells do not have long extending processes, and thus only those glial cells located in the close vicinity of the electroporation site have a chance to be labeled. It could also be due to a short waiting time as compared with over a couple of days in the dextran pressure-injection study (Hirase et al., 2004). Nevertheless, as our electroporation loading method can reveal the elaborate dendritic structure characteristic of a neuron, glial cells even labeled can be unambiguously identified and excluded from data analysis.
A perspective of future developments
Dextran conjugated dyes exhibit several valuable properties including low toxicity and immunogenicity (Haugland, 2002). Compared with the AM-ester form of Ca2+ dye, they are biologically inert and remain stable in the cytosol for long periods of time (Beierlein et al., 2004). These features make dextran-bound Ca2+ indicators ideal for long-term imaging of experience-dependent network functional plasticity. Once dextran Ca2+ dye is loaded into the brain by electroporation, a chronic imaging window can be set up over the skull. After a few days of recovery from surgery, Ca2+ imaging can be performed with single-cell resolution on an awake or even behaving animal without anesthesia. This will open entirely new avenues for functional analysis of local circuits at a natural alert brain state, which carries numerous advantages over imaging an anesthetized animal brain. Given that local electroporation can also load DNA plasmids into neurons (Haas et al., 2001), it appears feasible to combine dextran-dye Ca2+ imaging and gene delivery to achieve local genetic manipulation of neuronal circuits for gaining insights into how a circuit performs its functions and how these functions are linked to intracellular biochemical molecular networks.
An exciting prospect for the application of this method is to study the functional connectivity in an anatomically defined neuronal circuit. As we know, the functioning of a neural network is not simply determined by its hardwired interconnections. Signal traffic within a network can change dynamically in response to variable inputs or under different brain states. This leads to the emergence of transient “soft-wiring”, which is not accessible with traditional anatomical tracing experiments. In this study we have been able to image Ca2+ signals in vivo in presynaptic boutons as well as postsynaptic dendrites and spines (Figure 6). By tracing dextran-labeled axons and dendrites to their synaptic contact site, it is possible to image pre- and postsynaptic Ca2+ activity simultaneously for identifying dynamical functional connections. The feasibility of this approach has recently been demonstrated in an in vitro study, in which synaptic transmission was directly imaged in neocortical slices with intracellular diffusion of Ca2+ dyes through dual-cell recordings (Kaiser et al., 2004). Undertaking similar experiments in vivo with Ca2+ dye electroporation will elucidate how the “soft-wiring” of an established local circuit evolves dynamically for the processing of different neural signals. However, to ultimately realize such a goal, it is essential to develop some new laser-scanning mechanisms to significantly increase both two-photon Ca2+ imaging speed and single-pixel dwell time for photon detection (Zeng et al., 2006).
Experimental Procedures
Animal surgery
All experiments were performed in accordance with the guidelines of the Yale Institutional Animal Care and Use Committee. A total of 44 mice (1.5–3 months old) were anesthetized using i.p. application of either urethane at a dose of 1.2 g/kg of body weight or Nembutal at 50 mg/kg. The animal was kept on a heating pad with circulating water set at 40°C. The skull was thinned with a dental drill, and a small hole of about 100 μm diameter was opened without removing the dura. In some experiments with a large craniotomy, 1.5% agarose dissolved in a Ringer solution was applied to reduce brain pulsation. The Ringer solution was composed of 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 0.01 mM EDTA, 10 mM HEPES and 10 mM glucose (pH 7.5).
In our experience, keeping the exposed brain in a good and healthy condition during craniotomy was the most important factor in determining the success of labeling. Drill-induced overheat or tissue damage, dirty brain surface or subdural bleeding could cause the clogging of a penetrating pipette, which severely reduced the quality and efficiency of dye loading. We also monitored closely blood circulation on the brain surface, as slow or stopped blood flow was a major reason for poor and blurred image as well as high fluorescence background.
