

Abstract
Objective
To analyze the effect of white matter lesions in different brain regions on regional cortical glucose metabolism, regional cortical atrophy, and cognitive function in a sample with a broad range of cerebrovascular disease and cognitive function.
Methods
Subjects (n = 78) were recruited for a study of subcortical ischemic vascular disease (SIVD) and Alzheimer disease (AD) contributions to dementia. A new method was developed to define volumes of interest from high-resolution three-dimensional T1-weighted MR images. Volumetric measures of MRI segmented white matter signal hyperintensities (WMH) in five different brain regions were related to regional PET glucose metabolism (rCMRglc) in cerebral cortex, MRI measures of regional cortical atrophy, and neuropsychological assessment of executive and memory function.
Results
WMH was significantly higher in the prefrontal region compared to the other brain regions. In all subjects, higher frontal and parietal WMH were associated with reduced frontal rCMRglc, whereas occipitotemporal WMH was only marginally associated with frontal rCMRglc. These associations were stronger and more widely distributed in nondemented subjects where reduced frontal rCMRglc was correlated with WMH for all regions measured. In contrast, there was no relationship between WMH in any brain region and rCMRglc in either parietal or occipitotemporal regions. WMHs in all brain regions were associated with low executive scores in nondemented subjects.
Conclusions
The frontal lobes are most severely affected by SIVD. WMHs are more abundant in the frontal region. Regardless of where in the brain these WMHs are located, they are associated with frontal hypometabolism and executive dysfunction.
Vascular factors are increasingly recognized as important contributors to cognitive decline and dementia. Subcortical ischemic vascular disease (SIVD) accounts for 36 to 67% of all vascular dementia cases1 and is a major cause of vascular cognitive impairment and dementia in the elderly. There is also evidence that coexisting cerebrovascular disease can amplify the effects of Alzheimer disease (AD) pathology.2,3
While impairment of executive function in patients with SIVD has been an often described feature of the syndrome,4-7 the specific mechanisms of the cerebrovascular changes leading to brain dysfunction and cognitive symptoms remain to be elucidated. Prominent imaging findings seen in cognitively impaired patients with SIVD include MRI findings of subcortical lacunes and white matter signal hyperintensities (WMH) on T2-weighted images. In addition to lacunar infarctions it is generally assumed that ischemic WMH cause disturbances in prefrontal-subcortical loops involved in executive control.7-9 Accordingly, WMH have been shown to correspond to poor executive performance but not to global or memory function.10,11 The mechanisms underlying the relationship between WMH and executive dysfunction have not been adequately elucidated, however. Possibilities include cortical atrophy perhaps secondary to WMH,12-14 and disconnection or diaschisis of frontal lobe function caused by WMH. The latter hypothesis can be examined using PET measures of glucose metabolism.
We sought to examine the regional distribution of WMH and define their effects on regional cortical glucose metabolism and atrophy. We hypothesized that WMH within a specific brain region would have stronger effects on glucose metabolism in the same region than on glucose metabolism in different regions. Similarly, we anticipated finding regional predominance of WMH in the frontal lobes that would explain the predominant executive dysfunction seen in this syndrome. We developed a new method to define volumes of interest (VOIs) from high-resolution three-dimensional T1-weighted MR images. Volumetric measures of MRI segmented WMH in five different brain regions were related to regional PET glucose metabolism in cortical gray matter (CGM) voxels, MRI measures of frontal cortical atrophy, and neuropsychological measures of executive and memory function.
Methods
Subjects
Eighty-four subjects recruited from two academic dementia centers between 1999 and 2002 were evaluated as part of a multicenter collaborative study of contributions of subcortical cerebrovascular brain injury and AD to cognitive impairment and dementia. All subjects received a comprehensive clinical evaluation, an MRI scan, and a PET scan of the brain. Six subjects were excluded due to scans of poor technical quality leaving 78 subjects in the study. Subjects were diagnosed using National Institute of Neurologic and Communicative Disorders and Stroke–AD and Related Disorders Association diagnostic criteria15 for AD and California ADDTC criteria16 for ischemic vascular dementia (IVD). The levels of cognitive impairment included normal (Clinical Dementia Rating [CDR]17,18 score of 0.0), cognitively impaired not demented (CIND) (CDR = 0.5), and demented (CDR ≥ 1.0). The Institutional Review Boards at all participating institutions approved this study and subjects or their legal representatives gave written informed consent for participating.
The MRI study was performed within 0.33 ± 0.48 (mean ± SD) (range 0 to 2.5) years and the neuropsychological testing within 0.24 ± 0.28 (0 to 1.6) years from the PET study. None of the subjects had a history of head injury or MRI signs of cortical infarcts.
Neuropsychological tests
All participants received a standardized battery of neuropsychological tests in common clinical use, details of which have been reported previously.19 Neuropsychological variables were selected a priori and consisted of the MMSE,20 and composite measures of executive and memory function. The executive function measure (EXEC) is a composite derived from Dementia Rating Scale Initiation–Perseveration,21 Wechsler Memory Scale–Revised digit span backwards and visual span backwards,22 and the results of a controlled oral word fluency test (FAS).23 The Memory function (MEM) measure was derived using total recall on Trials 2 to 6 on the Word List Learning Test of the Memory Assessment Scales along with the scores for short and long delayed free recall from the same test.24 The composite measures were constructed using methods associated with item response theory, details of which have been described previously.25 Scores were transformed in reference to a large normal control group yielding a scale corresponding to a traditional measurement scale with a mean of 100 and a SD of 15.
