

Abstract
Following intraparenchymal injection of the dopamine (DA) neurotoxin 6-hydroxydopamine, we previously demonstrated passage of fluoresceinisothiocyanate labeled albumin (FITC-LA) from blood into the substantia nigra (SN) and striatum suggesting damage to the blood-brain barrier (BBB). The factors contributing to the BBB leakage could have included neuroinflammation, loss of DA neuron control of barrier function, or a combination of both. In order to determine which factor(s) was responsible, we assessed BBB integrity using the FITC-LA technique in wild-type (WT), tumor necrosis factor alpha (TNF-α) knockout (KO), and minocycline (an inhibitor of microglia activation) treated mice 72 hrs following treatment with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Compared with WT mice, TNF-α KO mice treated with MPTP, showed reduced FITC-LA leakage, decreased numbers of activated microglia, and reduced pro-inflammatory cytokines (TNF-α and interleukin 1β) associated with significant MPTP-induced DA neuron loss. In contrast, minocycline treated animals did not exhibit significant MPTP-induced DA neuron loss although their FITC-LA leakage, numbers of activated microglia, and MPTP-induced cytokines were markedly attenuated. Since both TNF-α KO and minocycline treatment attenuated MPTP-induced BBB dysfunction, microglial activation, and cytokine increases, but had differential effects on DA neuron loss, it appears that neuroinflammation and not DA neuron loss was responsible for disrupting the blood-brain barrier integrity.
Keywords: Parkinson’s disease, neuroinflammation, microglia, TNF-α, IL-1β, minocycline, endothelial cells
Introduction
Parkinson’s disease (PD) is marked by the progressive loss of dopamine (DA) neurons in the substantia nigra (SN) (Hastings and Zigmond, 1997). While the etiology of PD remains unclear, both genetic factors and environmental toxins have been proposed in its pathogenesis(Ladeby et al., 2005;McGeer and McGeer, 2004). Regardless of the underlying etiology, neuroinflammation [the some total of cellular changes (e.g. microglial activation) and secreted factors (e.g. proinflammatory cytokines or free radicals) that accompany an inflammatory response within the CNS] is thought to contribute to the loss of DA neurons seen in patients with PD (McGeer and McGeer, 2004;Ladeby et al., 2005). Neuroinflammation is also present in trauma, stroke, multiple sclerosis, epilepsy and bacterial meningitis (Mennicken et al., 1999;Phillis et al., 2006) in which damage to the blood brain barrier (BBB) has been reported (Huber et al., 2001). More recent studies point to microvascular changes in the SN and alterations in several markers of normal BBB integrity in PD patients (Barcia et al., 2004;Faucheux et al., 1999;Kortekaas et al., 2005). Whether actual changes in permeability, functionality, or physical damage of the BBB occur in PD is currently unknown.
We recently demonstrated that the DA neurotoxin, 6-hydroxydopamine (6-OHDA), compromised BBB integrity producing apparent leakage of both fluoresceinisothiocyanate -labeled albumin (FITC-LA) and horseradish peroxidase into the SN and striatum. This leakage was accompanied by loss of DA neurons, activation of microglia, up-regulation of p-glycoprotein and β-integrin on the endothelial cells that comprise the BBB, and attenuation of a DA-mediated behavior by domperidone, a DA antagonist that normally does not cross the BBB (Carvey et al., 2005). Since neuroinflammation including microglia activation and increased levels of proinflammatory cytokines including tumor necrosis factor-alpha (TNF-α) are present in patients with PD and in animal models of the disease (Aschner, 1998;Hirsch et al., 2005;Nagatsu and Sawada, 2005) and both are well known to affect the integrity of the BBB (Ryu and McLarnon, 2006;Tsao et al., 2001;Yenari et al., 2006) it is quite possible that 6-OHDA-induced neuroinflammation was responsible for the breakdown in barrier integrity. However, neurons containing biogenic amines are found in close proximity to brain capillaries (Rennels and Nelson, 1975; DiCarlo, Jr. et al., 1984;Kapadia and de Lanerolle, 1984), endothelial cells express both noradrenergic and serotoninergic transporters (Wakayama et al., 2002) and receptors (Wakayama et al., 2002;Kobayashi et al., 1985), and stimulation of the locus coeruleus increases BBB permeability (Raichle et al., 1975) suggesting that neurotransmitters may regulate BBB function as well. The BBB leakage we observed in rats treated with 6-OHDA could be the result of neuroinflammation, DA neuron loss, or a combination of both. Therefore, we designed a set of experiments to determine the relative contribution of each to the leakage of the BBB.
Materials and methods
Animals
A total of 88 male mice, 8 weeks of age and weighing 22–25 g at the start of the study were used. The TNF-α KO mice (B6;129S6-TnftmlGkl/J; n=22), WT background control mice (B6;129S6; n=22), and C57BL/6 (n=44) mice were purchased from Jackson Laboratory (Bar Harbor, ME). All mice were acclimated to the animal facility for at least two weeks prior to the start of the study. One day prior to MPTP treatment, the mice were moved to a controlled ventilated room and housed in ventilation chambers until sacrificed. Mice were allowed free access to food and water for the duration of the study. The protocols used in this study were approved by the Rush University Medical Center Institutional Animal Care and Utilization Committee and were compliant with all regulations at the institutional, state, and federal levels. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-HCl (Sigma, St. Louis, MO) handling and safety measures followed methods described by Przedborski et al. (Przedborski et al., 2001).
Study Design
Study One
TNF-α KO (n=22) and WT (n=22) mice were randomly assigned to one of two groups (Saline or MPTP) for a total of 4 groups designated as follows: WT/Sal = WT mice treated with saline (n=10); WT/MPTP = WT mice treated with MPTP (n=12); TNF-α KO/Sal = KO mice treated with saline (n=10); and TNF-α KO/MPTP = KO mice treated with MPTP (n=12). TNF-α KO and WT mice were injected (i.p.) with either saline or MPTP and sacrificed 72 hours later. MPTP-HCl (10mg/kg freebase) was injected (i.p.) in a saline vehicle four times at 1-hour intervals for a total of 40mg/kg over a 4 hour period. Saline treated mice followed the same injection protocol. After their injections, the mice were returned to their home-chambers, and sacrificed after 3 days.
Study Two
C57BL/6 mice received either saline or minocycline followed by either MPTP or saline injection. Mice (n=44) were randomly divided into four groups designated as follows: Sal/Sal = saline injections given in place of minocycline and MPTP (n=10); Sal/MPTP = mice treated with saline in place of minocycline, but treated with MPTP(n=12); Mino/Sal = mice treated with minocycline and given saline in place of MPTP(n=10); and Mino/MPTP = mice treated with minocycline and MPTP (n=12). Minocycline (90mg/kg per day, dissolved in 5% sucrose, Sigma, St. Louis, MO) was injected (i.p) for 3 consecutive days, with the first minocycline dose administered 30 minutes prior to the first MPTP injection and last dose administered 24 hrs prior to sacrifice. MPTP-HCL (10mg/kg per hour) was injected as described above.
In both studies, 5 mice from each treatment group were processed for immunohistochemistry and 5 mice were processed for biochemical analysis. Extra animals were added in all the MPTP groups to account for the anticipated 10% morbidity/mortality that is normally encountered in the MPTP protocol. Only animals that were overtly healthy (no apparent distress, normal appearance, and normal weight) were processed further. The dependent measures in each study were identical and consisted of visualization of BBB leakage throughout the brain, DA and microglia cell counts in the SN, and determination of levels of TNF-α and IL-1β protein in the SN and striatum.