Dye loading by local electroporation and pressure ejection
For dye loading, a glass pipette was used with a tip of 2–4 μm inner diameter. Five to ten percent (mass/volume) of dextran-conjugated Oregon Green 488 BAPTA-1 (OGB-1) (10,000 m.w.), dextran-conjugated Calcium Green-1 (CG-1) (3,000 or 10,000 m.w.), OGB-1 hexapotassium, CG-1 hexapotassium or X-rhod-1 tripotassium salt was dissolved in Ringer solution for filling dye-loading pipettes. All the Ca2+ dyes, either in salt form or dextran-conjugated, were purchased from Molecular Probes. The Kd value of dextran-conjugated Ca2+ dyes varies across different production lots, and was between 454 and 700 nM in this study. For the CG-1 dextrans, we did not notice any significant difference in labeling between 3,000 and 10,000 molecular weight. The pipette was inserted into the brain with a micromanipulator. The precise location of the barrel cortex for inserting a dye-loading pipette was determined by recording evoked field potentials in response to whisker deflection.
Local electroporation was generally carried out by applying positive current pulses to the dextran-dye filled glass pipette and negative current to the salt-dye filled pipette. However, we noticed that current pulses of either polarity were able to label neurons, especially when the salt-form Ca2+ dye was used (see the caption of Supplemental Movie D). This is likely due to the relatively large pipette tip (2–4 μm inner diameter), which may make these small dye molecules easily diffuse out of the pipette (e.g. in the time window between current pulse application). Once leaking into extracellular space, the dye is loaded into neurons by membrane electroporation, in which positive and negative current should have an equal effect, at least on those thin processes located close to the pipette tip. The current pulse amplitude was usually 1–5 μA, except for optimizing electroporation conditions. The pulse width was 200 ms in some initial experiments, but was later shortened to 25 ms to reduce fluorescence background at the electroporation site. Current pulses were generally delivered at 5 Hz for 2–4 min in the cerebellum or at 2 Hz for 10 min in the olfactory bulb and barrel cortex. The dye-loading parameters for each figure are provided either in figure legends or Supplemental Dataset. The current delivery circuit consisted of an electric stimulator connected to an isolator that was set at current-output mode (Master-8 and Iso-Flex, A.M.P.I) (Figure 1A). The actual waveform and amplitude of injected current were monitored on an oscilloscope by measuring the voltage drop across a 100-KΩ series resistor.
At this stage it was important to ensure that Ca2+ dye was indeed flowing out of the pipette into brain tissue. When using a small pipette tip and passing either large or long current pulses, the loading pipette could easily get clogged during electroporation. Therefore, the waveform and amplitude of current injection were monitored so that a clogging condition could be immediately noted. For a clogged pipette, temporarily reversing the current polarity sometimes helped eliminate the problem; if not, the pipette was replaced.
For dye pressure injection, a pipette with a tip of ~2.5 μm inner diameter was filled with a dextran dye solution, and ejection was made with a 5-sec pulse of 6 psi air pressure applied from a picospritzer to the back of the pipette.
Estimating intracellular Ca2+ dye concentration
To estimate the intracellular dye concentration loaded with dextran electroporation, we first carried out in vivo loading in the barrel cortex with 10% dextran conjugated CG-1, and then sacrificed the animal to make slice preparations. The location of the dextran-loaded area was identified in slices by fluorescence observation made briefly to minimize photo-bleaching. Thereafter, with infrared DIC video microscopy, an unlabeled neuron within or close to the loaded area was whole-cell recorded with a patch pipette filled with a solution (pH 7.3–7.4) of 130 mM K-gluconate, 10 mM HEPES, 10 mM Na-phosphocreatine, 0.2 mM EGTA, 4 mM Mg-ATP, 0.3 mM Na3-GTP and 25 μM CG-1 hexapotassium salt. Initially, we tested several different pipette concentrations from 5 to 200 μM, and found that 25 μM CG-1 matched closely the fluorescence intensity of dextran-labeled neurons. After comparing the brightness of the patched neuron with that of the neighboring neurons labeled with CG-1 dextran dye, the intracellular dye concentration of each dextran-labeled neuron was calculated assuming a linear relation between dye concentration and fluorescence intensity. In our calibration experiment, the assumption of linearity held roughly over a tested concentration range of 5 to 90 μM.