MRI data acquisition
The entire brain was imaged with a 1.5 T Magnetom VISION system (Siemens, Erlangen, Germany) equipped with a standard quadrature head coil. The brain imaging protocol involved 1) a sequence yielding proton density (PD) and T2-weighted spin-echo axial images (repetition time [TR]/echo time [TE]1/TE2 2500/20/80 msec; 1 number of excitations [NEX]), 3 mm slice thickness with no slice gap, in-plane resolution 0.94 × 0.94 mm2, and 2) a sequence yielding T1-weighted coronal three-dimensional images (TR/TE 10/4 msec; 1 NEX), 1 × 1 mm2 in-plane resolution, with contiguous 1.4 mm thick slices. The coronal three-dimensional T1 image was resliced in axial orientation yielding a voxel dimension of 0.94 × 0.94 × 1.6 mm (256 × 256 × 124 voxels) using previously described software.26
MRI data analysis
A previously described computerized segmentation algorithm was used to classify brain MRI pixels on the T2 image as CGM, subcortical gray matter, white matter, WMH, ventricular CSF, sulcal CSF, or subcortical gray matter lacunes.27
MR data analysis used a series of steps: 1) segmentation of brain from non-brain and coregistration of images, 2) definition of 10 VOIs, and 3) alignment of VOIs to the segmented dataset and calculation of tissue type volumes for the five VOIs used in analysis. These three steps are described below.
First, the brain was segmented from non-brain on the T1 image by a technician. The T1 and segmented MR datasets were then coregistered using an affine coregistration program developed in-house following the method given in reference 28.
Next, the high-resolution three-dimensional T1-weighted image was divided into 10 VOIs using software developed in-house. This program uses a ray-casting method to define three-dimensional VOIs using operator-defined planes for cutting the VOIs (figure 1, top). This procedure resulted in each brain hemisphere being divided into five VOIs: 1) orbitofrontal; 2) prefrontal; 3) dorsolateral frontal; 4) parietal; 5) occipitotemporal VOI (figure 1, bottom). Cerebellum and brainstem were excluded from the occipitotemporal VOI by manual editing as were possible parts of temporal poles included in the orbitofrontal VOI.
By aligning each VOI with the segmented dataset using the previously computed coregistration parameters, we classified voxels in the VOI as CGM, normal white matter, or WMH. Tissue type volumes for the corresponding right and left VOIs were added creating five bilateral VOIs, which were used in the data analysis. A frontal VOI was created by adding the tissue type volumes of the bilateral orbitofrontal, prefrontal, and dorsolateral VOIs.
Figure 1.
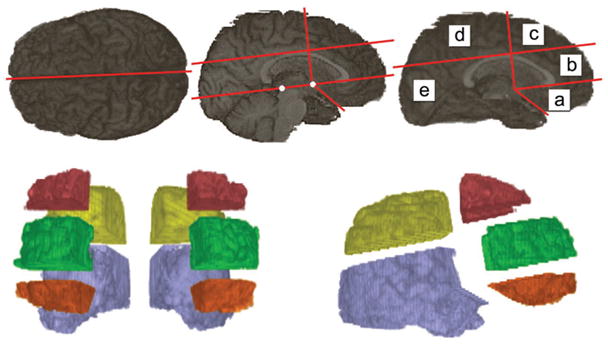
The definition of 10 volumes of interest from the high-resolution three-dimensional T1-weighted MR image. Anatomic landmarks used are anterior and posterior commissures (white dots), outer boundary of callosal body, and the point where the frontal and temporal lobes meet; a = orbitofrontal; b = prefrontal; c = dorsolateral frontal; d = parietal; e = occipitotemporal.
Regional WMH volume was converted to percent of total white matter volume in the same region to control for subject differences in VOI size. CGM volumes are expressed as percent of intracranial volume (ICV).
This VOI analysis was performed by one of the authors (M.T.) blind to all other data, with high reliability (intraclass correlations of 0.97 to 1.0 with the volume of WMH in each of 10 unilateral VOIs on three brains analyzed five times).
PET data acquisition
PET studies were performed on a Siemens-CTI ECAT EXACT (model 921) 47-section scanner in two-dimensional acquisition mode imaging the radiotracer, [18F]-FDG.29 All subjects were injected with 5 to 10 mCi of FDG and studied in a quiet room with eyes and ears unoccluded with a 40-minute emission acquisition in two-dimensional mode following a 20-minute transmission image.
The PET image was aligned to the segmented image using the coregistration method described above so that, for each VOI, radioactivity counts in CGM voxels could be extracted. In order to reduce partial volume effects, CGM counts were calculated as mean of the counts at the FWHM value of the histogram of all voxels within the gray matter volume. VOI PET counts were normalized to whole brain mean counts and used in data analyses as count ratios.
Statistics
WMH distributions within regions were positively skewed and were therefore log transformed. Repeated measures models, with post hoc pairwise contrasts between the prefrontal region and the other brain regions, were used to assess differences between the averages of the regional WMH. Analysis of variance (ANOVA) was used to test for differences in average WMH, glucose metabolism (rCMRglc), and CGM volumes between different cognitive groups. Linear regression models were used to investigate relationships between regional WMH, rCMRglc, CGM, and executive and memory function. Primary outcomes included rCMRglc and cognitive function. We began with simple unadjusted regression models, followed by multivariate models, when possible. Correlations between the independent variables were investigated to check for colinearity. Assumptions of all models were checked through graphical methods and were met by the data. A p value less than 0.05 was considered significant, although the Bonferroni correction was implemented to correct for multiple comparisons in the post hoc pairwise tests.
Results
Table 1 shows demographic data and results of cognitive testing for all subjects and for the cognitive subgroups. Nineteen of the demented subjects were diagnosed as AD, five as mixed AD and IVD, and two as IVD. Age was a nonsignificant covariate in all the statistical models used to study relationships among WMH, glucose metabolism, executive (EXEC), and memory (MEM) function measures.
Table 1.