FITC-LA Leakage
The leakage of FITC-LA (MW=69–70 KD, Sigma, St. Louis, MO) from vasculature into brain parenchyma was assessed as described previously (Carvey et al., 2005) to determine BBB integrity. Briefly, three days following the last MPTP or saline injections, the mice were anesthetized with pentobarbital (60mg/kg). Heparin (100 units/kg in Hank’s Balanced Salt Solution) was injected intracardially followed immediately by 5 ml FITC-LA (5mg/ml, in 0.1 M Phosphate-buffered saline (PBS) buffer) injected at a rate of 1.5ml/minute with the right atrium open. After perfusion, the brains were removed immediately and immersed into 4% paraformaldehyde. Three days later, the fixative was replaced with three changes of 30% sucrose in 0.1 M PBS buffer. Each brain was sectioned at 40 μm using a sliding microtome, divided into 6 consecutive free-floating series, and stored in cryoprotectant (0.05 M PBS with 30% sucrose and 30% ethylene glycol). One series of sections from each of the 5 mice from both studies were mounted onto gelatin-coated slides, dehydrated, and cover-slipped for confocal microscopy (Olympus) analysis of FITC-LA leakage.
Immunohistochemistry
A second series of brain sections from each animal in both studies was used for tyrosine hydroxylase (TH, a DA neuron marker) immunohistochemistry as described previously (Carvey et al., 2005). Briefly, sections were washed (6 × 10 minutes) in Tris buffered saline (TBS, pH 7.4) and incubated for 1 h in 0.05% TBS Triton-X-100 solution. Sections were then incubated 30 min in TBS solution with 2.13% sodium periodate to block endogenous peroxidase activity, and then incubated for 1 h with 3 % normal goat serum in TBS. Tissues were incubated overnight with the primary antibody to TH (rabbit:anti-mouse, Chemicon, 1:1,000 dilution). Immunolabeling was continued using biotinylated secondary antibody (goat:anti-rabbit, Vector Laboratories, 1:200) in TBS with 3% goat serum and incubated for 1 hour at room temperature. The antibody complex was amplified using an avidin-biotin kit (ABC-Elite kit; Vector) and visualized with 3,3′ –diaminobenzidine (DAB) with nickel enhancement.
A third series of sections from each animal of both studies was processed for CD11b immunohistochemistry to reveal activated microglial cells. The procedure for CD11b immunochemistry was similar to the THir staining except that the primary antibody to CD11b originated in rat (Rat:anti-mouse CD11b 1:200; Serotec) and the second antibody was goat:anti-rat IgG (1:200; Vector Laboratories).
Stereological assessment of THir cell and activated microglia cell counts
The estimation of the total number of THir neurons and activated microglia in the SN was performed using the computerized optical dissector method (MicroBrightField software) as described previously (Vu et al., 2000). Briefly, a 10X objective lens was used to define the contour around the entire SN (Ling et al., 2004) and a 100X lens was used for the tyrosine hydroxylase immunoreactive cells (THir cells) and activated microglia assessments. One series of sections from each animal was analyzed for THir and a second series for CD11b staining. The total number (N) of THir or activated microglia in the SN from each animal was estimated using the formula N = NV · VSN, where NV is the numerical density and VSN is the volume of the SN, as determined by Cavalieri’s principle.
ELISA for determination of TNF-α and IL-1β concentration in the SN and striatum
Mice were anesthetized with 60mg/kg pentobarbital. The brains were removed and immediately placed in cold 2-methylbutane, and then stored at −80°C. The TNF-α and IL-1β levels were determined for both studies as previously described (Ling et al., 2004). Briefly, brains were slightly thawed, the SNs and striata dissected out, then submerged in ice-cold 150μl homogenate buffer (Trizma/HCl pH 7.2, 0.1 M, sodium chloride 0.9%, protease inhibitor cocktail 1%), and sonicated. The homogenates were centrifuged at 13,000 × g for 30 minutes at 4°C. The concentrations of TNF-α and IL-1β in the supernatants were determined using commercial ELISA kit (BioSource International, Camarillo, CA). Standards, diluent buffers, or samples (100 μl) along with 50 μl of either biotinylated anti-TNF-α or anti-IL-1β solutions were pipetted into each well and then incubated for 2 hours at 37°C. After washing, streptavidin-HRP working solution was incubated for 1 hour at room temperature. Following washing, stabilized chromogen was incubated for 1 hour and the reaction was stopped with stop solution. The plates were read in a Dynatech ELISA plate reader (Dynatech Laboratories, Chanlity, VA) and the concentration of the TNF-α and IL-1β were determined against a seven-point standard curve. To standardized protein levels, the total protein concentration of each homogenate was determined using the BCA protein assay kit (Pierce, IL) and the quantity of TNF-α and IL-1β was expressed as pg/mg total protein.
Statistical analyses
All values were expressed as mean ± S.E.M. Separate Two-Way analyses of variance (ANOVA) were used to analyze the THir and activated microglia counts along with TNF-α and IL-1β levels in both studies. TNF-α genotype and MPTP treatment were factors in the TNF-α KO study. In the study of minocycline treatment, the two factors were minocycline and MPTP treatment. The least significant difference test (LSD) was used as the post-hoc test to determine individual group differences with significance set at p ≤ 0.05.
Results
MPTP increased FITC-LA leakage into the SN and striatum
FITC-LA was injected into the common carotid artery 72 hours following treatment with MPTP (a time at which active DA neurodegeneration was occurring (Sugama et al., 2003), and the brains were assessed using confocal microscopy to determine if this large molecular weight marker (MW range = 69–70kD) entered brain parenchyma. FITC-LA was confined exclusively to blood vessels in the SN and striatum in the control animals of both genotypes [WT/Sal (Fig. 1A,C); TNF-α KO/Sal (Fig. 1B,D)] and the C57 BL/6 minocycline study animals [Sal/Sal (Fig. 1I,K); Mino/Sal (Fig. 1J,L)] indicating an intact BBB. FITC-LA was also confined within vessels in the majority of the area within the SN and striatum of MPTP-treated animals (Fig. 1E,G,M,O). However, all control animals treated with MPTP (WT/MPTP and Sal/MPTP) also exhibited numerous patchy areas of leakage in both structures indicative of disruption of barrier integrity. In some areas, individual vessels were not recognizable because the leakage of the FITC-LA was so intense and wide spread. Interestingly, the locations of the leaks were not the same in every animal, but varied considerably from animal to animal. Moreover, the apparent magnitude of the leaks similarly varied from animal to animal such that in both the SN and striatum, some animals had areas of leakage in almost every section, whereas in other animals the number of leaks was markedly less. Regardless, every control animal exposed to MPTP exhibited leakage in some sections of the SN and striatum. The ability of MPTP to induce FITC-LA leakage did not appear to differ between the wild types of the two genotypes.
Figure 1.