In vivo electrophysiology and two-photon imaging
To assess whether electroporation compromised the function of neuronal circuits, local field potential was monitored before and after dextran-dye loading in the olfactory bulb and barrel cortex. The same glass pipette was used for both field-potential recording and dextran-dye electroporation, via switching the connection from an electric isolator to an amplifier headstage. This ensured that recorded field potential was able to reflect damages in the area to which electroporation was directly applied. Field potential in the olfactory bulb was induced by stimulating the LOT with a bipolar concentric electrode. Field potential in the barrel cortex was evoked by whisker deflection as described below.
In vivo imaging usually started at two or three hours after dye electroporation, using a custom-built two-photon system based on a mode-locked laser operating at 100-fs pulse width, 80-MHz pulse frequency and 810~830-nm wavelength (Tsunami and Millennia Xs, Spectra Physics) and an Olympus Fluoview scan box mounted on an upright BX50WI microscope. The laser power delivered to the brain was less than 120 mW. Ca2+ imaging were performed either in line-scan mode at a fastest rate of 2.02 ms/line or in frame-scan mode at two frames per second. Image analysis was carried out with Olympus Fluoview and Image-J software. Ca2+ response was presented as ΔF/Fo = (F−Fo)/Fo, where Fo is the average of baseline fluorescence before stimulation.
Whisker deflection, odor delivery and local electric stimulation
For whisker stimulation, the majority of whiskers on the contralateral side of the snout were deflected in caudal-to-rostral direction by a single air puff (50 ms, 80 psi) from 1-mm tubing connected to a picospritzer and nitrogen cylinder. For odor stimulation, a homologous series of aliphatic aldehydes was delivered in three-second pulses to the mouse nostrils using a custom-made olfactometer, either at a saturated vapor concentration or with different dilutions down to 1%. For local electric stimulation in the cerebellum and barrel cortex, a glass pipette (4–8 μm) filled with Ringer solution was inserted into a targeted area. Electric pulses were delivered with a width of 1 ms and amplitude of up to 200–300 μA.
Supplementary Material
Labeling of the mouse olfactory bulb with electroporation in the deep external plexiform layer using OGB-1 dextran conjugate (10,000 m.w.) (see the legend of Figure 3A for the dye loading parameters). The video shows up-and-down focusing of two-photon images from the superficial olfactory-glomerulus layer to the deep mitral cell soma layer. Many mitral cells were labeled, and their dendrites and glomerular tufts could be readily traced. A dense fluorescence spot on the right side reflects a specific olfactory glomerulus in which olfactory nerve terminals were genetically labeled with GFP.
A 3-D visualization of neuron labeling in the barrel cortex obtained by a single electroporation with CG-1 dextran conjugate (10,000 m.w.) (see the legend of Figure 3B for the dye loading conditions). Note that the full extension of neuronal dendrites was clearly visualized.
Labeling of the cerebellar Purkinje cells with CG-1 dextran conjugate (3,000 m.w.) (see Figure 3C1–3 legend for the dye loading parameters). Local dye electroporation was made in the superficial molecular layer at two different locations, producing two cross-like fluorescence patterns. Purkinje cell dendrites were visualized as parallel vertical fluorescent stripes, while two horizontal bundles were labeled parallel fibers. Two-photon imaging was gradually focused from the cerebellar surface deep into the Purkinje cell soma layer. The multiple flashes of fluorescence intensity were due to adjusting the laser power as the imaging depth gradually increased.
In vivo two-photon imaging of the barrel cortex, in which neurons were labeled via two separate electroporations: one with 5% OGB-1 hexapotassium salt (−3 μA current, 50 ms pulse duration, 600 pulses at 2 Hz) and the other with 5% X-rhod-1 tripotassium salt (+3 μA current, 50 ms pulse duration, 600 pulses at 2 Hz). Note that at least three neurons were double labeled, showing yellowish cell bodies; and that unlabeled neuronal somas were identifiable in deep imaging planes as many dark holes in fluorescence background due to the remnants of extracellular dyes.