Clinical characteristics of the study sample
| Characteristics | All, n = 78 | CN, n = 22 | CIND, n = 30 | Demented, n = 26 |
|---|---|---|---|---|
| M/F | 51/27 | 8/14 | 22/8 | 21/5 |
| Age, y, mean (SD) | 77.2 (7.7) | 76.6 (6.5) | 78.2 (7.7) | 76.5 (8.7) |
| MMSE, mean (SD) | 25.9 (4.9) | 29.6 (0.7) | 27.7 (2.7) | 20.7 (4.6) |
| Executive score, mean (SD) | 87.0 (21.6) | 102.4 (14.1) | 91.9 (20.7) | 68.5 (16.6) |
| Memory score, mean (SD) | 88.0 (28.2) | 112.0 (21.4) | 90.0 (19.3) | 57.8 (10.2) |
| Dementia etiology | ||||
| AD | 19 | |||
| Mixed AD 3 IVD | 5 | |||
| IVD | 2 |
CN = cognitively normal; CIND = cognitively impaired not demented; MMSE = Mini-Mental State Examination; AD = Alzheimers disease; IVD = ischemic vascular dementia.
Regional distribution of WMH and normalized glucose metabolism
WMH in different brain regions were highly correlated: rp ≥ 0.78 for frontal, parietal, and occipitotemporal WMH. Figures 2 and 3 illustrate the regional distribution of average WMH and average glucose metabolism (rCMRglc) in the different clinical groups. Average WMH was higher in the prefrontal VOI than in each of the other VOIs in all subjects and also within each clinical group (uncorrected p values < 0.0005, except for contrasts involving dorsolateral frontal [p = 0.003] and parietal [p = 0.007] in the cognitively normal group and parietal [p = 0.005] in the cognitively impaired, not demented group) (see figure 2). There were no significant differences in frontal, parietal, or occipitotemporal WMH between nondemented (n = 52) and demented (n = 26) subjects (p = 0.42 to 0.86) (ANOVA) and the range of WMH values was similar in both groups.
Figure 2.
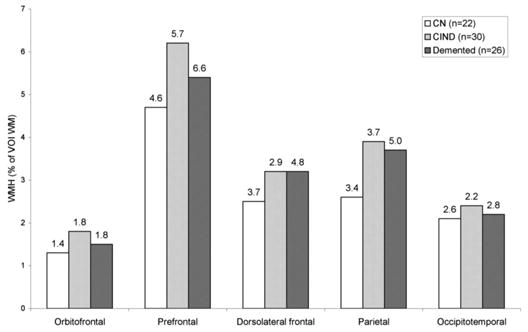
Regional distributions of white matter (WM) signal hyperintensities in different clinical groups. Values above bars are SD. White bars = cognitively normal (CN) (n = 22); light gray bars = cognitively impaired not demented (CIND) (n = 30); dark gray bars = demented (n = 26).
Figure 3.
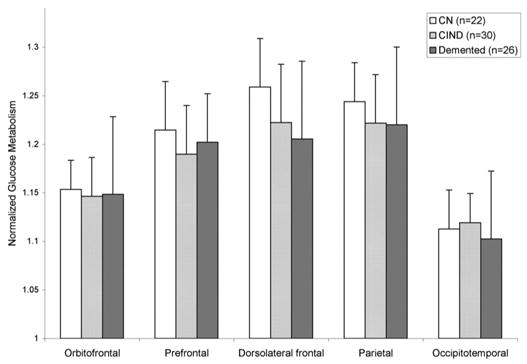
Regional distribution of normalized glucose metabolism. Error bars represent standard deviations. White bars = cognitively normal (CN) (n = 22); light gray bars = cognitively impaired not demented (CIND) (n = 30); dark gray bars = demented (n = 26).
Demented subjects had lower rCMRglc in the dorsolateral frontal VOI compared to cognitively normal subjects (p = 0.027) (ANOVA); otherwise, no differences were seen between the groups (see figure 3).
Regional WMH and frontal glucose metabolism
Table 2 summarizes the frontal rCMRglc by regional WMH regression analyses. Due to the high correlations between the WMH measures in the different brain regions, only simple regressions were fit. Frontal and parietal WMH volumes were significantly and negatively associated with glucose metabolism in the frontal region. Occipitotemporal WMH showed a similar relationship, although not statistically significant. These associations were stronger in nondemented subjects (figure 4). In contrast, there was no relationship between WMH in any brain region and glucose metabolism in either parietal or occipitotemporal lobes in all subjects or nondemented subjects. Adding frontal CGM to the model did not significantly change the results. The relationships between frontal lobe WMH and rCMRglc were not appreciably different in frontal lobe subregions (see table 2).
Table 2.
Frontal glucose metabolism by log-transformed regional WMH
| All subjects (n = 78)
|
Nondemented (n = 52)
|
Demented (n = 26)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WMH region | R2 | Est | SE | p Value | R2 | Est | SE | p Value | R2 | Est | SE | p Value |
| Frontal | 0.06 | −0.010 | 0.005 | 0.043 | 0.15 | −0.015 | 0.005 | 0.008 | 0.004 | −0.003 | 0.008 | 0.76 |
| Orbitofrontal | 0.06 | −0.008 | 0.004 | 0.058 | 0.16 | −0.012 | 0.004 | 0.007 | 0.01 | 0.005 | 0.01 | 0.67 |
| Prefrontal | 0.06 | −0.009 | 0.004 | 0.034 | 0.15 | −0.013 | 0.005 | 0.007 | 0.004 | −0.002 | 0.008 | 0.77 |
| Dorsolat. fr. | 0.05 | −0.007 | 0.004 | 0.072 | 0.08 | −0.010 | 0.005 | 0.061 | 0.02 | −0.005 | 0.007 | 0.48 |
| Parietal | 0.07 | −0.008 | 0.004 | 0.033 | 0.10 | −0.010 | 0.004 | 0.028 | 0.03 | −0.006 | 0.008 | 0.48 |
| Occ. temp. | 0.05 | −0.008 | 0.004 | 0.072 | 0.11 | −0.012 | 0.005 | 0.023 | 0.006 | −0.003 | 0.008 | 0.72 |
Bivariate linear least square regression. R2 is the coefficient of determination and gives a measure of the amount of variability in glucose metabolism that is accounted for by the WMH measures in the model.
