

Abstract
Interest in oral fluid as an alternative matrix for monitoring drug use is due to its ease-of-collection and non-invasiveness; however, limited data are available on the disposition of drugs into oral fluid. The objective of this research was to provide data on the presence and concentrations of heroin, cocaine and multiple metabolites in oral fluid after illicit opioid and cocaine use. Thrice weekly oral fluid specimens (N=403) from 16 pregnant opiate-dependent women were obtained with the Salivette® oral fluid collection device. Evidence of heroin (N=62) and cocaine (N=130) use was detected in oral fluid by LC-APCI-MS/MS. 6-Acetylmorphine (6-AM), heroin and morphine were the major opiates detected, with median concentrations of 5.2, 2.3, and 7.5 μg/L, respectively. Cocaine and benzoylecgonine (BE) had median concentrations of 6.4 and 3.4 μg/L. Application of the Substance Abuse Mental Health Services Administration (SAMHSA) recommended cutoffs for morphine and codeine (40 μg/L), 6-AM (4 μg/L) and cocaine and BE (8 μg/L), yielded 28 opiate- and 50 cocaine-positive specimens. Oral fluid is a promising alternative matrix to monitor opiate and cocaine use in drug testing programs. These data guide interpretation of oral fluid test results and evaluate currently proposed SAMHSA oral fluid testing cutoffs.
Keywords: oral fluid, cocaine, opiates, SAMHSA, recovery, LCMS
1. Introduction
Monitoring illicit drug use in drug treatment programs is traditionally performed by analysis of urine (Cone, 2001; Cone and Preston, 2002). In recent years, interest in other biological matrices as alternative drug-testing tools has grown, with oral fluid as one of the most promising new matrices (Choo and Huestis, 2004; Kidwell et al., 1998; Rivier, 2000; Schramm et al., 1992). This colorless, highly aqueous liquid is a natural ultrafiltrate of plasma (Haeckel and Hanecke, 1996). Substances are transported across epithelial membranes into oral fluid by passive diffusion across a concentration gradient. Drug transport into oral fluid is regulated by the physiochemical properties of the drug (molecular weight, dissociation constants, lipid solubility, and protein binding) and the membrane. Based on a modified version of the Henderson-Hasselbach equation, it is generally presumed that unbound weakly basic drugs will concentrate in oral fluid, while the opposite occurs for weakly acidic drugs (Pichini et al., 1996). Although less common, low-molecular mass compounds can also be transferred into oral fluid by active secretion or diffusion through pores in the membrane (Haeckel and Hanecke, 1996).
As an alternative matrix for drug testing, oral fluid offers some distinct advantages (Cone, 1993; Cone, 2001). The matrix is relatively clean and readily accessible for sampling. Collection is easy, non-invasive, and inexpensive. Sampling can be better supervised without invasion of privacy, reducing the opportunity for sample adulteration or substitution. Oral fluid testing also offers the possibility of direct comparison of unbound, pharmacologically active drug concentrations to pharmacodynamic effects. Disadvantages include variability of salivary flow and pH, and a shorter detection window than urine for active drugs. In addition, specimen collection can have a serious impact on analytical findings and smaller volumes of oral fluid are generally collected (Kato et al., 1993; O’Neal et al., 1999). Furthermore, there are few reports of drug concentration data to guide interpretation of oral fluid test results (Cooper et al., 2005). Finally, oral fluid drug concentrations often are lower than concentrations in traditional biofluids requiring new and sensitive analytical methodology (Rivier, 2000).
A number of illicit drugs have been detected in oral fluid including cocaine, amphetamine and derivatives, opioids, phencyclidine (PCP) and cannabinoids (Clauwaert et al., 2004; Cone, 1993; Cooper et al., ; Drummer, 2005; Idowu and Caddy, 1982; Schramm et al., 1992). Controlled drug administration studies have investigated the presence and pharmacokinetic characteristics of a number of these drugs in oral fluid (Cone, 1993; Cone et al., 1997; Jenkins et al., 1995; Jufer et al., 2000; Kacinko et al., 2004; Kato et al., 1993; Kim et al., 2002; Kintz et al., 1998; Kopecky et al., 1997; Moolchan et al., 2000; Navarro et al., 2001; O’Neal et al., 1999; Schepers et al., 2003; Skopp et al., 2001; Wang et al., 1994) Furthermore, oral fluid has been successfully applied in criminal justice programs (Yacoubian et al., 2001), workplace (Cone, 2001) and roadside drug testing (DUI) (Peel et al., 1984; Samyn and van Haeren, 2000), but little is known of the usefulness of oral fluid drug monitoring in a population of substance-abuse treatment patients. Schramm et al. assessed oral fluid as an alternative matrix for drug testing to serum and urine in volunteers attending a substance abuse treatment program, with self-reported cocaine-use within the previous 24h (Schramm et al., 1993). Oral fluid specimens collected with a special osmotic device were analyzed for cocaine and benzoylecgonine (BE) by radioimmunoassay (RIA) and GC-MS. Despite cross reactivity of the cocaine RIA kit for BE, the authors concluded that simultaneous measurement of cocaine and BE in oral fluid was suitable for screening recent cocaine use. Cooper et al. measured dihydrocodeine, codeine, morphine, 6-acetylmorphine (6-AM), cocaine and BE by GC-MS in oral fluid of donors being monitored in a drug misuse treatment program as part of the investigators’ effort to evaluate the Cozart® RapiScan screening device (Cooper et al., 2005).
Smith and Kidwell evaluated cocaine and BE concentrations in hair, oral fluid, skin secretion (mixture sebum and sweat), and urine of urban cocaine dependent treatment patients and their children (Smith and Kidwell, 1996). A SalivaSac™ pouch from BioQuant Inc. was utilized for oral fluid collection with subsequent analysis by RIA. Although most hair and skin secretion specimens contained measurable quantities of cocaine and BE, only two oral fluid specimens were positive (90 and 240 μg BE equivalents/L). Multiple patients, especially children, did not provide oral fluid specimens. It was concluded that cocaine use persisted during addiction treatment and that children of treatment seekers could test positive for cocaine in their hair and skin secretions due to environmental exposure. Finally, an assessment of the concordance of oral fluid and urine opiate results for participants in drug withdrawal therapy was made by Speckl et al (Speckl et al., 1999). Oral fluid specimens were collected with a Clin Rep® device from Recipe and analyzed by GC-MS for dihydrocodeine, codeine, 6-AM, and morphine. The authors found a good correlation between opiate results in both matrices, in specimens collected every 2–4 days over a 3-month period. They concluded that oral fluid was appropriate for analysis of drugs of abuse.
The objectives of this research were to provide data on the presence and concentrations of heroin, cocaine and metabolites in oral fluid after illicit opioid and cocaine use to guide interpretation of oral fluid test results, and to evaluate the proposed Substance Abuse Mental Health Services Administration (SAMHSA) oral fluid cutoffs (SAMHSA, 2004). Oral fluid was used to monitor opiate and cocaine use in patients enrolled in methadone maintenance treatment. Recovery of analytes from the Salivette® with neutral cotton oral fluid collection device was investigated. Identities and concentrations of opiates, cocaine, and metabolites in oral fluid after illicit drug use were determined by LC-APCI-MS/MS, preceded by protein precipitation.
2. Methods
2.1. Human participants
Sixteen pregnant heroin and/or cocaine addicts from the Center for Addiction and Pregnancy (CAP) at the Johns Hopkins Bayview Medical Center (JHBMC) participated in the study. Twelve African American and 4 Caucasian first-time drug treatment seekers had a mean + SD age of 29.5 ± 6.7 years (range 19 to 40 years) and were between 8 and 28 weeks of gestation. The women met DSM-IV diagnostic criteria for opiate and/or cocaine dependence and resided for several weeks at CAP for the initiation of methadone treatment and again during the postpartum period. Throughout gestation, participants received outpatient methadone-maintenance drug-abuse treatment, which included daily methadone (mean dose 75 ± 17 mg/day; range 45 – 110 mg/day), weekly individual and group counseling and specialized prenatal care. The protocol was approved by the JHBMC and the National Institute on Drug Abuse Institutional Review Boards. Participants provided written informed consent and received vouchers as part of a behavioral contingency treatment program.
