

Abstract
Mechanisms contributing to development of diabetic nephropathy (DN) remain unclear. High ambient glucose level transforms intracellular pathways, promoting stable phenotypic changes in the glomerulus such as mesangial cell hypertrophy, podocyte apoptosis, and matrix expansion. Insulin-like growth factors (IGFs) and the high affinity IGF binding proteins (IGFBPs) exert major effects on cell growth and metabolism. Compared with diabetic patients without microalbuminuria (MA), MA diabetic patients display perturbed GH-IGF-IGFBP homeostasis, including increased circulating IGF-I and IGFBP-3 protease activity, increased excretion of bioactive GH, IGF-I, and IGFBP-3, but decreased circulating IGFBP-3 levels. In diabetic animal models, expression of IGF-I and IGFBP-1 to -4 increases in key renal tissues and glomerular ulrafiltrate. Epithelial, mesangial, and endothelial cells derived from the kidney respond to IGF-I binding with increased protein synthesis, migration, and proliferation. This article reviews classic and emerging concepts for the roles of the GH-IGF-IGFBP axis in the etiopathophysiology, treatment, and prevention of diabetic renal disease.
Keywords: Diabetic nephropathy, IGF, IGFBPs, IGF receptor
Introduction
Diabetic nephropathy (DN) is defined as proteinuria resulting from reversible endovascular damage of the renal filtration capacity by long-standing diabetes mellitus. DN remains the most common origin for progression through chronic renal insufficiency to end-stage renal disease (ESRD) in developed countries. DN is the largest contributor to the total cost of diabetes care worldwide, accounting for approximately 40% of all new patients placed on dialysis therapy (1). The vascular damage from diabetes usually involves non-renal tissues too. Thus, diabetic patients with albuminuria possess higher risk of developing myocardial infarctions, cerebrovascular accidents, severe progressive retinopathy, and peripheral and autonomic neuropathy (1).
The earliest clinical marker of DN is the appearance of small amounts of serum proteins in the urine detected by microscopy or dipstick testing. The physiologic range of proteinuria in children is below 100 mg/m2 daily (<30 mg over 24 hours for adults). An intermediate degree of proteinuria ranges 100–1,000 mg/m2 daily (30–300 mg over 24 hours for adults), and the nephrotic range is above 1,000 mg/m2 daily (>300 mg over 24 hours for adults). Microalbuminuria (MA) can be defined as overnight albumin excretion at least 20 μg/min for two consecutive visits.
Prevalence of microalbuminuria (MA) in children with type 1 diabetes has been reported to vary between 7–20% (1,3). The growing magnitude of DN at all ages—associated with huge monetary and social costs—drive the search for early markers of hyperglycemia-induced damage and multiple efforts to prevent this disease.
DN results from the interplay of metabolic and haemodynamic factors in the renal microcirculation (1). Prior to the onset of overt proteinuria, specific changes occur in renal functions, causing renal hyperfiltration, hyperperfusion, and increasing capillary permeability to macromolecules. Diffuse expansion of the mesangial matrix is considered the hallmark pathological feature of established DN in humans (4, 5, 6, 7). Tyrosine kinase growth factors have emerged as logical mechanisms contributing to the etipathophysiology of diabetic kidney disease (8).
The growth hormone (GH)–insulin-like growth factor (IGF)–IGF binding protein (IGFBP) axis and related growth factor families [transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), and epidermal growth factor (EGF)] demonstrate significant actions on the development of experimental diabetic kidney disease through defined intra-renal systems. Recently, new data have emerged supporting the concept that these growth factors initiate the earliest renal changes associated with hyperglycemia (9, 10).
The IGF-IGFBP-IGFBP-rP superfamily
IGF-I, IGF-II, their high-affinity binding proteins (IGFBP-1 through -6), low affinity IGFBP related peptides (IGFBP-rP1 through -4), and IGFBP proteases comprise a complex system which exerts fundamental regulation on growth and carbohydrate metabolism. IGFs exert their mitogenic actions primarily via the type 1 IGF receptors (IGF-1R), heterotetrameric tyrosine kinases on the cell surface. IGFBPs represent the largest family of proteins that bind IGFs with high affinity and specificity. Cellular actions are determined by the level of free, unbound IGF ligand.
Levels of free IGFs in a system are determined by rates of IGF production, IGF clearance, and degree of affinity to the IGFBPs (regulated by IGFBP turnover) (12). Since 1993, several IGF-independent actions have been discovered for IGFBPs related to survival, proliferation, and metabolism. Although specific IGFBP receptors have not been identified, at least 14 IGFBP-specific binding partners have been reported. IGFBP-1 interacts with α5β1 integrin, influencing cell adhesion and migration. IGFBP-2, -3, -5, and -6 have heparin-binding domains and can bind glycosaminoglycans. IGFBP-3 and -5 have carboxyl-terminal basic motifs incorporating heparin-binding and additional basic residues that interact with the cell surface and matrix (transferrin, type 1 collagen) as well as the nuclear transporter importin-β. Binding to serine/threonine kinase receptors has been reported for both IGFBP-3 and -5. Other cell surface proteins interact with IGFBPs, but have not been characterized as functional receptors.
The most abundant IGFBP species in the human peripheral circulation, IGFBP-3 is a multi-functional protein that promotes apoptosis and insulin resistance in diverse cell types via IGF-dependent and IGF-independent mechanisms. In vitro IGFBP-3 binds retinoid X receptor (RXR)-α to modulate nuclear signaling, binds Smad proteins to regulate TGF-β signaling, and exerts specific phosphatase-like activity to deactivate several cytosolic phosphorelays which are activated by tyrosine kinase receptors (such as IGF-1R and the VEGF receptors) (61, 62). Since IGFBPs regulate cell functions by diverse mechanisms, manipulation of IGFBP-regulated pathways may offer diagnostic and therapeutic opportunities (13).
IGFBP proteases comprise another distinct level for the regulation of free IGFs and IGFBP actions. IGFBP proteolysis generates bioactive IGFBP fragments. Several IGFBP-related proteins (IGFBP-rP) share significant homology with IGFBPs but bind IGFs with lower affinity. IGFBP-rPs probably exert their greatest effects under the conditions of increased IGFBP proteolysis which generate IGFBP fragments.