WMH = white matter signal hyperintensities; Est = estimate; SE = standard error; Dorsolat. front. = dorsolateral frontal; Occ. temp = occipitotemporal.
Figure 4.
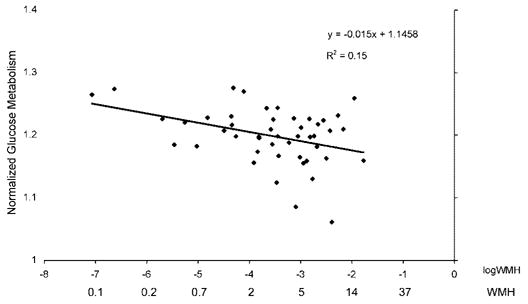
Frontal glucose metabolism by frontal white matter signal hyperintensities in nondemented subjects.
Cortical gray matter volume
Frontal CGM was lower in demented subjects (8.9 ± 0.9%) compared to cognitively normal (10.0 ± 1.2%) (p < 0.001) and CIND subjects (9.5 ± 1.0%) (p = 0.03). Also posterior CGM was lower in demented subjects (20.6 ± 2.3%) compared to cognitively normal (22.5 ± 2.1%) (p = 0.003) and CIND subjects (21.9 ± 1.8%) (p = 0.03). Increased WMH in the frontal region were associated with reduced frontal CGM in all subjects (R2 = 0.11, estimate [β] = −0.003, standard error [SE] = 0.001, p = 0.005) and also in nondemented (R2 = 0.11, β = −0.003, SE = 0.001, p = 0.02) and demented (R2 = 0.32, β = −0.004, SE = 0.001, p = 0.005) subjects.
The influence of imaging measures on cognitive function
Tables 3 and 4 show regression analyses using frontal WMH, rCMRglc, and CGM as independent variables explaining the two cognitive measures EXEC (see table 3) and MEM (see table 4). Once again, due to the high correlations between the WMH measures in the different regions, only one WMH measure could be included in each regression model.
Table 3.
Executive function (EXEC) by frontal WMH (on the log scale), frontal glucose metabolism (rCMRglc), and frontal CGM (bivariate or multivariate regression)
| All subjects (n = 78)
|
Nondemented (n = 52)
|
Demented (n = 26)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | R2 | Est | SE | p | R2 | Est | SE | p | R2 | Est | SE | p |
| Frontal WMH | 0.03 | −3.1 | 2.1 | 0.14 | 0.14 | −5.9 | 2.2 | 0.01 | 0.02 | −1.5 | 2.5 | 0.56 |
| Orbitofrontal | 0.03 | −2.2 | 1.9 | 0.25 | 0.006 | −1.0 | 3.3 | 0.75 | ||||
| Prefrontal | 0.12 | −4.8 | 2.0 | 0.02 | 0.03 | −1.8 | 2.4 | 0.47 | ||||
| Dorsolateral frontal | 0.12 | −5.1 | 2.1 | 0.02 | 0.007 | −0.8 | 2.2 | 0.73 | ||||
| Frontal rCMRglc | 0.16 | 183.4 | 49.9 | 0.0005 | 0.17 | 188.1 | 60.1 | 0.003 | 0.11 | 92.2 | 58.4 | 0.13 |
| Frontal CGM | 0.14 | 723.1 | 212.4 | 0.001 | 0.05 | 381.2 | 228.8 | 0.10 | 0.07 | 464.4 | 382.5 | 0.24 |
| Frontal WMH + rCMRglc | 0.12 | 0.02* | 0.21 | 0.008* | 0.07 | 0.52* | ||||||
| Frontal WMH + rCMRglc + CGM | 0.22 | 0.002* | 0.22 | 0.02* | 0.11 | 0.58* | ||||||
R2 is the coefficient of determination and gives a measure of the amount of variability in EXEC that is accounted for by the independent variables in the model.
p Values for comparing the presented model to the null model.
WMH = white matter signal hyperintensities; CGM = cortical gray matter; EST = estimate; SE = standard error.
Table 4.
Memory function (MEM) by log-transformed frontal WMH (on the log scale), frontal glucose metabolism (rCMRglc), and frontal CGM (bivariate or multivariate regression)
| All subjects (n = 78)
|
Nondemented (n = 52)
|
Demented (n = 26)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Region | R2 | Est | SE | p | R2 | Est | SE | p | R2 | Est | SE | p |
| Frontal WMH | 0.03 | −4.2 | 2.9 | 0.15 | 0.12 | −7.0 | 2.8 | 0.02 | 0.003 | −0.4 | 1.8 | 0.83 |
| Orbitofrontal | 0.01 | −1.9 | 2.4 | 0.44 | 0.0002 | −0.1 | 2.4 | 0.95 | ||||
| Prefrontal | 0.14 | −6.5 | 2.5 | 0.01 | 0.002 | −0.3 | 1.8 | 0.85 | ||||
| Dorsolateral frontal | 0.10 | −5.6 | 2.6 | 0.04 | 0.001 | −0.2 | 1.6 | 0.89 | ||||
| Frontal rCMRglc | 0.04 | 124.7 | 69.2 | 0.08 | 0.04 | 113.6 | 79.6 | 0.16 | 0.04 | −32.6 | 38.3 | 0.41 |
| Frontal CGM | 0.22 | 1173.4 | 263.7 | <0.0001 | 0.16 | 830.7 | 271.7 | 0.004 | 0.01 | 119.6 | 243.5 | 0.63 |
| Frontal WMH + CGM | 0.21 | 0.0008* | 0.18 | 0.017* | 0.02 | 0.83* | ||||||
| Frontal WMH + CGM + rCMRglc | 0.21 | 0.003* | 0.18 | 0.041* | 0.32 | 0.12* | ||||||
R2 is the coefficient of determination and gives a measure of the amount of variability in MEM that is accounted for by the independent variables in the model.
p Values for comparing the presented model to the null model.