2.2. Specimen collection
Volunteers participated in the study throughout gestation (mean number of weeks on study 17.0 ± 5.8; range 8.2 – 27.2 weeks) and visited the clinic seven days per week to receive methadone. A variety of biological specimens, including urine, oral fluid, sweat and hair, were collected at fixed times during the study, under direct observation by trained staff. Oral fluid specimens were collected three times per week (Monday, Wednesday, Friday) with the Salivette® device with neutral cotton from Sarstedt. As recommended by the manufacturer, the cotton was chewed for 1 min and placed in a sealed polypropylene container. Specimens were centrifuged on-site to release oral fluid from the cotton (5 min at 1000 × g) and frozen at −20°C until analysis. The mean number of oral fluid specimens collected and analyzed per subject was 27.8 ± 9.6 with a range of 9.0 – 49.0. The number of specimens collected was dependent on the enrollment period.
2.3. Chemicals and reagents
Normorphine, morphine, morphine-d6, codeine, 6-AM, 6-acetylmorphine-d6, heroin, heroin-d9, acetylcodeine, ecgonine methyl ester (EME), ecgonine methyl ester-d3, ecgonine ethyl ester (EEE), anhydroecgonine methyl ester (AEME), m-hydroxybenzoylecgonine, BE, benzoylecgonine-d8, norcocaine, norcocaine-d3, cocaine, norcocaethylene, cocaethylene, and cocaethylene-d3 were purchased from Cerilliant™ (Round Rock, Texas, USA). Papaverine, noscapine, ecgonine, norcodeine, m- and p-hydroxycocaine, benzoylnorecgonine, and p-hydroxybenzoylecgonine were obtained from Sigma Chemical Co. (St. Louis, Missouri, USA). Cocaine-d8 was purchased from High Standard Products Corporation (Westminster, California, USA), and acetylcodeine-d3 from Lipomed (Cambridge, Massachusetts, USA). Reagent grade ammonium formate and formic acid were purchased from Sigma Chemical Co. All solvents were of HPLC grade or higher.
2.4. Analytical procedures
Calibrators and clinical specimens were analyzed for opioids, cocaine, and metabolites by acetonitrile protein precipitation and LC-APCI-MS/MS according to a previously published procedure (Dams et al., 2003b). Oral fluid was collected from healthy volunteers with the Salivette collection device as described in the Specimen Collection section, and verified to be drug free (below the limit of detection (LOD) of the method) before used in the preparation of calibrators. In addition, matrix effects were evaluated and determined not to affect quantification (Dams et al., 2003a). Matrix matched calibrators were prepared at drug concentrations from 1 to 500 μg/L. Full calibration curves were run in the beginning and the end of each daily batch and averaged for analyte quantification.
Briefly, 10 μL of internal standard working solution (2-mg/L solution) was added to 200 μL of oral fluid and vortexed. Protein was precipitated and drugs extracted with 600 μL of acetonitrile. Drug analytes were concentrated by evaporation under nitrogen, followed by reconstitution in 100 μL mobile phase A and B (97:3, v/v). 50 μL of extract was injected into the LCQ Deca XP Ion Trap Mass Spectrometer interfaced to a Surveyor HPLC system (Thermo Electron Corporation, East Greenbush, NY). Data were acquired and analyzed using Xcalibur™ Software, version 1.2. Chromatographic separation was performed on a Synergi Polar RP column (150 × 2.0 mm, 4μm), fitted with a guard column with identical 4 × 2.0 mm packing material (Phenomenex, Torrance, CA, USA). Column oven temperature was maintained at 25 °C. Gradient elution with (A) 10 mM ammonium formate in water, 0.001% formic acid (pH=4.5) and (B) acetonitrile, at a flow rate of 300 μL/min was applied. The initial gradient conditions were 5% B, increasing to 26% B in 13 min, with a final composition of 90% B in 9 min.
MS data were collected in positive ion mode, with corona discharge needle voltage, 4.5kV; vaporizer temperature, 450 °C; sheath gas setting (high-purity nitrogen), 70; no auxiliary gas; and transfer capillary temperature, 220 °C. The electron multiplier voltage was set at 850 eV. Identification and quantification was based on selected reaction monitoring (SRM). Retention time, collision energy, and SRM transitions for all target analytes and internal standards has been previously reported (Dams et al., 2003b).
Calibration, using internal standardization with deuterated analogues, was done by linear regression analysis over a maximum linear range from 1 to 500 μg/L. Peak-area ratios of target analytes and internal standards were calculated for each concentration. Data were fit to a linear least-squares regression curve with a weighting factor of 1/x. Clinical specimens that exceeded the upper limit of linearity were diluted, re-extracted and analyzed. LOD was defined as the concentration with a signal-to-noise ratio of at least 3, and ranged from 0.25 – 5.0 μg/L for target analytes. Limit of quantification (LOQ), defined as the lowest standard with a signal-to noise ratio of at least 10, and acceptable imprecision and accuracy (both < 20.0%), was established between 1.0 – 10.0 μg/L. Intra- and inter-batch imprecision were less than 15.8% and 22.8%, respectively. Accuracy, calculated as the percent difference from the target value, was less than 20.8%. Relative extraction recovery with protein precipitation ranged from 85.5 to 114.5%. No matrix effect was observed for the target analytes throughout LC-MS analysis (Dams et al., 2003b).
2.5. Recovery study of the Salivette® device
In-vitro recovery from the Salivette® cotton was evaluated by fortifying 1 mL of drug-free oral fluid with 50, 250, or 400 μg/L of the target analytes. Salivette® cotton rolls were placed in the spiked human oral fluid until completely saturated, then placed in polypropylene containers and centrifuged for 5 min at 1000 g. Recovery from these samples were compared to recovery from spiked blank oral fluid samples that were not absorbed onto the Salivette® collection device. Two hundred microliters of each oral fluid recovery sample was pretreated with the acetonitrile protein precipitation procedure as previously described, after addition of 10 μL of a 2-mg/L internal standard solution. Extracts were reconstituted in 100 μL of a mixture of mobile phase A and B (97:3, v/v) and 50 μL was injected onto the LC-MS system. Each experiment was performed in triplicate. The mean and CV% of the percent recovery of target analytes from the Salivette® were calculated.
The volume of oral fluid recovered from the cotton was investigated by placing the wool in 0.5, 1.0, or 2.0 mL of oral fluid. After absorption of the liquid, the cotton was positioned in the Salivette® tube, centrifuged (5 minutes at 1000 × g), and recovered volume of oral fluid measured in 3 replicates. Mean volume recovered (and %loss) were determined.
2.6. Identification and quantification of opiates, cocaine, and metabolites in oral fluid
Specimens were considered positive if one of the analytes was present at a concentration equal to or higher than the method LOQ. Median, mean, and range of concentrations of opiate and cocaine analytes in positive oral fluid specimens were determined. Profile of analytes found and percentage positive for each combination of analytes were determined. Correlations between analyte concentrations within an individual were evaluated by least squares regression analysis. Statistical significance was assumed when P was ≤ 0.05. Representative patterns of positive and negative oral fluid results for illicit drug use during treatment were determined throughout the study period.
2.7. Comparison of oral fluid test results with method LOQs and proposed SAMHSA cutoff concentrations
The number of positive specimens at the LC-MS/MS method’s LOQ was compared to the number of positive oral fluid test results obtained with the SAMHSA-recommended cutoff concentrations for confirmatory drug testing in alternative matrices (Administration, 2004). The LOQ for all analytes was 1 μg/L. The proposed SAMHSA confirmation cutoff concentrations for opiates and cocaine in oral fluid are 4 μg/L for 6-AM, 40 μg/L for morphine and codeine and 8 μg/L for cocaine or BE. A 4 μg/L cutoff concentration for heroin in oral fluid also was evaluated.
3. Results
3.1. Recovery study of the Salivette® device
Table 1 shows recovery of opiates and cocaine analytes from the cotton of the Salivette® device. Recoveries ranged from 73.9 to 114.9 % for the majority of the analytes. Major losses were observed for papaverine (31–35% recovery), p-hydroxycocaine (47–50% recovery), and noscapine (56–63% recovery). Recoveries were reproducible (CV% ≤ 15.0%) and no apparent difference was observed between recoveries at low, medium or high concentrations (CV% < 9.2%).