The local IGF-IGFBP environment changes significantly in response to the diabetic mileau (13,15,16). Serum IGF-I levels correlate positively with serum IGFBP-3 and IGFBP-5 levels but negatively with serum IGFBP-1, IGFBP-4 and HbA(1c) levels in patients with diabetes (15,16).
The growing body of evidence from models of diabetic renal hypertrophy and diabetic glomerulosclerosis reveals: (1) changes in local IGF-I regulation, and (2) existence of a TGF-β loop in the diseased kidney (15). The specific mechanisms by which insulin and glucose mediate increases of IGF-I and IGFBPs in the kidney—related to the development of DN—remain the focus of intense investigation.
IGF actions in kidney cells
Proliferation of resident glomerular cells and the accumulation of mesangial matrix are histological abnormalities, which are observed during the course of many progressive glomerular diseases, including DN (17). Figure 1 summarizes the known effects of IGF-I in the kidney at the cellular level.
Fig. 1.
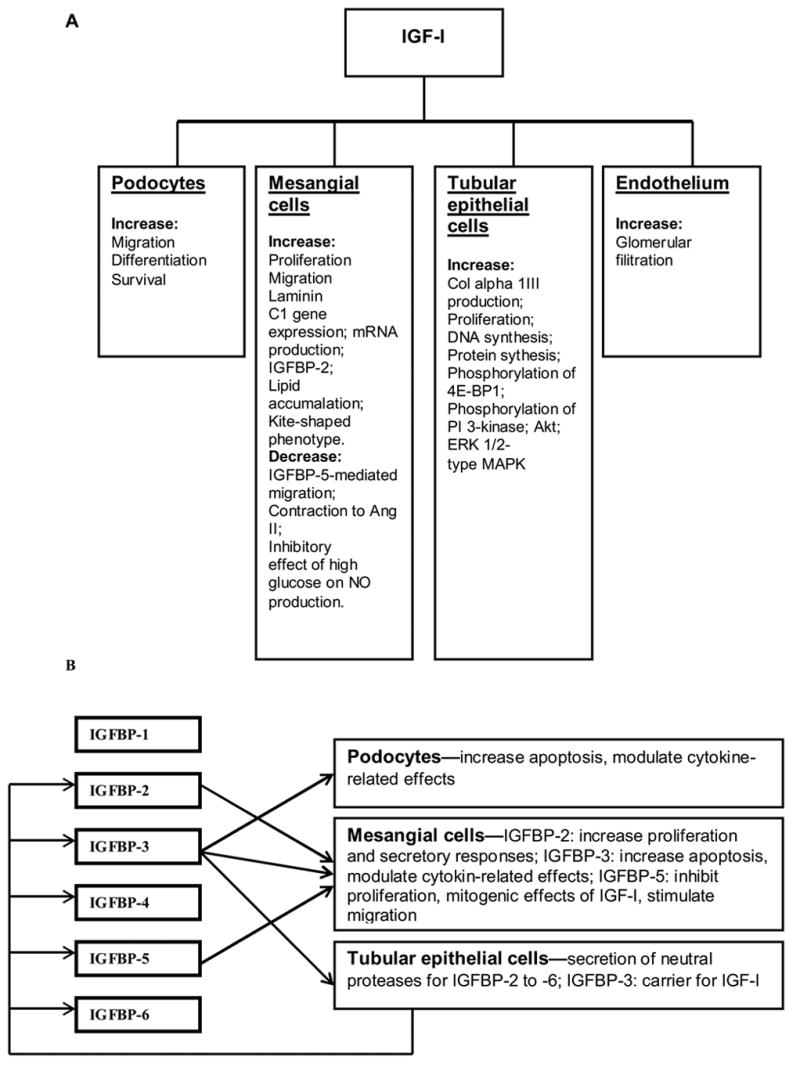
Summary of renal actions of IGF-I (A) and IGFBPs (B)
Mesangial cells (MC)
The MCs significantly contribute to the development of DN. IGF-I binding to its type 1 receptors does stimulate MC proliferation (18). IGF-I also stimulates MC migration by inducing a kite-shaped phenotype with β-actin-rich, leading lamella that propel the cell along an IGF-I concentration gradient (19). IGF-I-induced MC migration requires interaction between αvβ3 integrins and fibronectin (20). Treatment of MC with IGF-I results in intracellular lipid accumulation (21). IGF-I increased laminin C1 mRNA abundance in rat MC. The ability of high glucose concentrations and IGF-I to increase laminin C1 promoter activity in cultured MC—and the suppression of glucose actions by the nitric oxide (NO) donor SNAP—suggest how laminin gamma1 chain synthesis may be regulated in the glomerulus of a diabetic subject (22). It is known that nitric oxide NO contributes to the alterations in glomerular hemodynamics and extracellular matrix accumulation observed in DN. High glucose concentrations directly inhibit NO production by MC. One potential mechanism, which elucidates the development of DN could relate to IGF inhibitory effect of high glucose on NO production by MC (22).
IGFBPs significantly modify IGF action in the MC. We recently reported that IGFBP-3 mediates mesangial cell apoptosis, in part by blockade of Akt phosphorylation on threonine 308 (24). IGFBP-5 inhibits MC proliferation by direct activation of the IGFBP-5 receptor and by inhibiting the mitogenic effects of IGF-I (21). Chronic treatment of IGFBP-5 stimulates MC migration by a different mechanism from IGF-I. IGFBP-5 induces a spider-like morphology that migrates using a β1 integrin. IGFBP-5–independent effects are mediated by the heparin-binding domain, which binds to a putative IGFBP-5 receptor (25). Lipid accumulation within the MC interferes with the signal transduction response to IGFBP-5. IGFBP-5 fails to activate cdc42, a Rho GTPase required for IGFBP-5-mediated MC migration (21). In addition, MC treated with IGF-I show reduced contraction to angiotensin II (22). Hyperglycemia, as seen in diabetes, may increase MC IGF-I sensitivity by reducing IGFBP-2 expression, in turn increasing IGF-induced proliferative and secretory responses that contribute to the development of diabetic glomerulosclerosis (26).