WMH = white matter signal hyperintensities; CGM = cortical gray matter; Est = estimate; SE = standard error.
Executive function
Increased frontal WMH volume was associated with lower EXEC in nondemented subjects only (see table 3). Within the frontal region, this relationship was significant for prefrontal and dorsolateral frontal WMH, but not for the orbitofrontal region. Increases in both parietal (R2 = 0.19, β = −5.3, SE = 1.7, p = 0.003) and occipital WMH (R2 = 0.10, β = −4.6, SE = 2.1, p = 0.04) were associated with lower EXEC in nondemented subjects. In demented subjects, however, there were no significant relationships between frontal or posterior WMHs and EXEC. Frontal rCMRglc was related to EXEC in all subjects and also in nondemented subjects, with increases in glucose metabolism associated with higher executive scores (see table 3). This association was not significant in demented subjects. Increased frontal CGM was associated with higher EXEC in all subjects but not in the nondemented or demented groups taken separately. The combination of frontal WMH and rCMRglc explained 21% of the variability in EXEC in nondemented subjects (p = 0.008), with estimates similar to those in the bivariate models (for WMH, β = −4.13; for rCMRglc, β = 127.5). Adding frontal CGM to this model did not explain further variance in EXEC performance.
Memory function
Increased frontal WMH was associated with reduced MEM in nondemented subjects only (see table 4). Frontal rCMRglc was not associated with MEM in any group. Increased frontal CGM was associated with higher MEM in all subjects and nondemented subjects but not in demented subjects. The combination of frontal WMH and CGM explained 18% of the variability in MEM in the nondemented group (p = 0.017), with estimates similar to or reduced compared to the bivariate models (for WMH, β = −5.32; for CGM, β = 504.7). Adding frontal rCMRglc to the model did not change the R2 substantially (see table 4). Posterior CGM was not related to MEM in nondemented or demented subjects (R2 = 0.01 to 0.02, p = 0.30 to 0.67) (data not shown).
Twenty-five subjects had segmented subcortical gray matter lacunes in one or more locations. Excluding these subjects did not have any influence on the relationships found between WMH and glucose metabolism or EXEC.
Discussion
This study focused on the relationships between regional WMH, glucose metabolism, and cognitive function. The major findings are as follows: 1) WMHs were predominant in the frontal lobes. 2) Regardless of their regional distribution, WMH exerted their greatest metabolic effects on the frontal lobes. These effects were more prominent in nondemented subjects and were absent in demented subjects. 3) Executive function was strongly influenced by WMH and frontal glucose metabolism whereas cortical atrophy did not contribute to executive function. 4) Memory function was related to both frontal WMH and cortical atrophy but not to frontal glucose metabolism. In the aggregate, these findings support the view that WMH in any brain region cause frontal cortical hypo-metabolism that at least partially explains the executive dysfunction seen in vascular brain injury. The subcortical ischemic vascular pathology has a moderately strong influence on frontal cortical metabolism and cognitive function that is significant in nondemented subjects. However, in demented subjects who presumably have coexisting AD and IVD, the impact of AD pathology on cortical function appears to be of a considerably higher magnitude, making the additional effects of concomitant cerebrovascular pathology nonsignificant.
The relation between WMH and cortical metabolism has recently been studied with conflicting results, some reports showing that subjects with SIVD have reduced global30,31 or frontal31,32 cortical metabolism whereas other studies have failed to show that WMH are related to cortical metabolism33 or blood flow.34,35 These studies have used global and in most cases semiquantitative measures of white matter lesions. It is reasonable to assume that both the location and the volume of WMH may influence the effect of white matter damage on regional cortical metabolism and cognitive function. Thus, these factors should be taken into account when looking for relationships between subcortical structural abnormalities and cortical metabolism.
The methods used in this study have several advantages for analyzing WMH and cortical glucose metabolism. The segmentation of WMH on MRI gives a good estimate of WMH volume. The coregistration method and the VOI approach make a regional comparison possible. We used the coregistered segmented image to identify cortical gray matter voxels in the PET image to ensure a good measure of cortical gray matter glucose metabolism and to reduce the effect of cortical atrophy. Accordingly, the associations found between WMH and frontal rCMRglc were independent of frontal CGM.
The rationales behind evaluating the entire sample as a whole and the subsequent grouping of subjects in a nondemented (cognitively normal and CIND subjects) and a demented group (AD and IVD cases) are 1) the development of WMHs and their influence on cortical metabolism and cognitive function is a continuous process taking place in subjects with subcortical vascular disease throughout the whole spectrum of cognitive function, 2) all subjects included were selected to have signs or risk factors of cerebrovascular disease and therefore presumably the same etiology of WMH, and 3) the focus of this study was on pathophysiology and not on diagnostic issues.
WMH volume was significantly higher in the pre-frontal region than in any of the other regions. This is a novel finding and points at the prefrontal white matter as being particularly vulnerable to small-artery disease, which may relate to periventricular arterial borderzones highly susceptible to hypoperfusion.36,37
WMH in all regions were associated with reduced frontal glucose metabolism and cortical atrophy. A possible mechanism for these findings could be that lesions in the subcortical white matter cause structural or metabolic damage to axons that downregulate neuronal metabolism and cause cortical hypometabolism and even cell death. Accordingly, subcortical infarcts have been shown to produce cortical hypometabolism and hypoperfusion (diaschisis).38-40 These changes in brain structure may underlie, to some extent, the metabolic changes, although based on research from other samples, atrophy is not likely to be the only explanation for metabolic declines.41
The preferential effect on frontal cortex by WMH is unexplained, but could be related to substantial convergence of fiber pathways on the frontal lobes, so that lesions in any subcortical region might be expected to exert their effects on this large region of association cortex. This could also explain why WMH in both frontal and posterior regions were associated with frontal hypometabolism. Frontal WMH were more strongly related to frontal hypometabolism than posterior white matter lesions, possibly because the proportion of axons connecting to the frontal cortex is highest in the frontal white matter.