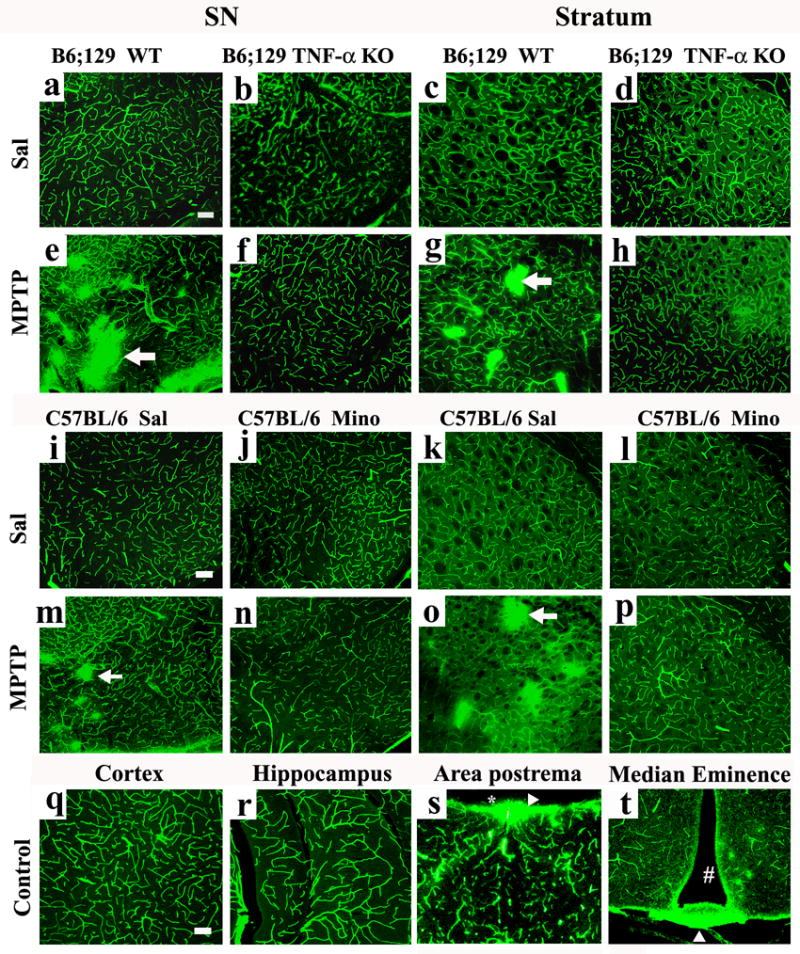
TNF-α KO and minocycline treatment attenuated the FITC-LA leakage in the SN and striatum after MPTP treatment. FITC-LA was visualized using confocal microscopy in the SN of WT/Sal (A) and TNF-α KO/Sal (B) as well as the striatum of WT/Sal (C) and TNF/Sal animals (D). FITC-LA leakage was evident in the SN of the WT/MPTP (E) and striatum of WT/MPTP (G) treated mice. Areas of leakage were not found in the SNs (F) or striata (H) of TNF-α KO/MPTP treated mice. Similar findings were seen in the minocycline treated animals. FITC-LA was confined within vessels in the SN of Sal/Sal (I) and Mino/Sal (J) as well as the striatum of Sal/Sal (K) and Mino/Sal (L) animals. FITC-LA leakage was evident in the SN of the Sal/MPTP (M) and striatum of Sal/MPTP (O) treated mice. Areas of leakage were not found in the SN (N) or striata (P) of Mino/MPTP treated mice. There was no FITC-LA leakage in the parietal cortex (Q) and hippocampus (R), in any of the animals regardless of treatment. However, leakage was always seen in the area postrema (S), and median eminence (T) areas devoid of a BBB. [# = the third ventricle and * = the fourth ventricle: Arrows indicate areas of FITC-LA leakage. Scale bar = 100um (A-T)].
If FITC-LA distribution reflects BBB integrity, then areas of the brain not protected by the BBB should also reveal leakage. This was indeed the case. The periventricular structures, such as the area postrema (Fig 1S) and the median eminence (Fig. 1T) showed extensive leakage into the brain parenchyma in all animals, regardless of their treatment condition. In contrast, leakage was not detected in areas outside the nigro-striatal pathway or periventricular organs in these same animals. Thus, in MPTP treated animals, leakage was not seen in the parietal cortex (Fig. 1Q) or hippocampus (Fig. 1R) even though leakage was detected in the SNs, striata and periventricular structures of these animals. This suggests that the perfusion pressure used to deliver the FITC-LA filled the brain’s vascular compartment completely without leakage resulting from excess perfusion pressure.
Both TNF-α KO and Minocycline attenuated BBB leakage
In order to determine if inflammation and/or DA neuron loss was responsible for the apparent BBB dysfunction, FITC-LA leakage was assessed in TNF-α KO animals as well as animals treated with minocycline. MPTP treatment increases TNF-α (Nagatsu et al., 2000) which could, in turn, disrupt BBB integrity given its well documented ability to do so (Farkas et al., 2006;Miller et al., 2005;Wong et al., 2004). MPTP also activates microglia (Sugama et al., 2003;Wu et al., 2002;Du et al., 2001;Orr et al., 2002) which release TNF-α (Hirsch et al., 2005;Huang et al., 2005;Nagatsu et al., 2000) as well as a number of other proinflammatory mediators that could disrupt barrier integrity (e.g., free radicals; (Abbott, 2000;Calingasan et al., 1998;Hirsch et al., 2005;Yenari et al., 2006). Since minocycline prevents microglial activation (Du et al., 2001;Hirsch et al., 2005;Zemke and Majid, 2004) by reducing ED-1 and other transcription factors involved in inflammatory processes (Lai and Todd, 2006) and TNF-α KO prevents release of a key proinflammatory cytokine implicated in barrier integrity, assessment of leakage in these animals would enable us to assess the role of neuroinflammation on the barrier dysfunction produced by MPTP. Of perhaps greater importance, however, is that TNF-α KO and minocycline produce differential effects on MPTP-induced DA neuron loss in the SN. Thus, TNF-α KO animals exhibit nigral DA neuron loss in response to MPTP (Rousselet et al., 2002;Ferger et al., 2004), while minocycline treatment prevents MPTP-induced DA neuron loss (Du et al., 2001;Tikka and Koistinaho, 2001). The use of these two models would therefore enable us to ascertain the relative contributions of neuroinflammation and DA neuron loss to the MPTP-induced BBB dysfunction.
FITC-LA leakages into the SNs and striata were markedly attenuated in both TNF-α KO and minocycline treated animals following MPTP. Thus, in both the SN (Fig. 1F,N) and the striatum (Fig. 1H,P), few areas of FITC-LA leakage were observed, and in many animals, no leakage at all was observed. Examination of the vessels in these animals revealed less distinct borders in many of the vessels suggesting a minor compromise of barrier integrity. However, no areas of overt entry into brain parenchyma were observed in the SNs or striata. These findings suggest that TNF-α and activated microglia participate in MPTP-induced barrier dysfunction.