Glomerular epithelial cells (podocytes)
Podocyte structural changes also contribute to the pathogenesis of albuminuria in diabetes. Hyperglycemic conditions limit the protective role of IGF-I against podocyte apoptosis. Longitudinal data provide evidence for an association between podocyte loss and albumin excretion rate, but whether cellular changes are a response to, a cause of, or concomitant with the progression of nephropathy remains uncertain (26). IGFBP-3 can facilitate podocyte apoptosis (Fig. 2).
Figure 2.
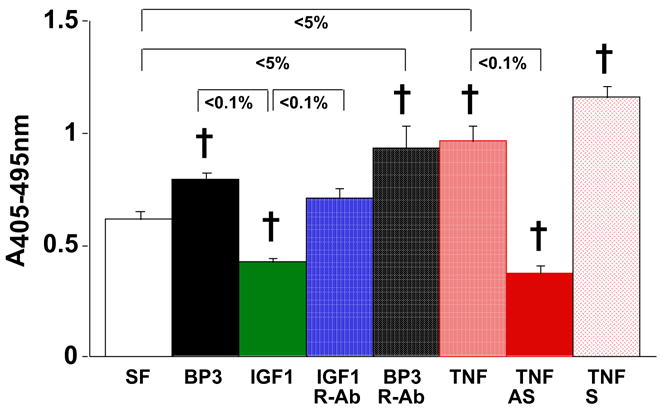
Oligonucleosome ELISA of rat glomeruli epithelial cell lysates, with Roche Cell Death Detection Kit performed according to the manufacturer’s instructions. Rat glomeruli epithlial cells were generously provided by Prof. H. Abboud, UTHSCSA Division of Nephrology. Monolayers were grown to 80–90% confluence in 60 mm dishes. Experimental media was replaced to serum-free (SF) conditions with standard (5 mM) glucose. Cells were harvested with trypsin-EDTA, pelleted, and washed with 1X phosphate-buffered saline. Under serum-free (SF) conditions, exogenous recombinant human IGFBP-3 exposure at 1 mg/mL for 24 h induces apoptosis in the presence of neutralizing IgG to the type 1 IGF receptor (n=8; †p<5% vs. SF control and other p values as shown by two-tailed Student’s t test). As described using mesangial cells in ref. 24, pre-treatment of the rat podocytes with anti-sense IGFBP-3 oligo (but not sense oligo) blocked apoptosis induced by tumor necrosis factor (TNF)-α. As observed with other cell types, IGFBP-3 can induce podocyte apoptosis acting independently of the type 1 IGF receptor.
Tubular epithelial cells
Chronic interstitial fibrosis, which follows the onset of glomerular proteinuria, significantly contributes to progressive renal failure in DN. Connective tissue growth factor (CTGF) is produced by tubular epithelia, has an IGF-binding domain, and does bind IGF-I in vitro. In NRK-49F cells, IGF-I increased the activity of CTGF toward the expression of collagen alpha type 1III (22). IGF-1R demonstrated in the Madin-Darby canine kidney cells are also involved in DNA synthesis and cell proliferation (28).
When added to cultures of opossum kidney cells, IGF-I is internalized and transported to distinct intracellular compartments that depend on the cell location within the monolayer. Similar to IGF-I, IGFBP-3 is also internalized and accumulates in the endosomal compartment in resting cells, whereas it is targeted to the nucleus in proliferating and apoptotic cells. IGFBP-3, which contains a nuclear localization signal, may ferry IGF-I to the nuclear compartment (28).
In murine proximal tubular epithelial cells, IGF-I stimulates protein synthesis, augments phosphorylation of 4E-BP1, and promotes the dissociation of eIF4E from 4E-BP1. IGF-I stimulates the activities of PI 3-kinase, Akt, and the ERK1/2-type MAPK. ERK activation by IGF-I is also PI 3-kinase dependent. Transfection with the Thr37,46-->Ala37,46 mutant of 4E-BP1 shows that phosphorylation of Thr37,46 residues is required for IGF-I induction of protein synthesis in these cells (29). MDCK cells also secrete several specific proteases, which cleave IGFBP-2, -3, -4, -5 and -6. The side-specific secretion of several distinct IGFBP proteases with partially overlapping IGFBP specificities may be another level in the regulation of IGF-dependent epithelial functions (31).
Endothelial cells
The alterations in the microvascular system of diabetes mellitus patients are responsible for the most devastating complications. In the kidney, microangiopathy thickens the glomerular capillary basement membrane, expands the mesangial matrix, and thickens the tubular basement membrane (32). Specific biochemical pathways linking hyperglycaemia to microvascular changes have been proposed. These pathways are linked to altered synthesis of growth factors. Makino et al. reported that both enhanced NO synthesis by ecNOS in afferent arterioles and glomerular endothelial cells, and increased expression of IGF-1R, can cause glomerular hyperfiltration (33).
GH-IGF-IGFBP superfamily action in the animal models of DN
GH and IGFs have measurable effects on the development of diabetic kidney disease in experimental animal models through changes in a complex intrarenal system (34). The glomeruli of mice transgenic for bovine GH are disproportionately enlarged as a function of either kidney or body weight. Glomerular size correlates with mesangial sclerosis and the urinary albumin/creatinine ratio. Of note, mice transgenic for IGF-I did not develop glomerulosclerosis, even though glomerular size significantly increased (34). Experimental diabetic kidney disease is characterized by renal hypertrophy associated with increased tissue concentrations of IGF-I. For diabetic kidney disease in the Psammomys model of T2DM, significant increases in kidney/body-weight ratio associated with elevated renal IGF-I protein level as well as mRNA and protein levels of renal IGFBP-1 (35). IGFBP-1 negatively feeds back on GH secretion (37). A line of transgenic mice expressing human IGFBP-1 mRNA in the liver developed glomerulosclerosis without glomerular hypertrophy. These changes are potentially related to a decrease in IGF-I availability and/or to an IGF-I–independent role of IGFBP-1 (37).