Table 1.
Percent recovery of opiates, cocaine, and metabolites from the Cozart Salivette® oral fluid collection device (%, n=3)
| Opiate analytes | 0.05 μg/L (CV%) | 0.25 μg/L (CV%) | 0.40 μg/L (CV%) |
|---|---|---|---|
|
|
|||
| Normorphine | 78.1 (10.3) | 93.8 (13.4) | 88.7 (12.1) |
| Morphine | 78.3 (7.8) | 73.9 (9.4) | 74.0 (9.9) |
| Norcodeine | 82.3 (8.3) | 95.3 (12.3) | 87.5 (11.8) |
| Codeine | 82.6 (10.0) | 89.6 (13.9) | 79.0 (11.2) |
| 6-AM | 86.9 (7.9) | 92.6 (8.0) | 88.6 (6.6) |
| Heroin | 79.2 (9.1) | 85.2 (8.0) | 81.9 (6.3) |
| Acetylcodeine | 93.3 (11.0) | 95.1 (9.4) | 91.1 (8.5) |
| Papaverine | 35.2 (8.6) | 34.1 (10.2) | 30.8 (12.4) |
| Noscapine | 62.6 (12.0) | 63.4 (15.0) | 56.4 (12.7) |
| Cocaine analytes | |||
| Ecgonine | 82.2 (12.5) | 91.1 (9.9) | 85.5 (11.1) |
| EME | 103.1 (10.3) | 106.1 (12.1) | 114.9 (8.2) |
| EEE | 96.8 (7.8) | 96.2 (10.3) | 105.5 (8.3) |
| p-hydroxybenzoylecgonine | 102.5 (8.6) | 103.8 (11.0) | 90.9 (6.9) |
| m-hydroxybenzoylecgonine | 94.1 (8.0) | 93.5 (6.4) | 87.2 (6.9) |
| Benzoylnorecgonine | 99.0 (4.7) | 113.0 (4.8) | 104.8 (5.6) |
| BE | 95.3 (5.8) | 98.2 (7.6) | 90.7 (6.3) |
| p-hydroxycocaine | 48.7 (8.5) | 49.6 (9.4) | 47.4 (11.3) |
| m-hydroxycocaine | 74.8 (7.0) | 76.2 (8.6) | 74.3 (6.4) |
| Norcocaine | 87.3 (3.1) | 94.1 (4.4) | 87.5 (6.0) |
| Cocaine | 81.7 (6.4) | 91.4 (5.6) | 84.6 (6.4) |
| Norcocaethylene | 81.0 (4.3) | 79.1 (4.8) | 81.8 (4.6) |
| Cocaethylene | 95.6 (4.4) | 98.8 (4.8) | 88.1 (3.5) |
Between 70 and 85% of the volume of oral fluid was recovered from the Salivette®. A constant %loss per volume was observed; 25–30% at 0.5 mL, 20–25% at 1.0 mL, and 15% at 2 mL.
3.2. Clinical specimens
A total of 403 oral fluid specimens were collected from 16 participants. Mean volume of oral fluid collected was 1.7 ± 0.7 mL, ranging from 0.3 to 3.0 mL. Mean pH of 403 oral fluid specimens was 6.1 ± 0.9 (range 4.3 – 9.4).
3.3. Heroin and metabolites in oral fluid
Oral fluid specimens were analyzed by LC-APCI-MS/MS for heroin and metabolites. Opiates were detected in 62 (15.4%) of 403 specimens. Of the 16 subjects, 4 had no opiate positive results. Eight subjects had less than 20% positive specimens; 3 subjects had between 20 and 35% opiate positive specimens and 1 subject had 91% positive results for opiates, often with high concentrations.
6-AM, heroin, and morphine were the primary opiates identified in oral fluid. 6-AM was detected in 45 (72.6%) of 62 positive oral fluid specimens, with a median concentration of 5.2 μg/L (range 1.4 to 434.0 μg/L). Heroin was identified in 26 (41.9%) specimens, followed by morphine in 16 (25.8%) of positive specimens. Median (range) concentrations were 2.3 μg/L (1.0 to 295.2 μg/L) for heroin and 7.5 μg/L (1.1 to 53.1 μg/L) for morphine. 6-AM concentrations were greater than heroin concentrations in 83% of positive specimens. Morphine concentrations were considerably lower than those of 6-AM or heroin. Codeine, acetylcodeine, norcodeine, normorphine, and noscapine also were detected in a small number (<10) of the 62 positive specimens and in considerably lower concentrations. Papaverine was not detected in oral fluid at the LOQ of 1 μg/L in any specimen. A detailed overview of the number of positive specimens, median, mean, and range of opiate concentrations in oral fluid is given in Table 2.
Table 2.
Opiate oral fluid concentrations in specimens (N= 62) collected from heroin-dependent pregnant women in methadone maintenance treatment
| Analytes | n | Median (μg/L) | Mean ± SD (μg/L) | Range (μg/L) |
|---|---|---|---|---|
| Normorphine | 1 | 1.8 | * | * |
| Morphine | 16 | 7.5 | 13.9 ± 15.6 | 1.1 – 53.1 |
| Norcodeine | 4 | 2.2 | 2.5 ± 0.9 | 1.8 – 3.6 |
| Codeine | 8 | 1.8 | 2.6 ± 1.6 | 1.3 – 6.4 |
| 6-AM | 45 | 5.2 | 35.8 ± 93.5 | 1.4 – 434.0 |
| Heroin | 26 | 2.3 | 24.3 ± 68.5 | 1.0 – 295.2 |
| Acetylcodeine | 6 | 1.8 | 5.1 ± 6.3 | 1.0 – 18.3 |
| Papaverine | 0 | * | * | * |
| Noscapine | 3 | 3.9 | 11.2 ± 13.6 | 2.7 – 26.9 |
n<2, no calculation possible
6-AM was identified exclusively in the largest percentage of positive specimens (29%) (Table 2); heroin and 6-AM were identified concurrently in 16.1%, heroin alone in 9.7%, and combinations of morphine, 6-AM, and heroin with and without acetylcodeine, in 8.1% and 6.5%, respectively. All other combinations were present in less than 5.0% of positive specimens.
Patterns of opiate concentrations in oral fluid collected during three study days per week across the course of the trial are illustrated for two representative subjects in Fig. 1. Concentrations of analytes are plotted against the number of days the subject provided specimens. For subject A, 6-AM and heroin were the only analytes detected in the low μg/L-range. In almost all positive specimens, 6-AM was present in higher concentration. No constant ratio or significant relationship (r=0.1, P > 0.05) between concentrations of analytes was apparent in this participant’s oral fluid specimens. Test results for subject A showed low concentrations of opiates in oral fluid throughout treatment, suggesting a low frequency and/or magnitude of drug use. For subject B, concentrations of 6-AM and heroin in oral fluid were much higher. Additionally, morphine, acetylcodeine, codeine, and noscapine also were identified. The subject appeared to abstain from opiate use in the beginning of treatment but occasionally used heroin later in gestation. Concentrations of analytes varied over time in similar patterns. 6-AM was always present in the highest quantity, while acetylcodeine, codeine, and noscapine were present in the lowest quantities. Significant correlations between concentrations of heroin, 6-AM, morphine, and acetylcodeine in oral fluid were observed (r > 0.94, P < 0.05).
Fig. 1.