Up-and-down focusing of a bundle of cerebellar parallel fibers labeled with CG-1 dextran conjugate (10,000 m.w.). Note that individual presynaptic boutons were linked with much fainter fluorescence threads of axons. The loading parameters were 3 μA current, 100 ms pulse duration and 1,800 pulses delivered at 5 Hz.
Acknowledgments
We thank Dr. Gordon Shepherd for critical reading of the manuscript, Dr. Minmin Luo for valuable technical advice, and three anonymous referees for their constructive comments. The work was supported by NIH grants (DC003918 & MH064214), the US-Japan Brain Research Collaborative Program and the National Science Foundation of China (#30328014 & #30370463). S.N. is a Patterson Trust Postdoctoral Fellow. A.V.M. is under the NIH Medical Scientist Training Program. M.L.F. is on an NIH training grant (NS007224).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beierlein M, Gee KR, Martin VV, Regehr WG. Presynaptic calcium measurements at physiological temperatures using a new class of dextran-conjugated indicators. J Neurophysiol. 2004;92:591–599. doi: 10.1152/jn.00057.2004. [DOI] [PubMed] [Google Scholar]
- Bonnot A, Mentis GZ, Skoch J, O’Donovan MJ. Electroporation loading of calcium-sensitive dyes into the CNS. J Neurophysiol. 2005;93:1793–1808. doi: 10.1152/jn.00923.2004. [DOI] [PubMed] [Google Scholar]
- Bozza T, McGann JP, Mombaerts P, Wachowiak M. In vivo imaging of neuronal activity by targeted expression of a genetically encoded probe in the mouse. Neuron. 2004;42:9–21. doi: 10.1016/s0896-6273(04)00144-8. [DOI] [PubMed] [Google Scholar]
- Brecht M, Roth A, Sakmann B. Dynamic receptive fields of reconstructed pyramidal cells in layers 3 and 2 of rat somatosensory barrel cortex. J Physiol. 2003;553:243–265. doi: 10.1113/jphysiol.2003.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpak S, Mertz J, Beaurepaire E, Moreaux L, Delaney K. Odor-evoked calcium signals in dendrites of rat mitral cells. Proc Natl Acad Sci U S A. 2001;98:1230–1234. doi: 10.1073/pnas.021422798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarbieux F, Audinat E, Charpak S. Action potential propagation in dendrites of rat mitral cells in vivo. J Neurosci. 2003;23:5553–5560. doi: 10.1523/JNEUROSCI.23-13-05553.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich RW, Korsching SI. Combinatorial and chemotopic odorant coding in the zebrafish olfactory bulb visualized by optical imaging. Neuron. 1997;18:737–752. doi: 10.1016/s0896-6273(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Milos RI, Grienberger C, Marandi N, Adelsberger H, Konnerth A. Eur J Physiol. 2006;453:385–396. doi: 10.1007/s00424-006-0150-x. [DOI] [PubMed] [Google Scholar]
- Haas K, Sin WC, Javaherian A, Li Z, Cline HT. Single-cell electroporation for gene transfer in vivo. Neuron. 2001;29:583–591. doi: 10.1016/s0896-6273(01)00235-5. [DOI] [PubMed] [Google Scholar]
- Haugland RP. Handbook of Fluorescent Probes and Research Products. 9. Eugene, OR: Molecular Probes, Inc; 2002. [Google Scholar]
- Heim N, Garaschuk O, Friedrich MW, Mank M, Milos RI, Kovalchuk Y, Konnerth A, Griesbeck O. Improved calcium imaging in transgenic mice expressing a troponin C-based biosensor. Nat Meth. 2007;2:127–129. doi: 10.1038/nmeth1009. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Svoboda K, Denk W, Tank DW. In vivo dendritic calcium dynamics in deep-layer cortical pyramidal neurons. Nat Neurosci. 1999;2:989–996. doi: 10.1038/14788. [DOI] [PubMed] [Google Scholar]
- Hirase H, Creso J, Singleton M, Bartho P, Buzsaki G. Two-photon imaging of brain pericytes in vivo using dextran-conjugated dyes. Glia. 2004;46:95–100. doi: 10.1002/glia.10295. [DOI] [PubMed] [Google Scholar]