Both TNF-α KO and minocycline reduced MPTP-induced inflammation
Although CD11b- immunoreactive (ir) cells (a marker for microglial cells) were detected in all animals, control animals exhibited only several hundred cells in their SNs and striata, the vast majority of which had small rounded cell bodies with few ramified processes typical of resting microglia. In contrast, MPTP treatment not only significantly increased the numbers of CD11b-ir cells in both TNF-α KO (F1,16=60.317; p<0.001) and minocycline treated animals (F1,16=62.762; p<0.001), but also induced dramatic changes in their morphology as well, such that the vast majority of the cells had large cell bodies with highly ramified processes typical of activated microglia (Fig. 2A,B). Stereological assessments of the CD11b-ir cell counts revealed that both TNF-α KO and minocycline treatment significantly attenuated the numbers of activated microglia (Fig. 2C). Thus, in the B6;129S6 (WT) mice, MPTP treatment increased the numbers of activated microglia 26 fold compared with WT/Sal (p<0.05), whereas the TNF-α KO/MPTP mice showed only a 2.6 fold increase (p<0.05). Similarly, MPTP induced an 18 fold (p<0.05) increase in numbers of CD11b-ir cells in Sal/MPTP mice compared with Sal/Sal treated (C57 BL/6) controls, whereas the minocycline treated animals (Mino/MPTP) revealed only a 1.07 fold increase (p>0.05) (Fig. 2D). Thus, both TNF-α KO and minocycline treatment markedly attenuated microglial activation as well as FITC-LA leakage suggesting that microglia activation was associated with the barrier dysfunction observed. Moreover, assessment of CD11b-ir cells in the patches of FITC-LA leakage revealed the almost universal presence of activated microglia (Fig. 3) further supporting their involvement in BBB dysfunction.
Figure 2.
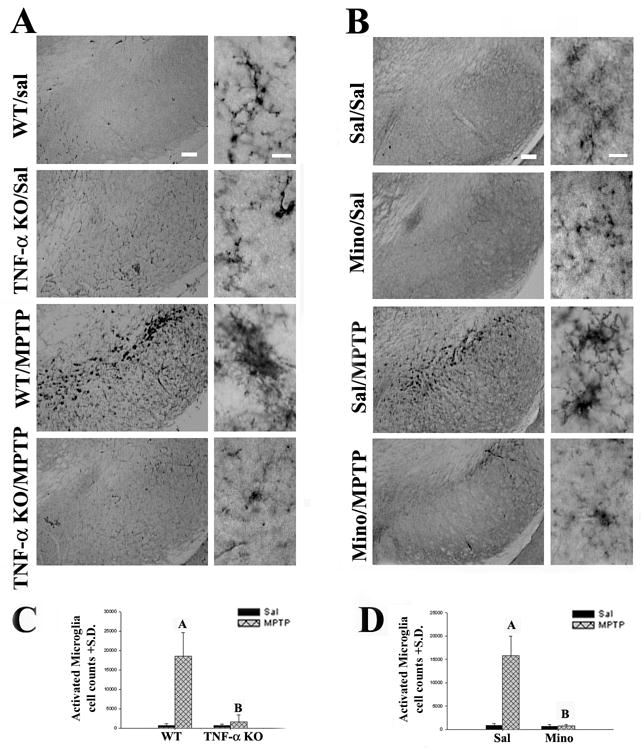
TNF-α KO and minocycline reduced microglial activation. (A) TNF-α KO inhibited MPTP-induced microglial activation. The shapes of microglia were not different in WT (Lane 1: SN of WT/Sal) and TNF-α KO mice (Lane 2: SN of TNF-α KO/Sal) treated with saline. Three days after the last MPTP administration, microglia became large, amoeboid, and had thick processes (Lane 3: SN of WT/MPTP). However, in TNF-α KO/MPTP mice the morphology of microglia remained small and ramified (Lane 4: SN of TNF-α KO/MPTP) comparable with those in WT/Sal mice. (B) Minocycline treatment inhibited MPTP-induced microglial activation. The shape of microglia showed no difference in the C57BL/6 mice between saline only (Lane 1: SN of Sal/Sal) and minocycline only (Lane 2: SN of Mino/Sal) treatment groups. Numerous activated microglia were present in the SN of mice 3 days after MPTP treatment (Lane 3: SN of Sal/MPTP). Minocycline treatment attenuated the MPTP-induced microglia activation (Lane 4: SN of Mino/MPTP). Microglia in the mice injected with both MPTP and minocycline had small cell bodies with ramified long, thin processes in the SN. (C) Quantification of CD11b-ir cells in the WT and TNF-α KO mice following the MPTP treatment. After the MPTP treatment, the number of activated microglia increased 26 fold in the SN of WT mice. However, TNF-α KO clearly prevented microglia activation. (Marker A: p<0.001 relative to WT/Sal group; Marker B: p<0.001 relative to WT/MPTP group, but it was undistinguishable from WT/Sal group, P>0.05). (D) Quantification of CD11b-ir cells in C57BL/6 mice with or without minocycline treatment following MPTP administration. MPTP treatment increased the number of activated microglia. Minocycline treatment (90mg/kg X3) attenuated this change. (Marker A: p<0.05 relative to Sal/Sal group; Marker B: p<0.05 relative to Sal/MPTP group, but it was indistinguishable from Sal/Sal group, P>0.05). Left (Scale bar=100 um) and right (Scale bar=10 um).
Figure 3.
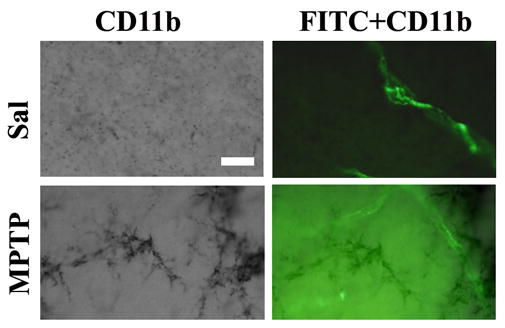
Areas of FITC-LA leakage contained activated microglia. Upper images show no FITC-LA leakage in areas of resting microglia whereas the lower row demonstrates FITC-LA leakage associated with marked microglial activation. (Bar=25um).
In addition to microglia, we also assessed the proinflammatory cytokines TNFα and interleukin 1 β (IL-1β) in these animals (Table I). As expected, TNF-α signal was not detectable in any tissue sample taken from TNF-α KO mice whereas both WT saline treated animals exhibited similar levels of TNF-α in the SNs and striata. MPTP treatment consistently increased levels of TNF-α in both the SNs (F1,8=6.474, p<0.05) and striata (F1,8=20.215, p<0.05) of WT/MPTP mice, compared with WT/Sal. Similar increases were seen in the C57 BL/6 animals treated with MPTP (Sal/MPTP) in both the SNs (F1,16=12.611; p<0.05) and striata (F1,16=14.162; p<0.05). However, these increases were significantly attenuated by minocycline treatment (Mino/MPTP) in both the SN (F1,16=15.433; p<0.05) and the striatum (F1,16=18.119; p<0.05).
Table 1.
TNF-α and IL-1 β level (pg/mg protein) ±S.D.