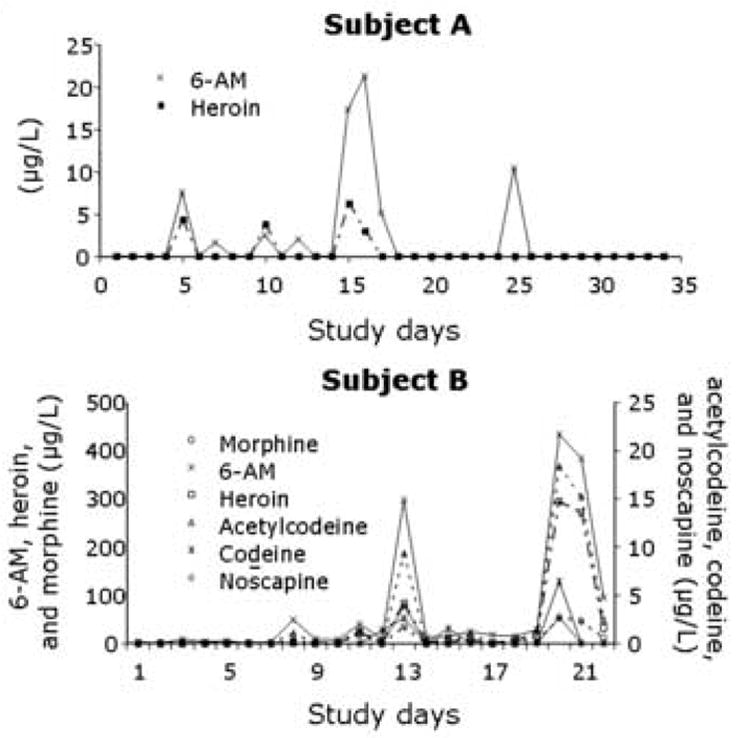
Concentration patterns of opiate analytes in oral fluid collected over time from heroin-dependent pregnant women enrolled in methadone maintenance treatment.
3.4. Cocaine and metabolites in oral fluid
Oral fluid specimens also were analyzed concurrently by LC-APCI-MS/MS for cocaine and metabolites. Of the 403 specimens, 130 (32.3%) tested positive (≥ LOQ) for one or more cocaine analytes. Of 16 subjects, two had no positive cocaine tests. Seven subjects tested positive for cocaine analytes in less than 20% of oral fluid specimens; 2 subjects had 30 to 35% cocaine positive specimens; 3 subjects had between 50 and 80% cocaine positive test results and, for 2 subjects, all oral fluid specimens tested positive for cocaine analytes. In 4 subjects, concurrent use of heroin and cocaine was noted.
Cocaine and BE were the primary cocaine analytes in oral fluid. Cocaine was detected in 111 (85.4%) of the 130 positive specimens with a median concentration of 6.4 μg/L (range 1.0 to 508.0 μg/L). BE was identified in 98 (75.4%) specimens, with a median concentration of 3.4 μg/L (range 1.0 to 157.2 μg/L). EME, a major metabolite of cocaine, and AEME, an indicator of smoking cocaine base, were detected in 10 (7.7%) and 5 (3.8%) positive specimens, respectively. Median and concentration ranges for these analytes were 51.1 μg/L (range 13.3 to 173.7 μg/L) for EME and 96.4 μg/L (range 24.7 to 142.5 μg/L) for AEME. Minor metabolites, i.e. norcocaine, benzoylnorecgonine, cocaethylene and ecgonine also were detected in low μg/L-concentrations in a limited number of specimens. Traces of m- and p-hydroxycocaine, below the method LOQ, also were observed. EEE, m- and p-hydroxybenzoylecgonine, and norcocaethylene were not identified. A detailed overview of positive specimens, median, mean, and range for cocaine analytes in oral fluid is given in Table 3.
Table 3.
Cocaine and metabolite oral fluid concentrations in specimens (N= 130) collected from heroin-dependent pregnant women in methadone maintenance treatment
| Analytes | n | Median (μg/L) | Mean ± SD (μg/L) | Range (μg/L) |
|---|---|---|---|---|
| Ecgonine | 1 | 25.9 | * | * |
| EME | 10 | 51.1 | 68.4 ± 54.9 | 13.3 – 173.7 |
| EEE | 0 | * | * | * |
| AEME | 5 | 96.4 | 93.9 ± 47.7 | 24.7 – 142.5 |
| p-hydroxybenzoylecgonine | 0 | * | * | * |
| m-hydroxybenzoylecgonine | 0 | * | * | * |
| Benzoylnorecgonine | 6 | 4.0 | 3.5 ± 1.3 | 1.6 – 4.7 |
| BE | 98 | 3.4 | 11.9 ± 26.8 | 1.0 – 157.2 |
| p-hydroxycocaine | 0 | TR** | * | * |
| m-hydroxycocaine | 0 | TR** | * | * |
| Norcocaine | 11 | 1.9 | 2.2 ± 1.0 | 1.4 – 5.0 |
| Cocaine | 111 | 6.4 | 29.7 ± 71.0 | 1.0 – 508.0 |
| Norcocaethylene | 0 | * | * | * |
| Cocaethylene | 2 | 1.5 | 1.5 ± 0.2 | 1.4 – 1.7 |
n<2, no calculation possible
TR, analyte identified but below LOQ
Cocaine and BE were identified concurrently in 50.8% of positive specimens; 21.5% tested positive for cocaine only, and 10.0% for BE only (Table 3). Other combinations of cocaine analytes in oral fluid were observed but prevalence was always 3.1% or lower.
Patterns of cocaine analyte concentrations in oral fluid across the course of the study are illustrated for two representative subjects in Fig. 2. For subject A, concentrations of cocaine, BE, EME, and AEME varied over time, but with a similar pattern. Cocaine concentrations were always present in the highest concentrations. Mean metabolite-parent concentration ratios were 0.3 ± 0.3 for BE, and 0.5 ± 0.3 for EME. A significant metabolite-parent concentration relationship was determined for each of the analytes (r > 0.75, P < 0.05). High levels of cocaine analytes in numerous specimens of subject A indicated continued cocaine use throughout treatment. Presence of AEME indicated “crack” (cocaine base) smoking. In contrast, subject B tested positive for cocaine and BE only. High concentrations of cocaine were detected on a number of study days while concentrations of BE were considerably lower (mean metabolite-parent ratio 0.3 ± 0.2). Analyte concentrations were significantly correlated (r > 0.9, P < 0.05).
Fig. 2.
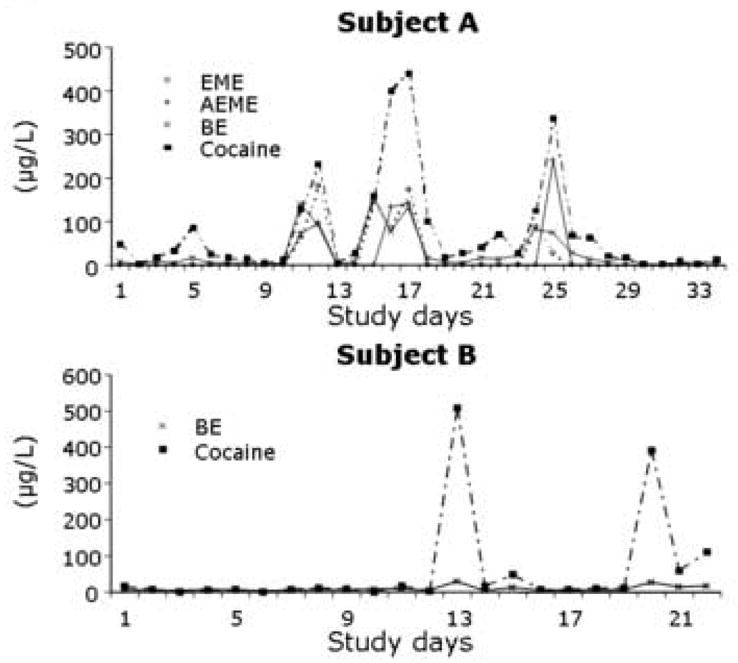
Concentration patterns of cocaine analytes in oral fluid collected over time from heroin-dependent pregnant women enrolled in methadone maintenance treatment.
3.5. Comparison of oral fluid test results with method LOQs and proposed SAMHSA cutoff concentrations
Fifty-six oral fluid specimens contained more than 1 μg/L of heroin, 6-AM, morphine, or codeine. Of these specimens, 28 (50%) were positive for opiates with the SAMHSA confirmatory cutoff concentrations for 6-AM (4 μg/L), and morphine and codeine (40 μg/L). 6-AM was present in all positive specimens, while morphine and codeine were detected above SAMHSA cutoff concentrations in zero and 2 (7%) specimens, respectively. Due to the prevalence of heroin in our oral fluid specimens, an additional 4 μg/L cutoff concentration for this analyte was evaluated. Thirteen (46%) of the SAMHSA positive specimens contained heroin above 4 μg/L, yet, in all cases 6-AM was present in equal or higher concentration. Applying a 4 μg/L cutoff concentration for heroin did not provide additional positive results.