IGF-I in experimental animals is localized exclusively to principal cells of cortical and medullary collecting ducts (38). IGF-1R mRNA and IGF-2R mRNA levels significantly increase in the diabetic kidney. IGF-1R is able to transduce mitogenic signals upon activation of its tyrosine kinase domain. Increased IGF-2R expression in the diabetic kidney may be important for the intracellular transport and packaging of lysosomal enzymes, although a role for this receptor in signal transduction cannot be excluded (39). Taking into consideration that IGF bioavailability is higher in DN, finding receptors overexpressed may explain the IGF hyperaction at the glomerular level.
Experimental diabetic animals treated with insulin reverse their renal hypertrophy. At the same time, the changes occurring in the renal IGF-I system can also reverse. However, it is still unclear whether the effect of insulin treatment is due to lowering of serum glucose level or increased circulating insulin levels (37). Landau et al. showed that in streptozotocin-induced diabetic rats, GH accelerated the course of diabetic kidney disease (40). In rats with DN (but not in normal animals), IGF-I–containing binding protein complexes undergo glomerular ultrafiltration, allowing the peptide to interact with IGF receptors in apical tubular membranes. By this route, ultrafiltered IGF-I may increase tubular epithelial cell sodium absorption in overt DN, thereby contributing to the sodium (and fluid) retention commonly observed in patients with severe DN. IGFBPs-1 to -4 are all present in diabetic rat glomerular ultrafiltrate, but IGFBP-2 levels are greater than those of each of the other IGFBPs (42). IGF axis changes in diabetic animals with kidney complications depend on age (42) and time course of diabetes (43,45).
Thus, in different animal models for DN, kidney hypertrophy, hyperfiltration, and albuminuria strongly associate with increased tissue levels of IGF-I and IGFBPs. Contributions vary from the other tissue components during development of DN. The tissue sensitivity to IGFs (depending on IGF-1R and IGF-2R expression) accelerates by GH treatment and partially reverses by insulin treatment.
Clinical research overview
The involvement of the GH-IGF axis in diabetic renal complications has been recognized for a long time. The kidney is a site of IGF-I production, which mediates effects on kidney growth and function (13,45). Advanced glycosylation end products are markedly increased in diabetic patients with ESRD. Free IGF-I and total IGFBP-3 levels in serum correlate inversely with hemoglobin A1c level (15). In adolescents with diabetes, MA is strongly associated with abnormalities of the GH-IGF-IGFBP system (45). MA patients display higher levels of urinary IGF-I, urinary GH, and plasma IGF-I than normoalbuminuric diabetic subjects (46).
Diabetic patients with MA have higher levels of urinary IGFBP-3 than those without MA. Spagnoli et al. (45) showed that urinary levels of IGFBP-3 directly correlate to urine albumin excretion (p < 0.001). The immunoreactive form of IGFBP-3 found in urine from patients with diabetes is an N-terminal 18 kD fragment (45). The urine of diabetic patients with micro- and macroalbuminuria contained little or no intact 40- to 46-kDa IGFBP-3, the usual glycosylated form in the circulation. In patients with DM, urinary IGFBP-3 protease activity in micro- and macroalbuminuric patients is significantly higher than that in normoalbuminuric patients (46).
Serum IGFBP-3 protease activity directly correlates to urinary IGFBP-3 levels (p < 0.001). Whether IGFBP-3 proteolysis (with production of low affinity IGFBP-3 fragments) facilitates the development of diabetic nephropathy remains to be shown (45). Shinada et al. (47) suggest that diabetic nephropathy is associated with IGFBP-3 proteolysis in urine. Since similar changes are not observed in patients’ sera, this likely reflects the changes in the kidney or urinary tract, resulting in increased local IGF bioavailability, and therefore may contribute to the structural changes of DN. Figure 3 summarizes these data. Increased urinary IGFBP-3 in patients with MA was related to incipient nephropathy, and not simply to poorer metabolic control. Indeed, patients with MA had higher levels of urinary IGFBP-3 even when compared to patients without MA matched for metabolic control (45). It is plausible that increased IGBPP-3 proteolysis within the kidney of patients with DN contributes to IGF bioavailability, MC proliferation, and matrix production. Fragments of IGFBP-3 retain biologic activity and facilitate albuminuria through increased apoptosis of glomerular epithelial and endothelial cells. Table 1 reviews published data on the IGF-IGFBP profiles in human subjects. Serum levels of IGF-I fall in diabetic people. This common finding in DM may be due to increased IGFBP-1 production by liver in response to increased circulating GH levels. Circulating IGF-I level is higher vs. DM patients without albuminuria. IGFBP-3 proteolysis would be expected to increase IGF-I turnover, thereby shortening IGF-I half-life.
Figure 3.

Changes in GH/IGF/IGFBP-3 axis in patient with DN
Table 1.
IGF-IGFBP axis profile in adults with DN vs. healthy people and diabetic patients without DN
| Serum levels | Urine levels | |
|---|---|---|
| IGF-I | ↓ H; ↑ DM | ↑ DM |
| IGF-II | ND | ND |
| IGFBP-1 | ↑ H | ND |
| IGFBP-2 | ND | ND |
| IGFBP-3 | ↓ H, DM | ↑ DM |
| IGFBP-4 | N for H | ND |
| IGFBP-5 | ↓ H | ND |
| IGFBP-6 | ND | ND |
H, healthy people; DM, diabetes without nephropathy; N, normal level; ND, no data in available literature.
Circulating IGF-I and IGFBP-1 levels reflect the partial GH resistance resulting from the malnourished state of patients with chronic renal failure. GH or rhIGF-I to overcome this resistance might improve the malnutrition by alleviating hypoproteinemia and normalizing the circulating amino acid profile in uremic patients on hemodialysis (19). DN may also contribute to bone loss from a disturbed IGF system (49,50). The contributions of perturbed IGF axis homeostasis in multiple damaged organs of patients with DN offer several therapeutic targets.
DN treatment & the GH/IGF/IGFBP axis
Therapies to restore the GH-IGF-IGFBP homeostasis have been reported in randomized clinical trials. Administration of recombinant human IGF-I (rhIGF-I) improved glycemic control as well as hepatic and muscle insulin sensitivity in adults with T2DM. RhIGF-I improves hyperglycemia by inhibiting hepatic gluconeogenesis (57). It remains unknown whether this approach to glycemic control is more beneficial for the kidneys than insulin administration alone.