| TNF-α | IL-1β | ||||
|---|---|---|---|---|---|
| N | SN | Striatum | SN | Striatum | |
| WT/Sal | 5 | 1.003±0.211 | 0.927±0.397 | 21.11±3.430 | 6.546±1.141 |
| TNF-α KO/Sal | 5 | Non-detectable | Non-detectable | 22.40±4.230 | 5.162±1.034 |
| WT/MPTP | 5 | 1.900±0.281A | 1.925±0.303A | 30.98±4.636A | 10.75±0.982A |
| TNF-α KO/MPTP | 5 | Non-detectable | Non-detectable | 24.94±3.125 | 8.120±1.033 |
| Sal/Sal | 5 | 1.213±0.277 | 1.190±0.214 | 20.90±4.239 | 5.622±0.424 |
| Mino/Sal | 5 | 0.736±0.292 | 0.939±0.276 | 16.75±4.754 | 4.820±0.772 |
| Sal/MPTP | 5 | 1.802±0.437B | 1.967±0.296B | 32.50±4.787B | 11.71±0.622B |
| Mino/MPTP | 5 | 1.160±0.232C | 1.127±0.344C | 25.05±4.266C | 8.849±2.524C |
Non-detectable:<0.7pg/mg protein
Marker A: p<0.05, compared to WT/Sal
Marker B: p<0.05, compared to Sal/Sal
Marker C: p<0.05, compared to Sal/MPTP
Similar findings were seen for IL-1 β (Table I). Normal levels of IL-1β were seen in both the SNs and striata of the WT, TNF-α KO mice, and C57 BL/6 animals. MPTP treatment consistently increased levels of IL-1β in both the SN (F1,16=12.683, p<0.05) and striatum (F1,16=50.098, p<0.05) of WT/MPTP mice. TNF-α KO attenuated the release of IL-1β in the striatum (F1,16=15.731, p<0.05) but not in the SN (F1,16=1.859, p>0.05). Similar increases of IL-1β were seen in the C57 BL/6 animals treated with MPTP (Sal/MPTP) in both the SN (F1,16=22.283, p<0.05) and striatum (F1,16=34.885, p<0.05). However, these increases were significantly attenuated by minocycline treatment (Mino/MPTP) in the SN (F1,16=7.146; p<0.05) and the striatum (F1,16=6.770; p<0.05). Thus, MPTP increased the levels of this proinflammatory cytokine whereas TNF-α KO and minocycline treatment markedly attenuated the MPTP-induced increases. These data suggest that the attenuation in FITC-LA leakage seen in the TNF-α KO and minocycline treated animals was also associated with reductions in these pro-inflammatory cytokines.
MPTP-induced DA neuron loss was not associated with BBB leakage
MPTP treatment led to an anticipated loss of THir neurons (a marker for DA neurons). Thus, stereological assessments of THir cells in the SN revealed a 35% reduction (F1,16=37.415; p<0.05) in the B6;129S6 control mice and a 44% reduction in the C57BL/6 controls (F1,16=14.856; p<0.05), which were not significantly different from one another (Fig. 4A; p>0.05). Compared with their wild type controls treated with MPTP, the TNF-α KO mice were equivalently sensitive to MPTP losing 30% of their THir cells (Fig 4B; F1,16=0.005, p=0.943). In contrast, minocycline treatment markedly attenuated the MPTP-induced THir cell loss (Fig. 4C; F1,16=4.707; p<0.05). There was no significant difference in THir cell counts between Mino/MPTP and Mino/Sal (p>0.05). These data confirm the previously reported differential effects on DA neuron loss (Tikka and Koistinaho, 2001;Rousselet et al., 2002;Leng et al., 2005;Ferger et al., 2004;Du et al., 2001). Most importantly, our studies demonstrate that the attenuation of FITC-LA leakage in these two sets of animals was independent from DA neuron loss suggesting inflammatory events, and not a DA neuron loss per se, were responsible for the barrier dysfunction induced by MPTP.
Figure 4.
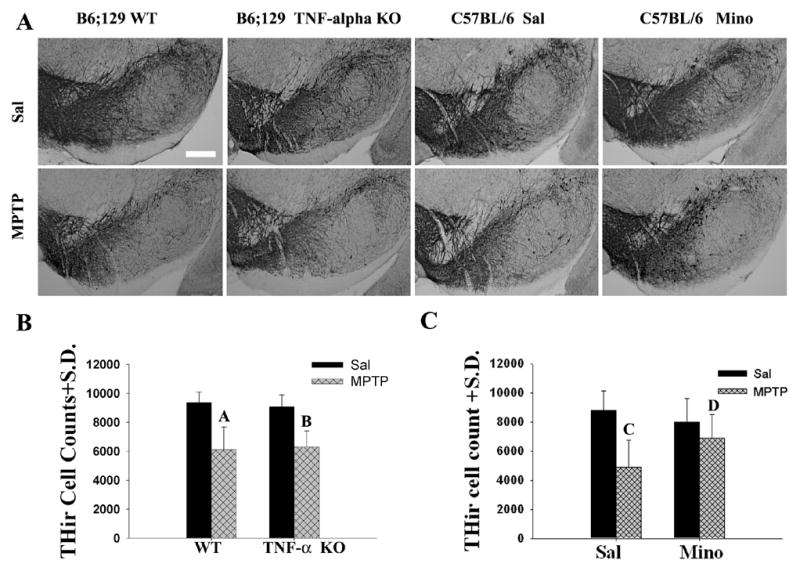
The loss of dopamine neurons was attenuated by minocycline treatment, not by TNF-α KO following MPTP treatment. (A) Photomicrographs of representative sections of THir staining in the SNs of the mice used to generate the bar graph of (B) and (C). SN of (a) WT/Sal, (b) TNF-α KO/Sal, (c) WT/MPTP, (d) TNF-α KO/MPTP, (e) Sal/Sal, (f) Mino/Sal, (g) MPTP/Sal, and (h) MPTP/Mino (Magnification bar=0.25mm). (B) Quantification of THir cells in the WT and TNF-α KO mice following MPTP treatment. MPTP similarly reduced the numbers of THir cells in the SN of both WT and TNF-α KO mice. The number of THir cells was not significantly different in WT/MPTP and TNF-α KO/MPTP mice [(p=0.774); Marked A and B, separately]. (C) Quantification of THir cells in C57BL/6 mice with or without minocycline treatment following MPTP administration. The number of THir cells was significantly different (p=0.044) between Sal/MPTP (Marker C) and Mino/MPTP mice (Marker D).
Discussion
The current study confirms and extends our previous work in rats in which injection of the DA neurotoxin 6-hydroxydopamine into the striatum or medial forebrain bundle produced similar patchy areas of leakage of FITC-LA and horseradish peroxidase as well as increases in β3 integrin expression (a marker of angiogenesis) in the SN and striatum (Carvey et al., 2005). In the present study, the DA neurotoxin (MPTP) was injected systemically so that the barrier dysfunction seen could not be a consequence of stereotaxic brain surgery. In addition, two different genotypes of mice exhibited barrier dysfunction using MPTP suggesting that the previous effects observed in rats were neither species nor toxin specific. Finally, the current study clearly demonstrated the validity of the FITC-LA injection procedure as a marker for barrier integrity, because areas devoid of a BBB exhibited leakage, regardless of treatment history, and detailed evaluation of several non-dopaminergic brain areas revealed complete vascular perfusion without evidence of leakage demonstrating specificity while ruling out a perfusion-based epiphenomenon. These data strongly argue that DA neurotoxins induce BBB dysfunction in well-established animal models of PD.
At this time, we do not know if the areas of leakage were static (i.e., an area developed a leak and remained leaky) or dynamic (i.e. an area developed a leak and repaired itself such that after several days, leakage would no longer be detected). Given that activated microglia were associated with areas of leakage and microglia are known to migrate (Cho et al., 2006;Carbonell et al., 2005), it is more likely that the pattern of leaks is dynamic. A dynamic pattern of leakage was further supported by the fact that the areas of leakage were different in every animal. This suggests that a “window” of increased leakage developed in an area and could subsequently close. In addition, at the present time we do not know if DA neurotoxins produce only “patchy” areas of severe dysfunction as indicated here with FITC-LA leakage (and as we observed in our previous study (Carvey et al., 2005)), or if subtle dysfunction exists throughout the SN and striatum, but have not yet been identified. The large molecular weight of FITC-LA may only identify areas of significant leakage and if markers of smaller molecular weight were used, a much less punctated pattern of leakage might have been observed. Regardless, it is becoming increasingly clear that the BBB dysfunction, although it may be punctate and dynamic in nature, is not temporary. We previously showed that FITC-LA leakage was still present 10 and 34 days following 6-OHDA treatment (Carvey et al., 2005). Whether or not barrier dysfunction continues for months after toxin exposure remains to be established. Regardless, these results argue that the BBB is somewhat long-lived and common to two different animal models of PD.