In 128 specimens, cocaine and/or BE were detected above the LOQs but only 50 specimens (39%) were positive with the 8 μg/L cutoff concentration proposed by SAMHSA. Twenty-four (48%) specimens contained cocaine and BE, 23 (46%) contained cocaine only and three (6%) BE only.
3.6. Comparison of LCMSMS oral fluid test results with method LOQs and qualitative urine screening tests with 300 μg/L cutoffs for opiates and cocaine
Test results for 403 paired quantitative oral fluid and qualitative urine specimen collections were compared. Qualitative urine test results were obtained with the On Trak TesTstik (Roche Diagnostics, Indianapolis, IN) with 300 μg/L cutoffs for cocaine and opiates. There were 62 opiate positive oral fluid tests and 37 positive opiate urine tests and 130 cocaine positive oral fluid tests and 22 positive cocaine urine tests.
4. Discussion
Monitoring illicit drug use in biological matrices is an objective tool to assess drug abstinence in substance-abuse treatment patients and to evaluate effectiveness of treatment interventions. In addition, oral fluid provides a useful matrix for monitoring drug use in workplace, criminal justice, military and driving under the influence of drugs settings. Each biological matrix, traditional or alternative, provides unique information and offers specific advantages and disadvantages. The ease and non-invasiveness of specimen collection has made oral fluid an interesting biological matrix. Oral fluid was selected as the most promising means of detecting drugged driving during the Roadside Testing Assessment (ROSITA) program (Verstraete and Pudda, 2000); however, current testing technology was found to be inadequate for accurate and sensitive detection of drug use. Small specimen volumes and low salivary drug concentrations create analytical challenges. By implementing new bio-analytical technology, namely LC-MS, a wider range of analytes, e.g. heroin and metabolites, could be studied simultaneously. LC-MS does not require derivatization or extensive sample preparation. In addition, it is a soft ionization technique that provides mainly protonated molecular ions and, depending on the type of mass analyzer, typical product ions. It is a universal, sensitive and specific bio-analytical tool that can broaden and improve our knowledge of detection and excretion profiles of drugs in biological matrices.
The oral fluid collection device can seriously impact analytical findings in oral fluid drug testing (Kato et al., 1993; O’Neal et al., 2000). Crouch recently suggested in a review of collection methods that drug and volume recovery be determined, since these variables greatly impact the validity of oral fluid test results (Crouch, 2005). Manufacturers do not publish oral fluid or analyte recoveries, placing the burden on individual investigators to document these important factors. In this study, the majority of analytes were reproducibly recovered from the Salivette® cotton collection device, with substantial adsorptive losses only for papaverine, p-hydroxycocaine, and noscapine. In addition, 70–85% of the volume of oral fluid was recovered after centrifugation. We concluded that Salivette® was a practical oral fluid collection device for monitoring opiate and cocaine use. Caution is required for interpretation of quantitative clinical data due to adsorptive losses for a limited number of analytes. Also, when utilizing Salivette® or other collection devices in clinical studies, evaluation of the in-vitro recovery of analytes of interest is a necessity. O’Neal et al. observed that the oral fluid collection method could have a serious influence on salivary codeine and morphine concentrations (O’Neal et al., 2000). For Salivette®, good recovery was reported for both analytes, but there was a constant loss of 10–25% of oral fluid in the cotton, in agreement with our results. Hold et al. reported adsorptive losses with Salivette® for a different class of drugs, i.e. β-adrenoceptor blocking drugs (Hold et al., 1995). They observed a negative correlation between recovery and lipophilicity of analytes, but not with stereochemical configuration. In a similar manner, we found that papaverine and noscapine, two lipophilic opiates, were most susceptible to adsorption on the Salivette® swab. This line of reasoning could not explain the poor recovery of p-hydroxycocaine, a more polar cocaine analyte. Finally, Bermejo et al., reported 30–40% losses of methadone and EDDP in oral fluid collected with Salivette® (Bermejo et al., 2000). This is in agreement with the finding that more lipophilic analytes have a higher risk of adsorption to cotton. It confirms our conclusion that for each new application, analyte recovery from the oral fluid collection device needs to be determined.
The smaller available volume of oral fluid, in comparison to urine, is often identified as one of the drawbacks of oral fluid testing (Kidwell, 1990). In this study, the mean volume of a specimen collected with Salivette® (n=403) was 1.7 ± 0.7 mL. This is a relatively large volume of oral fluid, allowing for standard screening and confirmatory analyses. In addition, by applying LC-MS we developed a simplified sample preparation method and limited the volume of matrix necessary for analysis. The mean pH of oral fluid specimens was 6.1 ± 0.9, which is consistent with the pH of unstimulated oral fluid (Haeckel and Hanecke, 1996).
Heroin and metabolites were detected by LC-MS in 62 oral fluid specimens collected from pregnant outpatient treatment subjects when applying the LOQs of the method. The major analytes detected were 6-AM, heroin, and morphine while minor metabolites were detected only in highly positive opiate specimens. Papaverine was never detected in oral fluid, which could be due to adsorptive losses with the Salivette® device. Detection of 6-AM and/or heroin in 84% of positive oral fluid specimens proved heroin usage, rather than licit codeine or morphine, or ingestion of opiate-containing foodstuffs. In contrast, only 37 urine specimens collected the same day in the same subjects as the oral fluid samples were positive at the 300 μg/L cutoff concentration for opiates in urine. This indicates that oral fluid testing is highly sensitive for detecting opiate usage.
Speckl et al. also detected 6-AM, morphine, codeine, and dihydrocodeine in oral fluid of participants in drug detoxification therapy, but heroin was not included in the assay and no quantitative GC-MS data were shown (Speckl et al., 1999). Cooper et al. collected oral fluid specimens with a Cozart® collection device from donors monitored during drug treatment (Cooper et al., 2005). Specimens were screened with a cutoff equal to or above 15 μg/L and confirmed with a GC-MS cutoff of 15 μg/L. 6-AM, dihydrocodeine, codeine, and morphine concentrations were higher than those in the present study, but direct comparison is difficult due to differences in cutoff concentrations, and the fact that the Cozart collection device dilutes specimens approximately 1:3. No other reports are available on the presence and concentrations of heroin and metabolites in oral fluid of substance-abuse treatment patients. The results of the present study are consistent with salivary opiate concentrations reported in oral fluid studies in a parole or probation population (Kidwell, 1990). These authors detected 6-AM, heroin and morphine in oral fluid by LC-Thermospray-MS, with maximum concentrations of 128, 48 and 43 μg/L respectively. 6-AM, morphine and codeine also were identified in oral fluid of individuals leaving a discotheque (Samyn and van Haeren, 2000). Maximum concentrations found in this population were approximately 10-fold higher for 6-AM and 150-fold higher for morphine and codeine, most likely due to the shorter time interval between drug use and oral fluid collection.
Although the dose of self-administered opiates and elapsed time between drug use and specimen collection is unknown in our study, our results also are consistent with controlled drug administration studies. Historically, morphine was detected first in oral fluid by immunoassay, following controlled intravenous administration of heroin (Gorodetzky and Kullberg, 1974). Later, heroin, 6-AM, and morphine were identified in oral fluid following parenteral (Cone, 1993), intranasal (Cone, 1993; Goldberger et al., 1993; Wang et al., 1994), intravenous (Jenkins et al., 1995) and smoked administration (Jenkins et al., 1995). Although the presence of minor metabolites in oral fluid was investigated, they could not be detected above the limits of detection of the applied methods (Jenkins et al., 1995; Wang et al., 1994). Concentrations were highly dependent on dose and oral contamination (Jenkins et al., 1995; Wang et al., 1994). Furthermore, re-contamination of the oral cavity by coughing or clearing the nasal passage was reported (Wang et al., 1994). In the present study, heroin and its major metabolites were identified in oral fluid in similar concentrations and patterns as those reported in controlled clinical studies. Additionally, by utilizing a sensitive LC-APCI-MS/MS method, with LOQs of 1 μg/L, we were able to quantify minor opiate analytes including codeine, norcodeine, normorphine, noscapine, and acetylcodeine in highly opiate positive specimens.