GH therapy safely and effectively reduces linear growth failure in children with ESRD not due to DN (51). In adults with DN, GH may exaggerate the pathological process via elevated IGF-I action. Transgenic mice expressing a GH antagonist are protected from diabetes and GH-induced nephropathy (55).
PTR-3173 is a novel somatostatin analogue which exerts a prolonged inhibitory action selectively on the GH-IGF axis, but not on insulin secretion. Landau et al. investigated the potential effect of this agent on development of markers for DN in the non-obese diabetic (NOD) mouse model of type 1 diabetes. GH antagonism by PTR-3173 blunted renal/glomerular hypertrophy, albuminuria, and glomerular filtration rate in diabetic NOD mice. These phenomena were apparently associated with the prevention of renal IGF-I accumulation (52, 52).
Furthermore, new data obtained in mice lacking the GH receptor (GHR)/GH-binding protein (GHBP) gene shown they are protected against diabetes-induced renal changes. The recent development of specific inhibitors of GH action (i.e., specific GHR antagonists, or GHRAs) offers the possibility that this group of inhibitors may be used as therapies in conditions where GH and IGFs exert pathophysiological roles, such as the late complications of diabetes (49). Recent data from studies in diabetic mice, treated with a GHRA (G120K-PEG) from the onset of diabetes, showed normalization of the diabetes-associated renal hypertrophy and glomerular enlargement. Most important, this GHRA lowered the diabetes-induced rise in urinary albumin excretion (UAE). In addition, late intervention with GHRAs—alone or in combination with angiotensin-converting enzyme inhibitors in NOD mice with manifest renal changes—showed regression in some diabetes-associated renal changes (e.g. UAE and renal hypertrophy). These promising, pre-clinical data strongly suggest that GHR blockade is a viable, new modality for treating or preventing diabetic renal complications. Studies are underway to characterize fully the clinical potential of GHRAs for DN and related conditions (46).
Comparing the effects of somatostatin analogue with an ACE inhibitor (ACEi), the standard of care for patients with DN, showed that somatostatin analogues exert beneficial effects in most parameters of diabetic kidney disease to the same extent as the ACEi. Indeed, the ACEi enalapril had no effect on renal hypertrophy and did not decrease deposition of mesangial type IV collagen (58). Bach et al. (54, 57) demonstrated that aminoguanidine ameliorates changes in the IGF system in experimental DN. Aminoguanidine inhibits the effects of diabetes on renal mRNA for IGF-I, IGFBP-1, and IGFBP-4, but not IGFBP-5. These results suggest that amelioration of changes in the renal IGF system by aminoguanidine may contribute to its reno-protective effects.
Summary
GH-IGF-IGFBP axis components play key roles in the maintenance of normal renal function and the development of DN. IGF-I action increases in the diabetic kidney in an autocrine/paracrine manner that promotes matrix production, mesangial cell proliferation and migration. Although this process is opposed by IGFBPs through binding to IGFs and IGF-independent mechanisms, the balance in DN favors IGF actions. This pathology is facilitated by over-expression of IGF-1R and reduced IGFBP-2 expression in the presence of high ambient glucose level. Over-expression of IGFBP-1 in diabetes may also contribute to glomerulosclerosis.
High ambient glucose also increases renal IGFBP-3 levels, a significant contributor to podocyte apoptosis. Strong evidence associates podocyte loss and albuminuria. Together with endothelial damage, podocyte loss leads to glomerular hyperfiltration and proteinuria. Tubular epithelial damage from high ambient glucose, hyperfiltration, and proteinuria contributes to the interstitial fibrosis causing renal failure. CTGF plays an important role too and is stimulated by renal IGF-I. IGFs also directly stimulate tubular epithelial cell DNA, protein synthesis and proliferation. Further research may validate viable approaches to these problems.
GH antagonism is emerging as a viable approach to prevent and treat DN in adults. RhIGF-I improves glycemic control but cannot replace exogenous insulin therapy in type 1 diabetic patients. Well established ACEi therapy for DN acts in part by inhibiting IGF-I-induced activation of bradykinin via Erk 1 and 2 (59). Several unanswered questions related to renal GH-IGF-IGFBP etiopathophysiology promise fruitful research for addressing the problems of DN, which is a growing problem across the lifespan.