- Kaiser KM, Lubke J, Zilberter Y, Sakmann B. Postsynaptic calcium influx at single synaptic contacts between pyramidal neurons and bitufted interneurons in layer 2/3 of rat neocortex is enhanced by backpropagating action potentials. J Neurosci. 2004;24:1319–1329. doi: 10.1523/JNEUROSCI.2852-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JN, Greenberg D, Helmchen F. Imaging input and output of neocortical networks in vivo. Proc Natl Acad Sci U S A. 2005;102:14063–14068. doi: 10.1073/pnas.0506029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Gee KR, Archer EA, Regehr WG. Monitoring presynaptic calcium dynamics in projection fibers by in vivo loading of a novel calcium indicator. Neuron. 2000;27:25–32. doi: 10.1016/s0896-6273(00)00006-4. [DOI] [PubMed] [Google Scholar]
- Lodovichi C, Belluscio L, Katz LC. Functional topography of connections linking mirror-symmetric maps in the mouse olfactory bulb. Neuron. 2003;38:265–276. doi: 10.1016/s0896-6273(03)00194-6. [DOI] [PubMed] [Google Scholar]
- Mori K, Kishi K, Ojima H. Distribution of dendrites of mitral, displaced mitral, tufted, and granule cells in the rabbit olfactory bulb. J Comp Neurol. 1983;219:339–355. doi: 10.1002/cne.902190308. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, Davison I, Delaney KR. Mitral cell presynaptic Ca2+ influx and synaptic transmission in frog amygdala. Neuroscience. 2001;104:137–151. doi: 10.1016/s0306-4522(01)00057-4. [DOI] [PubMed] [Google Scholar]
- Nagayama S, Takahashi YK, Yoshihara Y, Mori K. Mitral and tufted cells differ in the decoding manner of odor maps in the rat olfactory bulb. J Neurophysiol. 2004;91:2532–2540. doi: 10.1152/jn.01266.2003. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- O’Donovan MJ, Ho S, Sholomenko G, Yee W. Real-time imaging of neurons retrogradely and anterogradely labelled with calcium-sensitive dyes. J Neurosci Methods. 1993;46:91–106. doi: 10.1016/0165-0270(93)90145-h. [DOI] [PubMed] [Google Scholar]
- Ohki K, Chung S, Ch’ng YH, Kara P, Reid RC. Functional imaging with cellular resolution reveals precise micro-architecture in visual cortex. Nature. 2005;433:597–603. doi: 10.1038/nature03274. [DOI] [PubMed] [Google Scholar]
- Ohki K, Chung S, Kara P, Hubener M, Bonhoeffer T, Reid RC. Highly ordered arrangement of single neurons in orientation pinwheels. Nature. 2006 doi: 10.1038/nature05019. [DOI] [PubMed] [Google Scholar]
- Pinault D. A novel single-cell staining procedure performed in vivo under electrophysiological control: morpho-functional features of juxtacellularly labeled thalamic cells and other central neurons with biocytin or Neurobiotin. J Neurosci Meth. 1996;65:113–136. doi: 10.1016/0165-0270(95)00144-1. [DOI] [PubMed] [Google Scholar]
- Rall W, Shepherd GM. Theoretical reconstruction of field potentials and dendrodendritic synaptic interactions in olfactory bulb. J Neurophysiol. 1968;31:884–915. doi: 10.1152/jn.1968.31.6.884. [DOI] [PubMed] [Google Scholar]
- Rubin BD, Katz LC. Optical imaging of odorant representations in the mammalian olfactory bulb. Neuron. 1999;23:499–511. doi: 10.1016/s0896-6273(00)80803-x. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Chen WR, Greer CA. Olfactory bulb. In: Shepherd GM, editor. The Synaptic Organization of the Brain. Oxford University Press, Inc; 2004. pp. 165–216. [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Nimmerjahn A, Sarkisov DV, Helmchen F, Wang SS. In vivo calcium imaging of circuit activity in cerebellar cortex. J Neurophysiol. 2005;94:1636–1644. doi: 10.1152/jn.01013.2004. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 1997;385:161–165. doi: 10.1038/385161a0. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neurosci. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Svoboda K, Malinow R. Experience strengthening transmission by driving AMPA receptors into synapses. Science. 2003;299:1585–1588. doi: 10.1126/science.1079886. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Uchida N, Takahashi YK, Tanifuji M, Mori K. Odor maps in the mammalian olfactory bulb: domain organization and odorant structural features. Nat Neurosci. 2000;3:1035–1043. doi: 10.1038/79857. [DOI] [PubMed] [Google Scholar]