Cocaine and metabolites also were found in 130 oral fluid specimens. The major analytes identified were cocaine and BE, detected in approximately 98% of all cocaine positive specimens. Other metabolites identified in oral fluid were found in the following relative abundances: norcocaine>EME>benzoylnorecgonine>AEME>cocaethylene>ecgonine. As noted in opiate analysis, oral fluid primarily contained parent drug and major metabolites, with minor metabolites only detected in specimens with high parent drug concentrations. In contrast only 22 urine specimens collected from the same subjects on the same day were positive for cocaine metabolites at a 300 μg/L cutoff concentration. Oral fluid testing was shown to be more sensitive than urine testing for identifying cocaine usage at the respective specified cutoff concentrations and when considering all of the oral fluid cocaine analytes of interest.
Limited naturalistic data on the presence and concentration of cocaine and metabolites in oral fluid of specific populations are available. Peel et al. first identified cocaine in oral fluid of an impaired driver (Peel et al., 1984). Cocaine (De Giovanni et al., 2002) and BE (Schramm et al., 1993; Smith and Kidwell, 1996) also were detected in oral fluid from substance abuse treatment patients. Kidwell detected cocaine, BE, EME and ecgonine in oral fluid collected from a parole and probation population, in similar concentrations to those reported here (Kidwell, 1990). Additionally, cocaine and BE were the primary analytes detected, in agreement with our results. These analytes also were identified in oral fluid of suspected drug users, but due to a short self-reported 2–4 h elapsed time between drug use and specimen collection, concentrations greatly exceeded our results (22–3505 μg/L for cocaine and 0–2395 μg/L for BE) (Samyn and van Haeren, 2000). AEME was identified also in oral fluid from this population following self-reported smoking of cocaine. Clauwaert et al. used LC-TOF-MS to analyze 15 oral fluid specimens collected with a Salivette® device as part of method validation (Clauwaert et al., 2004). One specimen had cocaine, BE, and cocaethylene concentrations of 200, 59, and 10 μg/L, respectively, and all others had no detectable BE or cocaethylene and cocaine concentrations near the cutoff concentration of 10 μg/L. Cocaine concentrations were within the range observed in the present study, but metabolite concentrations are difficult to compare due to the higher cutoff utilized.
Similarly, the results of the present study are consistent with analyte profiles described in controlled cocaine administration studies. Cocaine, with or without metabolites, was identified in oral fluid after intravenous (Cone et al., 1988; Cone et al., 1997; Jenkins et al., 1995; Kato et al., 1993; Thompson et al., 1987), smoked (Cone et al., 1994; Cone et al., 1997; Jenkins et al., 1995), intranasal (Cone, 1993; Cone et al., 1997) and oral (Jufer et al., 2000) administration, as well as, during withdrawal (Cone and Weddington, 1989; Moolchan et al., 2000). The primary analyte was cocaine followed by BE and EME, the major metabolites of cocaine; AEME was identified in oral fluid after smoked cocaine exposure (Cone et al., 1997; Jenkins et al., 1995; Moolchan et al., 2000). Other minor metabolites detected in oral fluid were norcocaine (Jenkins et al., 1995; Jufer et al., 2000) and p-hydroxycocaine (Jufer et al., 2000). Concentrations of cocaine and metabolites were clearly dependent on dose and route of administration. Oral or nasal insufflation contaminated the oral cavity (Cone et al., 1997). Prolonged excretion was noted after chronic cocaine use (Cone and Weddington, 1989; Jufer et al., 2000; Moolchan et al., 2000). In the present study, all previously identified major and minor analytes were detected in oral fluid, in addition to benzoylnorecgonine and cocaethylene.
Simultaneous analysis of cocaine, heroin, and metabolites in oral fluid of pregnant substance abuse treatment patients revealed 32% cocaine and 15% opiate positive oral fluid specimens as compared to 5.5% and 9% cocaine and opiate positive urine screens. These differences in positivity rates are due to differences in cutoff concentrations, as well as the spectrum of analytes being tested. Therefore, the oral fluid tests were more sensitive than the urine tests for detecting drug usage at the defined cutoff concentrations. Another issue that must be considered is the fact that the urine tests are directed at measurement of benzoylecgonine and morphine, while the oral fluid LCMSMS method targeted low concentrations of the following opiates: morphine, normorphine, codeine, norcodeine, 6-acetylmorphine (6AM), heroin, 6 acetylcodeine, papaverine and noscapine and the following cocaine analytes: benzoylecgonine, m or p-hydroxy-benzoylecgonine, benzoylnorecgonine, cocaine, m or p-hydroxy-cocaine, norcocaine, cocaethylene, norcocaethylene, ecgonine, ecgonine methyl ester, ecgonine ethyl ester, and anhydroecgonine methyl ester (AEME). An additional advantage of the oral fluid test results is that important information about the drug ingested, i.e., heroin vs codeine, or the route of drug administration, i.e. AEME indicated the smoked route of cocaine administration, may be available. In addition, polydrug abuse was observed for four subjects who simultaneously abused cocaine and heroin. Limited information is available on this topic, but trends identified in the present study are consistent with previous observations for heroin addicts in methadone maintenance treatment (De Giovanni et al., 2002), subjects on parole or probation (Kidwell, 1990) and impaired drivers (Peel et al., 1984).
One of the strengths of this study is the comparison of oral fluid results at the low LOQs of the LCMSMS method and at the proposed SAMHSA guidelines for oral fluid testing. Application of the SAMHSA-recommended cutoff concentrations for confirmatory oral fluid analysis identified far fewer positive specimens than our method LOQs. Only 50% of opiate and 39% of cocaine positive specimens were detected with the cutoff concentrations proposed by SAMHSA. However, it is unclear whether the lower cutoff concentration employed in this study identified multiple drug use episodes or whether residual drug excretion produced serially consecutive positive tests. Clearly, the window of drug detection was expanded with the lower cutoff concentration. The smaller number of positive specimens with the recommended SAMHSA cutoff concentrations is noteworthy and documents the need for further controlled drug administration studies.
In contrast, at the 4 μg/L recommended cutoff concentration, 6-AM proved to be an excellent and sensitive biomarker for heroin abuse. Codeine and morphine did not contribute additionally since the concentrations of these analytes in oral fluid were frequently below the 40 μg/L recommended cutoff concentration, and 6-AM always was identified with these analytes in positive specimens. We also evaluated a 4 μg/L cutoff concentration for heroin. Heroin was detected above the cutoff concentration in 46% of opiate positive specimens, but in all specimens 6-AM was present in equal or higher concentrations. With the SAMHSA recommended 8 μg/L cutoff concentration for cocaine or BE, cocaine was identified in 94% of positive specimens, while BE was detected in only 52%. The results of the present study clearly support selection of 6-AM and cocaine as effective oral fluid biomarkers for heroin and cocaine use.
This study was an outpatient study of opiate-dependent women receiving methadone replacement and behavioral therapy to reduce illicit opiate and cocaine use. Therefore, it is not possible to define windows of drug detection, as is possible after controlled drug administration studies. Data on windows of drug detection for specific analytes and at specific cutoffs are available for some drugs and also may include evaluation of relationships to simultaneously collected blood specimens (Choo and Huestis, 2004; Kim et al., 2002; Schepers et al., 2003). In general, Verstraete reported that drugs of abuse could be detected in oral fluid for 5 to 48 hours (Verstraete, 2004). Of course, as previously described, cutoff concentrations, collection device specifications, analytes tested and detection methodology will influence detection rates. Furthermore, the dose administered also may influence detection windows. Doses in controlled drug administration studies are limited by concern for participant safety and ethical issues. Oral fluid data presented in this manuscript reflect doses self-administered by drug addicts in the community, and thus, provide a realistic view of opiate and cocaine analyte concentrations in oral fluid of individuals who abuse illicit drugs.
In addition, these oral fluid data do not provide information on the relationship between oral fluid and blood concentrations. In general, oral fluid test results are temporally correlated with plasma drug concentrations; however, dependent upon the lipophilicity and pKa of the drug and metabolites, concentrations in oral fluid can be much higher or lower than in plasma (Choo and Huestis, 2004; Kim et al., 2002; Schepers et al., 2003). In addition, if the route of drug administration is smoking, there can be significant contamination of the oral fluid immediately after dosing, as for example after smoking cannabis (Huestis and Cone, 2004). Furthermore, drugs and metabolites may bind differentially to the oral fluid collection device, again reducing the correlation between blood and oral fluid drug concentrations. Also, passive contamination of oral fluid by drug-laden smoke also may be a factor in reducing the correlation of drug concentrations in blood and oral fluid (Niedbala et al., 2004).