LIST OF ABBREVIATINS
- ACEi
Angiotensin-converting enzyme inhibitor
- CTGF
Connective tissue growth factor
- DN
Diabetic nephropathy
- 4E-BP1
Eukaryotic initiation factor 4E-binding protein
- ecNOS
Endothelial constitutive nitric oxide synthase
- EGF
Epidermal growth factor
- eIF4E
Eukaryotic initiation factor 4E
- ESRD
End-stage renal disease
- GH
Growth hormone
- IGF
Insulin-like growth factor
- IGFBP
IGF binding protein
- IGFBP-rP
IGFBP related peptides
- IGF-1R
Type 1 IGF receptors
- MA
Microalbuminuria
- MAPK
Mitogen-activated protein kinase
- MC
Mesangial cells
- MDCK
Madin-Darby Canine Kidney cell line
- NO
Nitric oxide
- NRK-49F cells
Rat renal interstitial cells
- PI 3 kinase
Phosphatidylinositol 3-kinase
- rhIGF-I
Recombinant Insulin-like growth factor
- Rho GTPase
Rho guanosine triphosphatases
- SNAP
Nitric oxide -donors S-nitroso-N-acetylpenicillamine
- T2DM
Type 2 diabetes mellitus
- TGF-β
Transforming growth factor-β
- VEGF
Vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chiarelli F, Trotta D, Verrotti A, Mohn A. Kidney involvement and disease in patients with diabetes. Panminerva Med. 2003;45:23–41. [PubMed] [Google Scholar]
- 2.Campbell F. Microalbuminuria and nephropathy in insulin-dependent diabetes mellitus. Arch Dis Child. 1995;73:4–7. doi: 10.1136/adc.73.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quattrin T, Waz WR, Duffy L, Sheldon MW, Campos SP, Albini CH, et al. Microalbuminuria in an adolescent cohort with insulin-dependent diabetes mellitus. Clinica Pediatrica. 1995;34:12–17. doi: 10.1177/000992289503400103. [DOI] [PubMed] [Google Scholar]
- 4.Mauer S, Steffes M, Brown D. The kidney in diabetes. Am J Med. 1981;70:603–612. doi: 10.1016/0002-9343(81)90582-9. [DOI] [PubMed] [Google Scholar]
- 5.Mauer S, Steffes M, Ellis E, Sutherland D, Brown DM, Goetz FC. Structural-functional relationships in diabetic nephropathy. J Clin Invest. 1984;74:1143–1155. doi: 10.1172/JCI111523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osterby R. A quantitative electron microscopic study of mesangial. regions in glomeruli from patients with short-term juvenile diabetes mellitus. Lab Invest. 1973;29:99–110. [PubMed] [Google Scholar]
- 7.Steffes MW, Osterby R, Chavers B, Mayer SM. Mesangial expansion as a central mechanism for loss of kidney function in diabetic patients. Diabetes. 1989;38:1077–1081. doi: 10.2337/diab.38.9.1077. [DOI] [PubMed] [Google Scholar]
- 8.Karl M, Potier M, Schulman I, Rivera A, Werner H, Fornoni A, et al. Autocrine activation of the local insulin-like growth factor I system is up-regulated by estrogen receptor (ER)-independent estrogen actions and accounts for decreased ER expression in type 2 diabetic mesangial cells. Endocrinology. 2005;146:889–900. doi: 10.1210/en.2004-1121. [DOI] [PubMed] [Google Scholar]
- 9.Flyvbjerg A. Putative pathophysiological role of growth factors and cytokines in experimental diabetic kidney disease. Diabetologia. 2000;43:1205–1223. doi: 10.1007/s001250051515. [DOI] [PubMed] [Google Scholar]
- 10.Flyvbjerg A, Frystyk J, Osterby R, Orskov H. Kidney IGF-I and renal hypertrophy in GH-deficient diabetic dwarf rats. Am J Physiol. 1992;262:E956–962. doi: 10.1152/ajpendo.1992.262.6.E956. [DOI] [PubMed] [Google Scholar]
- 11.Flyvbjerg A, Khatir D, Jensen L, Dagnæs-Hansen F, Gronbaek H, Rasch R. The involvement of growth hormone (GH), insulin-like growth factors (IGFs) and vascular endothelial growth factor (VEGF) in diabetic kidney disease. Curr Pharm Des. 2004;10:3385–3394. doi: 10.2174/1381612043383106. [DOI] [PubMed] [Google Scholar]
- 12.Ferry RJ, Jr, Cohen P. The insulin-like growth factor axis in pediatrics. Clin Pediatr Endocrinol. 1999;8:1–10. [Google Scholar]
- 13.Firth S, Baxter R. Cellular actions of the insulin-like growth factor binding proteins. Endocrine Reviews. 2002;23:824–854. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 14.Raz I, Wexler I, Weiss O, Flyvbjerg A, Segev Y, Rauchwerger A, et al. Role of insulin and the IGF system in renal hypertrophy in diabetic Psammomys obesus (sand rat) Nephrol Dial Transplant. 2003;18:1293–1298. doi: 10.1093/ndt/gfg170. [DOI] [PubMed] [Google Scholar]
- 15.Gambaro G, Baggio B. Growth factors and the kidney in diabetes mellitus. Crit Rev Clin Lab Sci. 1998;35:117–151. doi: 10.1080/10408369891234174. [DOI] [PubMed] [Google Scholar]
- 16.Garay-Sevilla M, Nava L, Malacara J, Nava LE, Wrobel-Zasada K, Castro-Rivas A, et al. Advanced glycosylation end products (AGEs), insulin-like growth factor-1 (IGF-1) and IGF-binding protein-3 (IGFBP-3) in patients with type 2 diabetes mellitus. Diabetes Metab Res Rev. 2000;16:106–113. doi: 10.3904/kjim.2007.22.3.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fornoni A, Li H, Foschi A, Stiker LJ. Hepatocyte growth factor, but not insulin-like growth factor I, protects podocytes against cyclosporin A-induced apoptosis. Am J Pathol. 2001;158:275–280. doi: 10.1016/S0002-9440(10)63966-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abrass C, Raugi G, Gabourel L, Lovet DH. Insulin and insulin-like growth factor I binding to cultured rat glomerular mesangial cells. Endocrinology. 1988;123:2432–2439. doi: 10.1210/endo-123-5-2432. [DOI] [PubMed] [Google Scholar]
- 19.Sanaka T. Nutritional effect of dialysis therapy. Artif Organs. 2003;27:224–226. doi: 10.1046/j.1525-1594.2003.07215.x. [DOI] [PubMed] [Google Scholar]