The MPTP-induced barrier dysfunction appeared to be a consequence of neuroinflammation independent of DA neuron loss. Although MPTP produced neuroinflammation as evidenced by increased numbers of activated microglia and increases in TNF-α and IL-1β in both genotypes, the TNF-α KO animals exhibited significant DA neuron losses whereas the animals treated with minocycline did not. Both TNF-α KO and minocycline treatment dramatically attenuated the FITC-LA leakage in which a clear-cut dissociation between DA neuron loss and barrier dysfunction was shown. However, we must consider the differential effects of MPTP on the BBB that was observed could simply reflect strain differences used in the two different studies. In addition, there is the possibility that TNF-α KO and minocycline treatment could have had differential effects on the expression of receptors on the endothelial cells or other soluble factors produced by MPTP exposure that prevented BBB dysfunction independent of DA neuron death. While not inherently apparent from the present results other factors originating from the dieing or compromised DA neurons could also be involved in the dysfunction of the BBB. Regardless, although biogenic amine containing neurons are often seen in close proximity to endothelial cells that also express noradrenergic and serotonergic receptors, and norepinephrine containing cell of the locus coeruleus have been shown to regulate barrier function (Kobayashi et al., 1985;Raichle et al., 1975;Wakayama et al., 2002), there was no evidence of dopaminergic control of barrier leakage in the SN and striatum in the current study, after three days. We have previously shown that 6OHDA produces BBB dysfunction after 10 and 34 days (Carvey et al., 2005), therefore there is a possibility that neuroinflammation can initiate a first phase of BBB dysfunction (i.e. 3 days) that is further perpetuated by the ensuing DA neuron loss (between 3 and 10 days). Detailed time course studies would be needed to address this possibility. Regardless neuroinflammatory events by themselves appear to lead to barrier dysfunction in the absence of DA neuron loss. Whether or not the magnitude of inflammation is correlated with the degree of BBB dysfunction cannot be determined with the current study design. Future studies should be performed to ascertain the levels of inflammation produced and correlate these with the degrees of BBB dysfunction. Regardless, the current results do not negate the probability that the DA neuron degeneration led to the inflammation, but rather, that DA neuron loss by itself did not lead to the barrier dysfunction. Moreover, since MPTP is widely known to primarily affect catecholaminergic systems in general and the nigro-striatal pathway in particular, it was not surprising to observe here as well as in our previous study that the significant BBB dysfunction was confined to the SN and striatum.
TNF-α by itself activates microglia (John et al., 2003) and our results show that TNF-α KO inhibits microglia activation. Minocycline is a semisynthetic tetracycline derivative that exerts anti-inflammatory effects and inhibits microglial activation by decreasing IL-1 β and nitric oxide release (Du et al., 2001;Tikka and Koistinaho, 2001). Moreover, our data showed that minocycline also decreased the TNF-α release. Activated microglia release numerous neuroinflammatory substances that can potentially disrupt barrier function including prostanoids, proteases, nitric oxide (NO), superoxide, and the proinflammatory cytokines TNFα and IL-1β (Dringen, 2005;Lynch et al., 2004;Tsao et al., 2001;Wong et al., 2004;Yenari et al., 2006). Although any of these factors could have contributed to the barrier breakdown in the current study, the MPTP-induced increase in TNF-α and IL-1β in conjunction with FITC-LA leakage and the attenuation in these increases in the TNF-α KO and minocycline treated animals argues for their potential involvement in the barrier disruption. TNF-α and IL-1β are known to decrease electrical resistance and increase permeability in BBB in vitro models (Didier et al., 2003;Miller et al., 2005). TNF-α causes a redistribution of cadherin and junctional adhesion molecule (JAM) leading to a rearrangement of microfilaments, and a down-regulation of occludin expression increasing BBB permeability (Ozaki et al., 1999;Petrache et al., 2003;Kniesel and Wolburg, 2000;Mankertz et al., 2000). IL-1 β also increases BBB permeability by decreasing expression of occludin and zonula occludens-1 proteins leading to apparent redistribution of the adherens junction protein vinculin (Bolton et al., 1998). IL-1β may also induce cyclooxygenase-2 (COX-2) synthesis and activate NFκB in brain endothelial cells, which can be blocked by specific inhibitors of NFκB activation (Kortekaas et al., 2005;Laflamme et al., 1999;Nadjar et al., 2005). However, although the current evidence suggests the involvement of TNF-α and IL-1β, we cannot, at this time, rule out other inflammogens associated with microglial activation.
Several studies have argued against or have not found BBB dysfunction in PD. Haussermann et al., (2001) found no changes in blood cerebral spinal fluid (CSF) barrier function by examining CSF/serum ratios and oligoclonal bands in PD patients. If the BBB dysfunction is not universal, but rather punctate as our data suggests, major changes such as these would not be detected. Similarly, Kurkowska-Jastrzebska et al., (1999) argued that the DA neurotoxin MPTP does not disrupt BBB function. Yet, although they showed that IgG was restricted to the inside of the blood vessels, they reported that mononuclear cells infiltrated the SN and striatum, which would suggest BBB dysfunction. O’callaghan et al., (1990) used a single small dose of MPTP (12.5mg/kg, s.c.) that did not produce DA neuron loss and reported no dysfunction of the BBB. Furthermore, Canudas et al., (2000) injected MPP+ into the left SN of the rat that produced only a small lesion, yet assessed BBB integrity in the striatum with a crude index of BBB integrity (albumin staining) that could have easily been missed because of the small size of the SN injury. It is also important to note that the animal studies using MPTP or its metabolite MPP+ just described, were not designed to assess BBB integrity, but rather focused on other effects of MPTP. Moreover, they used global indices of BBB integrity, which could have readily missed the punctuate leakage that seems to characterize the DA neurotoxin exposed BBB. In contrast, the study by Faucheux et al., (1999) showed an increase in vascular density in the SN, but not the VTA of PD patients and Barcia et al., (2004) discussed evidence of microangiogenesis in the PD brain which is often associated with barrier dysfunction. Kortekaas et al., (2005) used [(11)C]-verapamil imaging, and demonstrated an 18% increase in brain uptake in the mesencephalon of PD patients relative to aged controls. This increase may have reflected alterations in P-gp function or simply increased leakage into brain. Notably, Barcia et al., (2004) also found an increase in the number of blood vessels indicative of microangiogenesis that follows barrier damage in close proximity to degenerating DA neurons in non-human primates. Moreover, the increase in vessels was highly correlated with increases in vascular endothelial growth factor (VEGF) probably caused by neo-microangiogenesis that similarly accompanies barrier brea kdown following an inflammatory event. Thus, this emerging literature supports barrier dysfunction in patients with PD and animal models of this disorder.