The high correlations between WMH in different regions indicate that cerebrovascular disease is generalized. It is thus also possible that the correlation between posterior WMH and frontal metabolism is driven by a correlation with frontal WMH that reflects this generalized process. Another possible explanation for the relation between WMH and cortical hypometabolism could be concomitant microvascular changes in the cerebral cortex more directly related to cortical metabolism with white matter lesions representing only a general marker of cerebrovascular disease. Accordingly, cortical microinfarctions have been reported to be an important feature in vascular dementia.42 This process is beyond the resolution of current MR techniques. However, a regional frontal effect of white matter lesions on cortical metabolism in our study may speak against this assumption since cortical microinfarctions are symmetrically distributed throughout frontal and posterior cortical regions.42
In the demented subjects studied, WMH was not related to frontal glucose metabolism. The amount and distribution of WMH in the demented subjects was similar to that of the nondemented subjects. It is therefore not likely that a restricted range of WMH volume in the demented group explains these differences. A probable explanation for these findings is that AD was the dominating cause of dementia in this sample: 25 of the dementia subjects were diagnosed with AD or mixed AD and IVD while only 2 subjects were diagnosed with IVD. In AD, the pathophysiologic mechanism is localized to the cerebral cortex with decreased glucose metabolism and atrophy. The process involves the medial temporal lobes and neocortex.43 The impact of AD pathology on cortical function is probably of a higher magnitude than that of subcortical ischemic vascular pathology. Hence, in AD subjects with concomitant cerebrovascular disease, it is more likely that the AD pathology in the frontal cortex determines the frontal cortical function in an individual, and not the subcortical WMH volume. AD pathology is probably also present in some of the subjects included in the nondemented group. The effect of subclinical AD on frontal metabolism and executive function may be of such a low magnitude that it is still possible to distinguish the specific effects of SIVD in these subjects.
Frontal, but also posterior WMH was related to executive function in nondemented subjects only. Within the frontal region, the dorsolateral and pre-frontal WMH volumes were the only significant predictors whereas the orbitofrontal region had a much weaker and insignificant correlation. These results indicate that WMHs are indeed related to the executive dysfunction in SIVD, which corresponds to previous reports10,11 and also to the fact that it is mainly the dorsolateral prefrontal circuit that is responsible for executive function whereas dysfunction in the inferior orbitofrontal circuit is characterized by behavioral disinhibition such as poor impulse control and emotional lability,44 Again, posterior WMHs seem to have a remote effect on the frontal lobes, causing frontal cognitive dysfunction.
The effect of frontal glucose metabolism on executive function in this group was of a similar magnitude to the effect of WMH, whereas frontal CGM did not contribute to executive function. In a multivariate model, the combination of frontal glucose metabolism and frontal WMH only marginally increased the summary of fit R2 compared to each variable taken separately. This is evidence that in SIVD, the executive dysfunction is at least partially explained by reduced frontal glucose metabolism caused by WMH. Addition of CGM had no effect on the model, probably because the effect of atrophy is already accounted for by WMH and rCMRglc.
Frontal WMH was associated with memory dysfunction in nondemented subjects. In this group also frontal but not posterior cortical atrophy was associated with memory dysfunction. These associations were not found in demented subjects. Memory impairment is a well-known feature of SIVD even if it is less prominent than in AD.5 It mainly involves working memory causing forgetfulness, and problems with spontaneous recall. Our results converge with the general knowledge that dorsolateral pre-frontal loops are involved in maintaining working memory,45-47 the failure of which may contribute to episodic memory impairment. Thus, also memory disturbance in SIVD may be explained by underlying subcortical vascular disease causing frontal cortical dysfunction among individuals without hippocampal dysfunction caused by AD (i.e., the dementia group).
The relations between white matter lesions, frontal glucose metabolism, executive function, and cortical atrophy reported in the present study are of only moderately strong magnitude with R2 values around 0.2. A 10% increase in frontal WMH was associated with a decrease of 0.0015 units in frontal rCMRglc in the nondemented group, which equals 3.5% of the sample SD. A 10% increase in frontal WMH was associated with a decrease in EXEC by 0.6 units and in MEM by 0.7 units, i.e., a reduction of approximately 5% of the SD of a cognitively normal group. The majority of subjects had signs of limited subcortical vascular disease and only two subjects were diagnosed as IVD compared to 24 subjects with AD or mixed AD and IVD. This could indicate that the burden of SIVD in the subjects studied was low. It is possible that we would have found a stronger relationship between WMH and frontal glucose metabolism if subjects with more severe SIVD had been included. This hypothesis warrants further study. The relations found over the whole distribution of WMH imply, however, that the negative effect of WMH on frontal glucose metabolism is continuous and that it is not just a threshold effect showing only in subjects with severe cerebrovascular disease.
In this study, we measured rCMRglc under resting conditions. Correlations between WMH, frontal glucose metabolism, and executive function would probably be considerably higher in a PET activation study. Accordingly, correlation coefficients reach 0.8 between activated rCMRglc and memory performance.46 Activation studies may help explain relationships between glucose metabolism, cognition, and WMH in the future. The normalization of regional CMRglc to global values in this study rather than determining absolute values might result in a loss of power to discriminate VOI changes in disease and to detect correlations. The fact that the demented cohort in this study failed to show any evidence of focal parieto-temporal rCMRglc loss that characterizes AD may support this notion. Another reason for this lack of rCMRglc changes typical for AD in the demented group may be the fact that the WMH depressed frontal glucose metabolism to such an extent that relative reductions in posterior brain regions were not as prominent, which is a previously noted finding.32
We did not include subcortical lacunar infarctions in the assessment of SIVD. A previous report from our group concluded that the cognitive impairment in SIVD is relatively unrelated to volume and location of lacunar infarctions.12 Also, the results reported in this study did not change if subjects without lacunar infarctions (n = 25) were excluded.