- 20.Abrass C, Berfield A, Andress D. Heparin binding domain of insulin-like growth factor binding protein-5 stimulates mesangial cell migration. Am J Physiol. 1997;273:F899–F906. doi: 10.1152/ajprenal.1997.273.6.F899. [DOI] [PubMed] [Google Scholar]
- 21.Berfield A, Andress D, Abrass C. IGF-1-induced lipid accumulation impairs mesangial cell migration and contractile function. Kidney Int. 2002;62:1229–1237. doi: 10.1111/j.1523-1755.2002.kid578.x. [DOI] [PubMed] [Google Scholar]
- 22.Phillips S, DeRubertis F, Craven P. Regulation of the laminin C1 promoter in cultured mesangial cells. Diabetes. 1999;48:2083–2089. doi: 10.2337/diabetes.48.10.2083. [DOI] [PubMed] [Google Scholar]
- 23.Trachtman H, Koss I, Bogart M, Abramowitz J, Futterweit S, Franki N, et al. High glucose enhances growth factor-stimulated nitric oxide production by cultured rat mesangial cells. Res Commun Mol Pathol Pharmacol. 1998;100:213–225. [PubMed] [Google Scholar]
- 24.Vasylyeva TL, Chen X, Ferry RJ., Jr Insulin-like growth factor binding protein-3 mediates cytokine-induced mesengial cell apoptosis. Growth Hormone & IGF Research. 2005;15(3):207–214. doi: 10.1016/j.ghir.2005.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berfield A, Spicer D, Abrass C. Insulin-like growth factor I (IGF-I) induces unique effects in the cytoskeleton of cultured rat glomerular mesangial cells. J Histochem Cytochem. 1997;45:583–593. doi: 10.1177/002215549704500410. [DOI] [PubMed] [Google Scholar]
- 26.Horney M, Shirley D, Kurtz D, Rosenzweig SA. Elevated glucose increases mesangial cell sensitivity to insulin-like growth factor I. Am J Physiol. 1998;274:F1045–1053. doi: 10.1152/ajprenal.1998.274.6.F1045. [DOI] [PubMed] [Google Scholar]
- 27.White K, Bilous R, Marshall S, El Nahas M, Remuzzi G, Piras G, et al. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes. 2002;51:3083–3089. doi: 10.2337/diabetes.51.10.3083. [DOI] [PubMed] [Google Scholar]
- 28.Sukegawa I, Hizuka N, Takano K, Asakawa K, Shizume K. Characterization of insulin-like growth factor I receptors on Madin-Darby canine kidney (MDCK) cell line. Endocrinol Jpn. 1987;34:339–346. doi: 10.1507/endocrj1954.34.339. [DOI] [PubMed] [Google Scholar]
- 29.Li W, Fawcett J, Widmer HR, Fielder PJ, Rabkin R, Keller GA. Nuclear transport of insulin-like growth factor-I and insulin-like growth factor binding protein-3 in opossum kidney cells. Endocrinology. 1997;138:1763–1766. doi: 10.1210/endo.138.4.5176. [DOI] [PubMed] [Google Scholar]
- 30.Senthil D, Choudhury G, Abboud H, Sonenberg N, Kasinath BS. Regulation of protein synthesis by IGF-I in proximal tubular epithelial cells. Am J Physiol Renal Physiol. 2002;283:F1226–1236. doi: 10.1152/ajprenal.00109.2002. [DOI] [PubMed] [Google Scholar]
- 31.Shalamanova L, Kubler B, Scharf JG, Braulke T. MDCK cells secrete neutral proteases cleaving insulin-like growth factor-binding protein-2 to -6. Am J Physiol Endocrinol Metab. 2001;281:E1221–1229. doi: 10.1152/ajpendo.2001.281.6.E1221. [DOI] [PubMed] [Google Scholar]
- 32.Tsilibary E. Microvascular basement membranes in diabetes mellitus. J Pathol. 2003;200:537–546. doi: 10.1002/path.1439. [DOI] [PubMed] [Google Scholar]
- 33.Makino H, Sugimoto H, Shikata K. Diabetic nephropathy—recent advances in its mechanism and treatment. Nippon Rinsho. 1999;57:590–600. [PubMed] [Google Scholar]
- 34.Flyvbjerg A. Potential use of growth hormone receptor antagonist in the treatment of diabetic kidney disease. Growth Horm IGF Res. 2001;11:S115–119. doi: 10.1016/s1096-6374(01)80019-8. [DOI] [PubMed] [Google Scholar]
- 35.Doi T, Striker LJ, Gibson CC, Agodoa LY, Brinster RL, Striker GE. Glomerular lesions in mice transgenic for growth hormone and insulinlike growth factor-I. I. Relationship between increased glomerular size and mesangial sclerosis. Am J Pathol. 1990;137:541–552. [PMC free article] [PubMed] [Google Scholar]
- 36.Raz I, Wexler I, Weiss O, Flyvbjerg A, Segev Y, Rauchwerger A, et al. Role of insulin and the IGF system in renal hypertrophy in diabetic Psammomys obesus (sand rat) Nephrol Dial Transplant. 2003;18:1293–1298. doi: 10.1093/ndt/gfg170. [DOI] [PubMed] [Google Scholar]
- 37.Cingel-Ristic V, Schrijvers BF, van Vliet AK, Rash R, Han V, Drop SL, et al. Kidney growth in normal and diabetic mice is not affected by human insulin-like growth factor binding protein-1 administration. Exp Biol Med. 2005;230:135–143. doi: 10.1177/153537020523000208. [DOI] [PubMed] [Google Scholar]
- 38.Doublier S, Seurin D, Fouqueray B, Verpont C, Callard P, Striker LJ, et al. Glomerulosclerosis in mice transgenic for human insulin-like growth factor-binding protein-1. Kidney Int. 2000;57:2299–2307. doi: 10.1046/j.1523-1755.2000.00090.x. [DOI] [PubMed] [Google Scholar]
- 39.Bortz J, Rotwein P, DeVol D, Bechtel PJ, Hansen VA, Hammerman MP. Focal expression of insulin-like growth factor I in rat kidney collecting duct. J Cell Biol. 1988;107:811–819. doi: 10.1083/jcb.107.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werner H, Shen-Orr Z, Stannard B, Burguera B, Roberts CT, Jr, LeRoith D. Experimental diabetes increases insulin-like growth factor I and II receptor concentration and gene expression in kidney. Diabetes. 1990;39:1490–1497. doi: 10.2337/diab.39.12.1490. [DOI] [PubMed] [Google Scholar]
- 41.Landau D, Israel E, Rivkis I, Kachko L, Schrijvers BF, Flyvbjerg A, et al. The effect of growth hormone on the development of diabetic kidney disease in rats. Nephrol Dial Transplant. 2003;18:694–702. doi: 10.1093/ndt/gfg142. [DOI] [PubMed] [Google Scholar]