- Vercelli A, Repici M, Garbossa D, Grimaldi A. Recent techniques for tracing pathways in the central nervous system of developing and adult mammals. Brain Res Bull. 2000;51:11–28. doi: 10.1016/s0361-9230(99)00229-4. [DOI] [PubMed] [Google Scholar]
- Wachowiak M, Cohen LB. Representation of odorants by receptor neuron input to the mouse olfactory bulb. Neuron. 2001;32:723–735. doi: 10.1016/s0896-6273(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Wilent WB, Contreras D. Synaptic responses to whisker deflections in rat barrel cortex as a function of cortical layer and stimulus intensity. J Neurosci. 2004;24:3985–3998. doi: 10.1523/JNEUROSCI.5782-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Chen WR. Dynamic gating of spike propagation in the mitral cell lateral dendrites. Neuron. 2002;34:115–126. doi: 10.1016/s0896-6273(02)00628-1. [DOI] [PubMed] [Google Scholar]
- Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- Zeng S, Lv X, Zhan C, Chen WR, Xiong W, Jacques SL, Luo Q. Simultaneous compensation for spatial and temporal dispersion of acousto-optical deflectors for two-dimensional scanning with a single prism. Opt Lett. 2006;31:1091–1093. doi: 10.1364/ol.31.001091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Labeling of the mouse olfactory bulb with electroporation in the deep external plexiform layer using OGB-1 dextran conjugate (10,000 m.w.) (see the legend of Figure 3A for the dye loading parameters). The video shows up-and-down focusing of two-photon images from the superficial olfactory-glomerulus layer to the deep mitral cell soma layer. Many mitral cells were labeled, and their dendrites and glomerular tufts could be readily traced. A dense fluorescence spot on the right side reflects a specific olfactory glomerulus in which olfactory nerve terminals were genetically labeled with GFP.
A 3-D visualization of neuron labeling in the barrel cortex obtained by a single electroporation with CG-1 dextran conjugate (10,000 m.w.) (see the legend of Figure 3B for the dye loading conditions). Note that the full extension of neuronal dendrites was clearly visualized.
Labeling of the cerebellar Purkinje cells with CG-1 dextran conjugate (3,000 m.w.) (see Figure 3C1–3 legend for the dye loading parameters). Local dye electroporation was made in the superficial molecular layer at two different locations, producing two cross-like fluorescence patterns. Purkinje cell dendrites were visualized as parallel vertical fluorescent stripes, while two horizontal bundles were labeled parallel fibers. Two-photon imaging was gradually focused from the cerebellar surface deep into the Purkinje cell soma layer. The multiple flashes of fluorescence intensity were due to adjusting the laser power as the imaging depth gradually increased.
In vivo two-photon imaging of the barrel cortex, in which neurons were labeled via two separate electroporations: one with 5% OGB-1 hexapotassium salt (−3 μA current, 50 ms pulse duration, 600 pulses at 2 Hz) and the other with 5% X-rhod-1 tripotassium salt (+3 μA current, 50 ms pulse duration, 600 pulses at 2 Hz). Note that at least three neurons were double labeled, showing yellowish cell bodies; and that unlabeled neuronal somas were identifiable in deep imaging planes as many dark holes in fluorescence background due to the remnants of extracellular dyes.
Up-and-down focusing of a bundle of cerebellar parallel fibers labeled with CG-1 dextran conjugate (10,000 m.w.). Note that individual presynaptic boutons were linked with much fainter fluorescence threads of axons. The loading parameters were 3 μA current, 100 ms pulse duration and 1,800 pulses delivered at 5 Hz.