Footnotes
Supported by research grants from the National Institute on Aging AG10129 and AG12435, by the California Department of Health Services Alzheimer’s Disease Program, and by grants from the Edit Jacobson Foundation, the John and Brit Wennerström Foundation, the Hjalmar Svensson Foundation, the Rune and Ulla Ahlmlöv Foundation, the Swedish Medical Society, Göteborg Medical Society, the Swedish Association of Neurologically Disabled, the P-O Ahl Foundation, the Hellmut Hertz Foundation, Adlerbertska forskningsstiftelsen, Göteborg University, and the Swedish Brain Foundation.
References
- 1.Chui H. Dementia due to subcortical ischemic vascular disease. Clin Cornerstone. 2001;3:40–51. doi: 10.1016/s1098-3597(01)90047-x. [DOI] [PubMed] [Google Scholar]
- 2.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markes-bery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study [see comments] JAMA. 1997;277:813–817. [PubMed] [Google Scholar]
- 3.Nagy Z, Esiri MM, Jobst KA, et al. The effects of additional pathology on the cognitive deficit in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:165–170. doi: 10.1097/00005072-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Cummings JL. Frontal-subcortical circuits and human behavior. Arch Neurol. 1993;50:873–880. doi: 10.1001/archneur.1993.00540080076020. [DOI] [PubMed] [Google Scholar]
- 5.Lafosse JM, Reed BR, Mungas D, Sterling SB, Wahbeh H, Jagust WJ. Fluency and memory differences between ischemic vascular dementia and Alzheimer’s disease. Neuropsychology. 1997;11:514–522. doi: 10.1037//0894-4105.11.4.514. [DOI] [PubMed] [Google Scholar]
- 6.Erkinjuntti T, Inzitari D, Pantoni L, et al. Research criteria for subcortical vascular dementia in clinical trials [in process citation] J Neural Transm Suppl. 2000;59:23–30. doi: 10.1007/978-3-7091-6781-6_4. [DOI] [PubMed] [Google Scholar]
- 7.Roman GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC. Subcortical ischaemic vascular dementia. Lancet Neurology. 2002;1:426–436. doi: 10.1016/s1474-4422(02)00190-4. [DOI] [PubMed] [Google Scholar]
- 8.Ishii N, Nishihara Y, Imamura T. Why do frontal lobe symptoms predominate in vascular dementia with lacunes? Neurology. 1986;36:340–345. doi: 10.1212/wnl.36.3.340. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe N, Linn R, Babikian VL, Knoefel JE, Albert ML. Frontal systems impairment following multiple lacunar infarcts. Arch Neurol. 1990;47:129–132. doi: 10.1001/archneur.1990.00530020025010. [DOI] [PubMed] [Google Scholar]
- 10.O’Brien JT, Wiseman R, Burton EJ, et al. Cognitive associations of subcortical white matter lesions in older people. Ann NY Acad Sci. 2002;977:436–444. doi: 10.1111/j.1749-6632.2002.tb04849.x. [DOI] [PubMed] [Google Scholar]
- 11.Mori E. Impact of subcortical ischemic lesions on behavior and cognition. Ann NY Acad Sci. 2002;977:141–148. doi: 10.1111/j.1749-6632.2002.tb04809.x. [DOI] [PubMed] [Google Scholar]
- 12.Fein G, Di Sclafani V, Tanabe J, et al. Hippocampal and cortical atrophy predict dementia in subcortical ischemic vascular disease. Neurology. 2000;55:1626–1635. doi: 10.1212/wnl.55.11.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mungas D, Jagust WJ, Reed BR, et al. MRI predictors of cognition in subcortical ischemic vascular disease and Alzheimer’s disease. Neurology. 2001;57:2229–2235. doi: 10.1212/wnl.57.12.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pohjasvaara T, Mantyla R, Salonen O, et al. How complex interactions of ischemic brain infarcts, white matter lesions, and atrophy relate to poststroke dementia. Arch Neurol. 2000;57:1295–1300. doi: 10.1001/archneur.57.9.1295. [DOI] [PubMed] [Google Scholar]
- 15.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 16.Chui HC, Victoroff JI, Margolin D, Jagust W, Shankle R, Katzman R. Criteria for the diagnosis of ischemic vascular dementia proposed by the State of California Alzheimer’s Disease Diagnostic and Treatment Centers. Neurology. 1992;42(3 Pt 1):473–480. doi: 10.1212/wnl.42.3.473. [DOI] [PubMed] [Google Scholar]
- 17.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 18.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 19.Mungas D, Reed BR, Jagust WJ, et al. Volumetric MRI predicts rate of cognitive decline related to AD and cerebrovascular disease. Neurology. 2002;59:867–873. doi: 10.1212/wnl.59.6.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folstein MF, Robins LN, Helzer JE. The Mini-Mental State Examination. Arch Gen Psychiatry. 1983;40:812. doi: 10.1001/archpsyc.1983.01790060110016. [DOI] [PubMed] [Google Scholar]
- 21.Mattis S. Dementia Rating Scale Professional Manual. Odessa, FL: Psychological Assessment Resources; 1988. [Google Scholar]
- 22.Wechsler D. Wechsler Memory Scale–Revised (WMS-R) San Antonio, TX: The Psychological Corporation; 1987. [Google Scholar]
- 23.Benton AL, Hamsher KD. Multilingual Aphasia Examination. Iowa City, IA: University of Iowa; 1976. [Google Scholar]