In conclusion, oral fluid is a promising alternative matrix to monitor opiate and cocaine use. Specimen collection with Salivette® was practical, easy and provided sufficient sample volume for oral fluid drug analysis in a drug treatment population. Recovery from the cotton was reproducible and adequate for most analytes. 6-AM, heroin, and morphine were the major opiates identified, while cocaine and BE were the primary cocaine analytes detected in oral fluid. Minor analytes and metabolites were detected only in oral fluid specimens with high concentrations of opiates or cocaine. Applications of the SAMHSA-recommended cutoff concentrations for confirmatory oral fluid analysis enabled identification of only 50% of opiate and 39% of cocaine positive specimens. 6-AM and cocaine proved to be excellent biomarkers in oral fluid for heroin and cocaine use, respectively. Further research is necessary to evaluate the effectiveness of the recommended cutoff concentrations for oral fluid drug testing in this population.
Acknowledgments
This research was supported by the NIH, National Institute on Drug Abuse Intramural Research Program and NIDA extramural research grant DA12403-02. We thank all those who helped in the course of this study, including the F.S.R.-Flanders (Belgium) for providing travel grant V 4.037.01N, Erin Kolbrich, Matthew Kearney, Wesen Nebro, and Debbie Price for assisting in sample handling and CAP personnel for collection of specimens.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bermejo AM, Lucas ACS, Tabernero MJ. Saliva/plasma ratio of methadone and EDDP. Journal of Analytical Toxicology. 2000;24:70–72. doi: 10.1093/jat/24.1.70. [DOI] [PubMed] [Google Scholar]
- Choo RE, Huestis MA. Oral fluid as a diagnostic tool. Clinical Chemistry Laboratory Medicine. 2004;42:1273–1287. doi: 10.1515/CCLM.2004.248. [DOI] [PubMed] [Google Scholar]
- Clauwaert K, Decaestecker T, Mortier K, Lambert W, Deforce D, Van Peteghem C, Van Bocxlaer J. The determination of cocaine, benzoylecgonine, and cocaethylene in small-volume oral fluid samples by liquid chromatography-quadrupole-time-of-flight mass spectrometry. Journal of Analytical Toxicology. 2004;28:655–659. doi: 10.1093/jat/28.8.655. [DOI] [PubMed] [Google Scholar]
- Cone EJ. Saliva testing for drugs of abuse. Annals of the New York Academy of Sciences. 1993;694:91–127. doi: 10.1111/j.1749-6632.1993.tb18346.x. [DOI] [PubMed] [Google Scholar]
- Cone EJ. Legal, workplace, and treatment drug testing with alternate biological matrices on a global scale. Forensic Science International. 2001;121:7–15. doi: 10.1016/s0379-0738(01)00446-7. [DOI] [PubMed] [Google Scholar]
- Cone EJ, Hillsgrove M, Darwin WD. Simultaneous measurement of cocaine, cocaethylene, their metabolites, and “Crack” pyrolysis products by gas chromatography-mass spectrometry. Clinical Chemistry. 1994;40:1299–1305. [PubMed] [Google Scholar]
- Cone EJ, Kumor K, Thompson LK, Sherer M. Correlation of saliva cocaine levels with plasma levels and with pharmacologic effects after intravenous cocaine administration in human subjects. Journal of Analytical Toxicology. 1988;12:200–206. doi: 10.1093/jat/12.4.200. [DOI] [PubMed] [Google Scholar]
- Cone EJ, Oyler J, Darwin WD. Cocaine disposition in saliva following intravenous, intranasal, and smoked administration. Journal of Analytical Toxicology. 1997;21:465–475. doi: 10.1093/jat/21.6.465. [DOI] [PubMed] [Google Scholar]
- Cone EJ, Preston KL. Toxicologic aspects of heroin substitution treatment. Therapeutic Drug Monitoring. 2002;24:193–198. doi: 10.1097/00007691-200204000-00001. [DOI] [PubMed] [Google Scholar]
- Cone EJ, Weddington WW., Jr Prolonged occurrence of cocaine in human saliva and urine after chronic use. Journal of Analytical Toxicology. 1989;13:65–68. doi: 10.1093/jat/13.2.65. [DOI] [PubMed] [Google Scholar]
- Cooper G, Wilson L, Reid C, Main L, Hand C. Evaluation of the Cozart RapiScan drug test system for opiates and cocaine in oral fluid. Forensic Science International. 2005;150:239–243. doi: 10.1016/j.forsciint.2004.12.042. [DOI] [PubMed] [Google Scholar]
- Crouch DJ. Oral fluid collection: the neglected variable in oral fluid testing. Forensic Science International. 2005;150:165–173. doi: 10.1016/j.forsciint.2005.02.028. [DOI] [PubMed] [Google Scholar]
- Dams R, Huestis MA, Lambert WE, Murphy CM. Matrix effect in bio-analysis of illicit drugs with LC-MS/MS: influence of ionization type, sample preparation, and biofluid. Journal of the American Society of Mass Spectrometry. 2003a;14:1290–1294. doi: 10.1016/S1044-0305(03)00574-9. [DOI] [PubMed] [Google Scholar]
- Dams R, Murphy CM, Choo RE, Lambert WE, De Leenheer AP, Huestis MA. LC-atmospheric pressure chemical ionization-MS/MS analysis of multiple illicit drugs, methadone, and their metabolites in oral fluid following protein precipitation. Analytical Chemistry. 2003b;75:798–804. doi: 10.1021/ac026111t. [DOI] [PubMed] [Google Scholar]
- De Giovanni N, Fucci N, Chiarotti M, Scarlata S. Cozart Rapiscan System: our experience with saliva tests. Journal of Chromatography B. 2002;773:1–6. doi: 10.1016/s0378-4347(01)00522-9. [DOI] [PubMed] [Google Scholar]
- Drummer OH. Review: Pharmacokinetics of illicit drugs in oral fluid. Forensic Science International. 2005;150:133–142. doi: 10.1016/j.forsciint.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Goldberger BA, Darwin WD, Grant TM, Allen AC, Caplan YH, Cone EJ. Measurement of heroin and its metabolites by isotope-dilution electron-impact mass spectrometry. Clinical Chemistry. 1993;39:670–675. [PubMed] [Google Scholar]
- Gorodetzky CW, Kullberg MP. Validity of screening methods for drugs of abuse in biological fluids II. Heroin in plasma and saliva. Clinical Pharmacology & Therapeutics. 1974;15:579–587. doi: 10.1002/cpt1974156579. [DOI] [PubMed] [Google Scholar]
- Haeckel R, Hanecke P. Application of saliva for drug monitoring. An in vivo model for transmembrane transport. European Journal of Clinical Chemistry and Clinical Biochemistry. 1996;34:171–191. [PubMed] [Google Scholar]
- Hold KM, de Boer D, Zuidema J, Maes RAA. Evaluation of the Salivette as sampling device for monitoring B-adrenoceptor blocking drugs in saliva. Journal of Chromatography. 1995;663:103–110. doi: 10.1016/0378-4347(94)00431-4. [DOI] [PubMed] [Google Scholar]
- Huestis MA, Cone EJ. Relationship of Delta 9-tetrahydrocannabinol concentrations in oral fluid and plasma after controlled administration of smoked cannabis. Journal of Analytical Toxicology. 2004;28:394–399. doi: 10.1093/jat/28.6.394. [DOI] [PubMed] [Google Scholar]
- Idowu OR, Caddy B. A review of the use of saliva in the forensic detection of drugs and other chemicals. Journal of Forensic Science Society. 1982;22:123–135. doi: 10.1016/s0015-7368(82)71460-4. [DOI] [PubMed] [Google Scholar]