There are numerous implications to PD if areas of active inflammation lead to focal areas of barrier dysfunction in patients. Areas of leakage in focal neuroinflammatory loci would afford the opportunity to target deliver anti-inflammatory agents that normally do not cross the BBB or deliver a variety of therapeutics to those areas using nanoparticles. Notwithstanding the effect of these agents on areas of the brain not protected by a BBB, this strategy would target areas ostensibly undergoing active DA neurodegeneration potentially slowing disease progression. On the other hand, BBB dysfunction could allow inhomogeneous entry of antiparkinsonian drugs into the SN and striatum that could contribute to dyskinesias due to dopaminergic “hotspots” that have been implicated in this DA agonist induced side effect (Bankiewicz et al., 2006). Alternatively, levodopa decarboxylase inhibitors including carbidopa and benserazide, which normally do not cross the BBB, may do so in these areas of leakage. This would inhibit conversion of levodopa to DA in these areas creating hot spots of increased DA activity in intact areas. In addition, focal BBB dysfunction would increase entry of elements of the peripheral vasculature into the SN and striatum that could similarly contribute to disease progression. Moreover, environmental toxins that may not cross the BBB readily would concentrate in areas of BBB dysfunction. Taken together, these results suggest that detailed imaging assessments of BBB integrity should be performed in patients with PD to determine the role, if any, the compromised BBB integrity plays in disease progression and side effects.
Acknowledgments
This work was supported by NINDS NS045316, NIEHS 012307, W81XWH-04-01-0365 and the Michael J. Fox Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. 2000;20:131–147. doi: 10.1023/a:1007074420772. [DOI] [PubMed] [Google Scholar]
- 2.Aschner M. Astrocytes as mediators of immune and inflammatory responses in the CNS. Neurotoxicology. 1998;19:269–281. [PubMed] [Google Scholar]
- 3.Bankiewicz KS, Daadi M, Pivirotto P, Bringas J, Sanftner L, Cunningham J, Forsayeth JR, Eberling JL. Focal striatal dopamine may potentiate dyskinesias in parkinsonian monkeys. Exp Neurol. 2006;197:363–372. doi: 10.1016/j.expneurol.2005.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barcia C, Emborg ME, Hirsch EC, Herrero MT. Blood vessels and parkinsonism. Front Biosci. 2004;9:277–282. doi: 10.2741/1145. [DOI] [PubMed] [Google Scholar]
- 5.Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience. 1998;86:1245–1257. doi: 10.1016/s0306-4522(98)00058-x. [DOI] [PubMed] [Google Scholar]
- 6.Calingasan NY, Park LC, Calo LL, Trifiletti RR, Gandy SE, Gibson GE. Induction of nitric oxide synthase and microglial responses precede selective cell death induced by chronic impairment of oxidative metabolism. Am J Pathol. 1998;153:599–610. doi: 10.1016/S0002-9440(10)65602-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canudas AM, Friguls B, Planas AM, Gabriel C, Escubedo E, Camarasa J, Camins A, Pallas M. MPP(+) injection into rat substantia nigra causes secondary glial activation but not cell death in the ipsilateral striatum. Neurobiol Dis. 2000;7:343–361. doi: 10.1006/nbdi.2000.0308. [DOI] [PubMed] [Google Scholar]
- 8.Carbonell WS, Murase S, Horwitz AF, Mandell JW. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: an in situ time-lapse confocal imaging study. J Neurosci. 2005;25:7040–7047. doi: 10.1523/JNEUROSCI.5171-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS, Snyder J, Zhu YG, Ling ZD. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosci. 2005;22:1158–1168. doi: 10.1111/j.1460-9568.2005.04281.x. [DOI] [PubMed] [Google Scholar]
- 10.Cho BP, Song DY, Sugama S, Shin DH, Shimizu Y, Kim SS, Kim YS, Joh TH. Pathological dynamics of activated microglia following medial forebrain bundle transection. Glia. 2006;53:92–102. doi: 10.1002/glia.20265. [DOI] [PubMed] [Google Scholar]
- 11.DiCarlo LA, Jr, Botvinick EH, Canhasi BS, Schwartz AS, Chatterjee K. Value of noninvasive assessment of patients with atypical chest pain and suspected coronary spasm using ergonovine infusion and thallium-201 scintigraphy. Am J Cardiol. 1984;54:744–748. doi: 10.1016/s0002-9149(84)80201-5. [DOI] [PubMed] [Google Scholar]
- 12.Didier N, Romero IA, Creminon C, Wijkhuisen A, Grassi J, Mabondzo A. Secretion of interleukin-1beta by astrocytes mediates endothelin-1 and tumour necrosis factor-alpha effects on human brain microvascular endothelial cell permeability. J Neurochem. 2003;86:246–254. doi: 10.1046/j.1471-4159.2003.01829.x. [DOI] [PubMed] [Google Scholar]
- 13.Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal. 2005;7:1223–1233. doi: 10.1089/ars.2005.7.1223. [DOI] [PubMed] [Google Scholar]
- 14.Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farkas E, Sule Z, Toth-Szuki V, Matyas A, Antal P, Farkas IG, Mihaly A, Bari F. Tumor necrosis factor-alpha increases cerebral blood flow and ultrastructural capillary damage through the release of nitric oxide in the rat brain. Microvasc Res. 2006 doi: 10.1016/j.mvr.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Faucheux BA, Bonnet AM, Agid Y, Hirsch EC. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet. 1999;353:981–982. doi: 10.1016/S0140-6736(99)00641-8. [DOI] [PubMed] [Google Scholar]
- 17.Ferger B, Leng A, Mura A, Hengerer B, Feldon J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J Neurochem. 2004;89:822–833. doi: 10.1111/j.1471-4159.2004.02399.x. [DOI] [PubMed] [Google Scholar]
- 18.Hastings TG, Zigmond MJ. Loss of dopaminergic neurons in parkinsonism: possible role of reactive dopamine metabolites. J Neural Transm Suppl. 1997;49:103–110. doi: 10.1007/978-3-7091-6844-8_11. [DOI] [PubMed] [Google Scholar]
- 19.Haussermann P, Kuhn W, Przuntek H, Muller T. Integrity of the blood-cerebrospinal fluid barrier in early Parkinson’s disease. Neurosci Lett. 2001;300:182–184. doi: 10.1016/s0304-3940(01)01574-9. [DOI] [PubMed] [Google Scholar]
- 20.Hirsch EC, Hunot S, Hartmann A. Neuroinflammatory processes in Parkinson’s disease. Parkinsonism Relat Disord. 2005;11(Suppl 1):S9–S15. doi: 10.1016/j.parkreldis.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y, Erdmann N, Peng H, Zhao Y, Zheng J. The role of TNF related apoptosis-inducing ligand in neurodegenerative diseases. Cell Mol Immunol. 2005;2:113–122. [PubMed] [Google Scholar]
- 22.Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24:719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- 23.John GR, Lee SC, Brosnan CF. Cytokines: powerful regulators of glial cell activation. Neuroscientist. 2003;9:10–22. doi: 10.1177/1073858402239587. [DOI] [PubMed] [Google Scholar]
- 24.Kapadia SE, de Lanerolle NC. Immunohistochemical and electron microscopic demonstration of vascular innervation in the mammalian brainstem. Brain Res. 1984;292:33–39. doi: 10.1016/0006-8993(84)90887-4. [DOI] [PubMed] [Google Scholar]