- 42.Wang S, Lapage J, Hirschberg R. Glomerular ultrafiltration of IGF-I may contribute to increased renal sodium retention in diabetic nephropathy. J Lab Clin Med. 1999;134:154–160. doi: 10.1016/s0022-2143(99)90120-8. [DOI] [PubMed] [Google Scholar]
- 43.Phillip M, Werner H, Palese T, Kowarski AA, Stannard B, Bach LA, et al. Differential accumulation of insulin-like growth factor-I in kidneys of pre- and postpubertal streptozotocin-diabetic rats. J Mol Endocrinol. 1994;12:215–224. doi: 10.1677/jme.0.0120215. [DOI] [PubMed] [Google Scholar]
- 44.Miyatake N, Shikata K, Wada J, Sugimoto H, Takahashi SI, Makino H. Differential distribution of insulin-like growth factor-1 and insulin-like growth factor binding proteins in experimental diabetic rat kidney. Nephron. 1999;81:317–323. doi: 10.1159/000045299. [DOI] [PubMed] [Google Scholar]
- 45.Park I, Kiyomoto H, Alvarez F, Xu YC, Abboud HE, Abboud SL. Preferential expression of insulin-like growth factor binding proteins-1, -3, and -5 during early diabetic renal hypertrophy in rats. Am J Kidney Dis. 1998;32:1000–1010. doi: 10.1016/s0272-6386(98)70075-7. [DOI] [PubMed] [Google Scholar]
- 46.Spagnoli A, Chiarelli F, Vorwerk P, Boscherini B, Rosenfeld RG. Evaluation of the components of insulin-like growth factor (IGF)-IGF binding protein (IGFBP) system in adolescents with type 1 diabetes and persistent microalbuminuria: relationship with increased urinary excretion of IGFBP-3 18 kD N-terminal fragment. Clin Endocrinol. 1999;51:587–596. doi: 10.1046/j.1365-2265.1999.00842.x. [DOI] [PubMed] [Google Scholar]
- 47.Verrotti A, Cieri F, Petitti MT, Morgese G, Chiarelli F. Growth hormone and IGF-I in diabetic children with and without microalbuminuria. Diabetes Nutr Metab. 1999;12:271–276. [PubMed] [Google Scholar]
- 48.Shinada M, Akdeniz A, Panagiotopoulos S, Jerums G, Bach LA. Proteolysis of insulin-like growth factor-binding protein-3 is increased in urine from patients with diabetic nephropathy. J Clin Endocrinol Metab. 2000;85:1163–1169. doi: 10.1210/jcem.85.3.6486. [DOI] [PubMed] [Google Scholar]
- 49.Jehle PM, Jehle DR, Mohan S, Bochm BO. Serum levels of insulin-like growth factor system components and relationship to bone metabolism in Type 1 and Type 2 diabetes mellitus patients. J Endocrinol. 1998;159:297–306. doi: 10.1677/joe.0.1590297. [DOI] [PubMed] [Google Scholar]
- 50.Gambaro G, Baggio B. Growth factors and the kidney in diabetes mellitus. Crit Rev Clin Lab Sci. 1998;35:117–151. doi: 10.1080/10408369891234174. [DOI] [PubMed] [Google Scholar]
- 51.Bambola O, Kaskel F. Growth hormone therapy in chronic kidney disease. Growth, Genetics and Hormones. 2005;12:7–14. [Google Scholar]
- 52.Landau D, Segev Y, Afargan M. A novel somatostatin analogue prevents early renal complications in the nonobese diabetic mouse. Kidney Int. 2001;60:505–512. doi: 10.1046/j.1523-1755.2001.060002505.x. [DOI] [PubMed] [Google Scholar]
- 53.Segev Y, Landau D, Rasch R, Flyvbjerg A, Phillip M. Growth hormone receptor antagonism prevents early renal changes in nonobese diabetic mice. J Am Soc Nephrol. 1999;10:2374–2381. doi: 10.1681/ASN.V10112374. [DOI] [PubMed] [Google Scholar]
- 54.Bach LA. IGF-I and IGF binding proteins in diabetes-related kidney growth. Growth Regul. 1992;2:30–39. [PubMed] [Google Scholar]
- 55.Orskov H. Somatostatin, growth hormone, insulin-like growth factor-1, and diabetes: friends or foes? Metabolism. 1996;45:91–95. doi: 10.1016/s0026-0495(96)90094-3. [DOI] [PubMed] [Google Scholar]
- 56.Chen NY, Chen WY, Bellush L, Yang CW, Striker LJ, Striker GE, et al. Effects of streptozotocin treatment in growth hormone (GH) and GH antagonist transgenic mice. Endocrinology. 1995;136:660–667. doi: 10.1210/endo.136.2.7835300. [DOI] [PubMed] [Google Scholar]
- 57.Bach L, Dean R, Youssef S, Cooper ME. Aminoguanidine ameliorates changes in the IGF system in experimental diabetic nephropathy. Nephrol Dial Transplant. 2000;15:347–354. doi: 10.1093/ndt/15.3.347. [DOI] [PubMed] [Google Scholar]
- 58.Pennisi P, Gavrilova O, Setser-Portas J, Jou W, Santopietro S, Clemmons D. Recombinant Human Insulin-Like Growth Factor-I (rhIGF-1) treatment inhibits gluconeogenesis in a transgenic mouse model of type 2 Diabetes Mellitus (DM) Endocrinology. 2006 doi: 10.1210/en.2005-1556. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 59.Segev Y, Eshet R, Rivkis I, Hayat C, Kachko L, Phillip M, et al. Comparison between somatostatin analogues and ACE inhibitor in the NOD mouse model of diabetic kidney disease. Nephrol Dial Transplant. 2004;19:3021–3028. doi: 10.1093/ndt/gfh528. [DOI] [PubMed] [Google Scholar]
- 60.Alric Pecher C, Cellier E, Schanstra JP, Poirier B, Chevalier J, et al. Inhibition of IGF-I-induced Erk 1 and 2 activation and mitogenesis in mesangial cells by bradykinin. Kidney Int. 2002;62:412–421. doi: 10.1046/j.1523-1755.2002.00475.x. [DOI] [PubMed] [Google Scholar]
- 61.Chen X, Ferry RJ., Jr Novel actions of IGFBP-3 on intracellular signaling pathways in insulin-secreting cells. Growth Horm IGF Res. 2006;16:41–48. doi: 10.1016/j.ghir.2005.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franklin SL, Ferry RJ, Jr, Cohen P. Rapid insulin-like growth factor (IGF)-independent effects of IGF binding protein-3 on endothelial cell survival. J Clin Endocrinol Metab. 2003;88:900–907. doi: 10.1210/jc.2002-020472. [DOI] [PMC free article] [PubMed] [Google Scholar]