- Jenkins AJ, Oyler JM, Cone EJ. Comparison of heroin and cocaine concentrations in saliva with concentrations in blood and plasma. Journal of Analytical Toxicology. 1995;19:359–374. doi: 10.1093/jat/19.6.359. [DOI] [PubMed] [Google Scholar]
- Jufer RA, Wstadik A, Walsh SL, Levine BS, Cone EJ. Elimination of cocaine and metabolites in plasma, saliva, and urine following repeated oral administration to human volunteers. Journal of Analytical Toxicology. 2000;24:467–477. doi: 10.1093/jat/24.7.467. [DOI] [PubMed] [Google Scholar]
- Kacinko SL, Barnes AJ, Kim I, Moolchan ET, Wilson L, Cooper GA, Reid C, Baldwin D, Hand CW, Huestis MA. Performance characteristics of thet Cozart RapiScan Oral Fluid Drug Testing System for opiates in comparison to ELISA and GC/MS following controlled codeine administration. Forensic Science International. 2004;141:41–48. doi: 10.1016/j.forsciint.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Kato K, Hillsgrove M, Weinhold L, Gorelick DA, Darwin WD, Cone EJ. Cocaine and metabolite excretion in saliva under stimulated and nonstimulated conditions. Journal of Analytical Toxicology. 1993;17:338–341. doi: 10.1093/jat/17.6.338. [DOI] [PubMed] [Google Scholar]
- Kidwell DA. Analysis of cocaine, heroin, and their metabolites in saliva. Naval Research Laboratory. 1990:1–14. [Google Scholar]
- Kidwell DA, Holland JC, Athanaselis S. Testing for drugs of abuse in saliva and sweat. Journal of Chromatography. 1998;713:111–135. doi: 10.1016/s0378-4347(97)00572-0. [DOI] [PubMed] [Google Scholar]
- Kim I, Barnes AJ, Oyler JM, Schepers R, Joseph RE, Jr, Cone EJ, Lafko D, Moolchan ET, Huestis MA. Plasma and oral fluid pharmacokinetics and pharmacodynamics after oral codeine administration. Clinical Chemistry. 2002;48:1486–1496. [PubMed] [Google Scholar]
- Kintz P, Cirimele V, Ludes B. Codeine testing in sweat and saliva with the Drugwipe. International Journal of Legal Medicine. 1998;111:82–84. doi: 10.1007/s004140050119. [DOI] [PubMed] [Google Scholar]
- Kopecky EA, Jacobson S, Klein J, Kapur B, Koren G. Correlation of morphine sulfate in blood plasma and saliva in pediatric patients. Therapeutic Drug Monitoring. 1997;19:530–534. doi: 10.1097/00007691-199710000-00008. [DOI] [PubMed] [Google Scholar]
- Moolchan ET, Cone EJ, Wstadik A, Huestis MA, Preston KL. Cocaine and metabolite elimination patterns in chronic cocaine users during cessation: plasma and saliva analysis. Journal of Analytical Toxicology. 2000;24:458–466. doi: 10.1093/jat/24.7.458. [DOI] [PubMed] [Google Scholar]
- Navarro M, Pichini S, Farre M, Ortuno J, Roset PN, Segura J, de La Torre R. Usefulness of saliva for measurement of 3,4-methylenedioxymehamphetamine and its metabolites: correlation with plasma drug concentrations and effect of salivary pH. Clinical Chemistry. 2001;47:1788–1795. [PubMed] [Google Scholar]
- Niedbala S, Kardos K, Salamone S, Fritch D, Bronsgeest M, Cone EJ. Passive cannabis smoke exposure and oral fluid testing. Journal of Analytical Toxicology. 2004;28:546–552. doi: 10.1093/jat/28.7.546. [DOI] [PubMed] [Google Scholar]
- O’Neal CL, Crouch DJ, Rollins DE, Fatah A, Cheever ML. Correlation of saliva codeine concentrations with plasma concentrations after oral codeine administration. Journal of Analytical Toxicology. 1999;23:452–459. doi: 10.1093/jat/23.6.452. [DOI] [PubMed] [Google Scholar]
- O’Neal CL, Crouch DL, Rollins DE, Fatah AA. The effects of collection methods on oral fluid codeine concentrations. Journal of Analytical Toxicology. 2000;24:536–542. doi: 10.1093/jat/24.7.536. [DOI] [PubMed] [Google Scholar]
- Peel HW, Perrigo BJ, Mikhael NZ. Detection of drugs in saliva of impaired drivers. Journal of Forensic Sciences. 1984;29:185–189. [PubMed] [Google Scholar]
- Pichini S, Altieri I, Zuccaro P, Pacifici R. Drug monitoring in nonconventional biological fluids and matrices. Clinical Pharmacokinetics. 1996;30:211–228. doi: 10.2165/00003088-199630030-00003. [DOI] [PubMed] [Google Scholar]
- Rivier L. Techniques for analytical testing of unconventional samples. Bailliere’s Clinical Endocrinology and Metabolism. 2000;14:147–165. doi: 10.1053/beem.2000.0060. [DOI] [PubMed] [Google Scholar]
- Samyn N, van Haeren C. On-site testing of saliva and sweat with Drugwipe and determination of concentrations of drugs of abuse in saliva, plasma and urine of suspected users. International Journal of Legal Medicine. 2000;113:150–154. doi: 10.1007/s004140050287. [DOI] [PubMed] [Google Scholar]
- Schepers RJF, Oyler JM, Joseph RE, Jr, Cone EJ, Moolchan ET, Huestis MA. Methamphetamine and amphetamine pharmacokinetics in oral fluid and plasma after controlled oral methamphetamine administration to human volunteers. Clinical Chemistry. 2003;49:121–132. doi: 10.1373/49.1.121. [DOI] [PubMed] [Google Scholar]
- Schramm W, Craig PA, Smith RH, Berger GE. Cocaine and benzoylecgonine in saliva, serum, and urine. Clinical Chemistry. 1993;39:481–487. [PubMed] [Google Scholar]
- Schramm W, Smith RH, Craig PA, Kidwell DA. Drugs of abuse in saliva: A review. Journal of Analytical Toxicology. 1992;16:1–9. doi: 10.1093/jat/16.1.1. [DOI] [PubMed] [Google Scholar]
- Skopp G, Potsch L, Klinder K, Richter B, Aderjan R, Mattern R. Saliva testing after single and chronic administration of dihydrocodeine. International Journal of Legal Medicine. 2001;114:133–140. doi: 10.1007/pl00007717. [DOI] [PubMed] [Google Scholar]
- Smith FP, Kidwell DA. Cocaine in hair, saliva, skin swabs, and urine in cocaine users’ children. Forensic Science International. 1996;83:179–189. doi: 10.1016/s0379-0738(96)02035-x. [DOI] [PubMed] [Google Scholar]
- Speckl IM, Hallbach J, Guder WG, Meyer LV, Zilker T. Opiate detection in saliva and urine--a prospective comparison by gas chromatography-mass spectrometry. Clinical Toxicology. 1999;37:441–445. doi: 10.1081/clt-100102434. [DOI] [PubMed] [Google Scholar]
- Substance Abuse Mental Health Services Administration (SAMHSA) Draft Guidelines for Federal Workplace Drug Testing Program. 2004 http://workplace.samhsa.gov/
- Thompson LK, Yousefnejad D, Kumor K, Sherer M, Cone EJ. Confirmation of cocaine in human saliva after intravenous use. Journal of Analytical Toxicology. 1987;11:36–38. doi: 10.1093/jat/11.1.36. [DOI] [PubMed] [Google Scholar]
- Verstraete A, Pudda M. General conclusions and recommendations. 2000 Status P. Rosita. [Google Scholar]
- Verstraete AG. Detection times of drugs of abuse in blood, urine, and oral fluid. Therapeutic Drug Monitoring. 2004;26:200–205. doi: 10.1097/00007691-200404000-00020. [DOI] [PubMed] [Google Scholar]
- Wang WL, Darwin WD, Cone EJ. Simultaneous assay of cocaine, heroin and metabolites in hair, plasma, saliva and urine by gas chromatography-mass spectrometry. Journal of Chromatography. 1994;660:279–290. doi: 10.1016/0378-4347(94)00309-2. [DOI] [PubMed] [Google Scholar]
- Yacoubian GS, Wish ED, Perez DM. A comparison of saliva testing to urinalysis in an arrrestee population. Journal of Psychoactive Drugs. 2001;33:289–294. doi: 10.1080/02791072.2001.10400576. [DOI] [PubMed] [Google Scholar]