- 25.Kniesel U, Wolburg H. Tight junctions of the blood-brain barrier. Cell Mol Neurobiol. 2000;20:57–76. doi: 10.1023/a:1006995910836. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi I, Osawa M, Ohta K, Maruyama S. L-dopa-induced asterixis. Folia Psychiatr Neurol Jpn. 1985;39:507–513. doi: 10.1111/j.1440-1819.1985.tb00804.x. [DOI] [PubMed] [Google Scholar]
- 27.Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, Hendrikse NH. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57:176–179. doi: 10.1002/ana.20369. [DOI] [PubMed] [Google Scholar]
- 28.Kurkowska-Jastrzebska I, Wronska A, Kohutnicka M, Czlonkowski A, Czlonkowska A. The inflammatory reaction following 1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol. 1999;156:50–61. doi: 10.1006/exnr.1998.6993. [DOI] [PubMed] [Google Scholar]
- 29.Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 30.Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-kappaB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol. 2006;84:49–59. doi: 10.1139/Y05-143. [DOI] [PubMed] [Google Scholar]
- 32.Leng A, Mura A, Feldon J, Ferger B. Tumor necrosis factor-alpha receptor ablation in a chronic MPTP mouse model of Parkinson’s disease. Neurosci Lett. 2005;375:107–111. doi: 10.1016/j.neulet.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 33.Ling Z, Chang QA, Tong CW, Leurgans SE, Lipton JW, Carvey PM. Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally. Exp Neurol. 2004;190:373–383. doi: 10.1016/j.expneurol.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 34.Lynch NJ, Willis CL, Nolan CC, Roscher S, Fowler MJ, Weihe E, Ray DE, Schwaeble WJ. Microglial activation and increased synthesis of complement component C1q precedes blood-brain barrier dysfunction in rats. Mol Immunol. 2004;40:709–716. doi: 10.1016/j.molimm.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Mankertz J, Tavalali S, Schmitz H, Mankertz A, Riecken EO, Fromm M, Schulzke JD. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci. 2000;113 ( Pt 11):2085–2090. doi: 10.1242/jcs.113.11.2085. [DOI] [PubMed] [Google Scholar]
- 36.McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord. 2004;10(Suppl 1):S3–S7. doi: 10.1016/j.parkreldis.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 37.Mennicken F, Maki R, de Souza EB, Quirion R. Chemokines and chemokine receptors in the CNS: a possible role in neuroinflammation and patterning. Trends Pharmacol Sci. 1999;20:73–78. doi: 10.1016/s0165-6147(99)01308-5. [DOI] [PubMed] [Google Scholar]
- 38.Miller F, Fenart L, Landry V, Coisne C, Cecchelli R, Dehouck MP, Buee-Scherrer V. The MAP kinase pathway mediates transcytosis induced by TNF-alpha in an in vitro blood-brain barrier model. Eur J Neurosci. 2005;22:835–844. doi: 10.1111/j.1460-9568.2005.04273.x. [DOI] [PubMed] [Google Scholar]
- 39.Nadjar A, Tridon V, May MJ, Ghosh S, Dantzer R, Amedee T, Parnet P. NFkappaB activates in vivo the synthesis of inducible Cox-2 in the brain. J Cereb Blood Flow Metab. 2005;25:1047–1059. doi: 10.1038/sj.jcbfm.9600106. [DOI] [PubMed] [Google Scholar]
- 40.Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson’s disease. J Neural Transm Suppl. 2000:143–151. [PubMed] [Google Scholar]
- 41.Nagatsu T, Sawada M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des. 2005;11:999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- 42.O’callaghan JP, Miller DB, Reinhard JF., Jr Characterization of the origins of astrocyte response to injury using the dopaminergic neurotoxicant, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 1990;521:73–80. doi: 10.1016/0006-8993(90)91526-m. [DOI] [PubMed] [Google Scholar]
- 43.Orr CF, Rowe DB, Halliday GM. An inflammatory review of Parkinson’s disease. Prog Neurobiol. 2002;68:325–340. doi: 10.1016/s0301-0082(02)00127-2. [DOI] [PubMed] [Google Scholar]
- 44.Ozaki H, Ishii K, Horiuchi H, Arai H, Kawamoto T, Okawa K, Iwamatsu A, Kita T. Cutting edge: combined treatment of TNF-alpha and IFN-gamma causes redistribution of junctional adhesion molecule in human endothelial cells. J Immunol. 1999;163:553–557. [PubMed] [Google Scholar]
- 45.Petrache I, Crow MT, Neuss M, Garcia JG. Central involvement of Rho family GTPases in TNF-alpha-mediated bovine pulmonary endothelial cell apoptosis. Biochem Biophys Res Commun. 2003;306:244–249. doi: 10.1016/s0006-291x(03)00945-8. [DOI] [PubMed] [Google Scholar]
- 46.Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res Brain Res Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem. 2001;76:1265–1274. doi: 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- 48.Raichle ME, Hartman BK, Eichling JO, Sharpe LG. Central noradrenergic regulation of cerebral blood flow and vascular permeability. Proc Natl Acad Sci U S A. 1975;72:3726–3730. doi: 10.1073/pnas.72.9.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rennels ML, Nelson E. Capillary innervation in the mammalian central nervous system: an electron microscopic demonstration. Am J Anat. 1975;144:233–241. doi: 10.1002/aja.1001440208. [DOI] [PubMed] [Google Scholar]
- 50.Rousselet E, Callebert J, Parain K, Joubert C, Hunot S, Hartmann A, Jacque C, Perez-Diaz F, Cohen-Salmon C, Launay JM, Hirsch EC. Role of TNF-alpha receptors in mice intoxicated with the parkinsonian toxin MPTP. Exp Neurol. 2002;177:183–192. doi: 10.1006/exnr.2002.7960. [DOI] [PubMed] [Google Scholar]
- 51.Ryu JK, McLarnon JG. Minocycline or iNOS inhibition block 3-nitrotyrosine increases and blood-brain barrier leakiness in amyloid beta-peptide-injected rat hippocampus. Exp Neurol. 2006;198:552–557. doi: 10.1016/j.expneurol.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 52.Sugama S, Yang L, Cho BP, DeGiorgio LA, Lorenzl S, Albers DS, Beal MF, Volpe BT, Joh TH. Age-related microglial activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration in C57BL/6 mice. Brain Res. 2003;964:288–294. doi: 10.1016/s0006-8993(02)04085-4. [DOI] [PubMed] [Google Scholar]
- 53.Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- 54.Tsao N, Hsu HP, Wu CM, Liu CC, Lei HY. Tumour necrosis factor-alpha causes an increase in blood-brain barrier permeability during sepsis. J Med Microbiol. 2001;50:812–821. doi: 10.1099/0022-1317-50-9-812. [DOI] [PubMed] [Google Scholar]
- 55.Vu TQ, Ling ZD, Ma SY, Robie HC, Tong CW, Chen EY, Lipton JW, Carvey PM. Pramipexole attenuates the dopaminergic cell loss induced by intraventricular 6-hydroxydopamine. J Neural Transm. 2000;107:159–176. doi: 10.1007/s007020050014. [DOI] [PubMed] [Google Scholar]
- 56.Wakayama K, Ohtsuki S, Takanaga H, Hosoya K, Terasaki T. Localization of norepinephrine and serotonin transporter in mouse brain capillary endothelial cells. Neurosci Res. 2002;44:173–180. doi: 10.1016/s0168-0102(02)00120-7. [DOI] [PubMed] [Google Scholar]
- 57.Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. 2004;190:446–455. doi: 10.1016/j.expneurol.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 58.Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate amage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37:1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 60.Zemke D, Majid A. The potential of minocycline for neuroprotection in human neurologic disease. Clin Neuropharmacol. 2004;27:293–298. doi: 10.1097/01.wnf.0000150867.98887.3e. [DOI] [PubMed] [Google Scholar]