- 24.Williams JM. Memory Assessment Scales. Odessa, FL: Psychological Assessment Resources; 1991. [Google Scholar]
- 25.Mungas D, Reed BR, Kramer JH. Psychometrically matched measures of global cognition, memory, and executive function for assessment of cognitive decline in older persons. Neuropsychology. 2003;17:380–392. doi: 10.1037/0894-4105.17.3.380. [DOI] [PubMed] [Google Scholar]
- 26.Klein GJ, Teng X, Jagust WJ, et al. A methodology for specifying PET VOI’s using multimodality techniques. IEEE Trans Med Imaging. 1997;16:405–415. doi: 10.1109/42.611350. [DOI] [PubMed] [Google Scholar]
- 27.Tanabe JL, Amend D, Schuff N, et al. Tissue segmentation of the brain in Alzheimer disease. AJNR Am J Neuroradiol. 1997;18:115–123. [PMC free article] [PubMed] [Google Scholar]
- 28.Maes F, Collignon A, Vandermeulen D, Marchal G, Suetens P. Multimodality image registration by maximization of mutual information. IEEE Trans Med Imaging. 1997;16:187–198. doi: 10.1109/42.563664. [DOI] [PubMed] [Google Scholar]
- 29.Wienhard K, Eriksson L, Grootoonk S, Casey M, Pietrzyk U, Heiss WD. Performance evaluation of the positron scanner ECAT EXACT. J Comput Assist Tomogr. 1992;16:804–813. doi: 10.1097/00004728-199209000-00024. [DOI] [PubMed] [Google Scholar]
- 30.Sultzer DL, Mahler ME, Cummings JL, Van Gorp WG, Hinkin CH, Brown C. Cortical abnormalities associated with subcortical lesions in vascular dementia. Clinical and position emission tomographic findings. Arch Neurol. 1995;52:773–780. doi: 10.1001/archneur.1995.00540320049012. [DOI] [PubMed] [Google Scholar]
- 31.Kwan LT, Reed BR, Eberling JL, et al. Effects of subcortical cerebral infarction on cortical glucose metabolism and cognitive function. Arch Neurol. 1999;56:809–814. doi: 10.1001/archneur.56.7.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeCarli C, Murphy DG, Tranh M, et al. The effect of white matter hyperintensity volume on brain structure, cognitive performance, and cerebral metabolism of glucose in 51 healthy adults. Neurology. 1995;45:2077–2084. doi: 10.1212/wnl.45.11.2077. [DOI] [PubMed] [Google Scholar]
- 33.Sabri O, Ringelstein EB, Hellwig D, et al. Neuropsychological impairment correlates with hypoperfusion and hypometabolism but not with severity of white matter lesions on MRI in patients with cerebral microangiopathy. Stroke. 1999;30:556–566. doi: 10.1161/01.str.30.3.556. [DOI] [PubMed] [Google Scholar]
- 34.Ott BR, Faberman RS, Noto RB, et al. A SPECT imaging study of MRI white matter hyperintensity in patients with degenerative dementia. Dement Geriatr Cogn Disord. 1997;8:348–354. doi: 10.1159/000106654. [DOI] [PubMed] [Google Scholar]
- 35.O’Sullivan M, Lythgoe DJ, Pereira AC, et al. Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology. 2002;59:321–326. doi: 10.1212/wnl.59.3.321. [DOI] [PubMed] [Google Scholar]
- 36.De Reuck J. The human periventricular arterial blood supply and the anatomy of cerebral infarctions. Eur Neurol. 1971;5:321–334. doi: 10.1159/000114088. [DOI] [PubMed] [Google Scholar]
- 37.Moody DM, Bell MA, Challa VR. Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency: an anatomic study. AJNR Am J Neuroradiol. 1990;11:431–439. [PMC free article] [PubMed] [Google Scholar]
- 38.Baron JC, D’Antona R, Pantano P, Serdaru M, Samson Y, Bousser MG. Effects of thalamic stroke on energy metabolism of the cerebral cortex. A positron tomography study in man. Brain. 1986;109(Pt 6):1243–1259. doi: 10.1093/brain/109.6.1243. [DOI] [PubMed] [Google Scholar]
- 39.Perani D, Papagno C, Cappa S, Gerundini P, Fazio F. Crossed aphasia: functional studies with single photon emission computerized tomography. Cortex. 1988;24:171–178. doi: 10.1016/s0010-9452(88)80027-3. [DOI] [PubMed] [Google Scholar]
- 40.Pappata S, Mazoyer B, Tran Dinh S, Cambon H, Levasseur M, Baron JC. Effects of capsular or thalamic stroke on metabolism in the cortex and cerebellum: a positron tomography study. Stroke. 1990;21:519–524. doi: 10.1161/01.str.21.4.519. [DOI] [PubMed] [Google Scholar]
- 41.Meltzer CC, Cantwell MN, Greer PJ, et al. Does cerebral blood flow decline in healthy aging? A PET study with partial-volume correction. J Nucl Med. 2000;41:1842–1848. [PubMed] [Google Scholar]
- 42.Esiri MM, Wilcock GK, Morris JH. Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry. 1997;63:749–753. doi: 10.1136/jnnp.63.6.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–408. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- 44.Tekin S, Cummings JL. Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002;53:647–654. doi: 10.1016/s0022-3999(02)00428-2. [DOI] [PubMed] [Google Scholar]
- 45.Cummings JL. Anatomic and behavioral aspects of frontal-subcortical circuits. Ann NY Acad Sci. 1995;769:1–13. doi: 10.1111/j.1749-6632.1995.tb38127.x. [DOI] [PubMed] [Google Scholar]
- 46.Reed BR, Eberling JL, Mungas D, Weiner MW, Jagust W. Memory failure has different mechanisms in subcortical stroke and Alzheimer’s disease. Ann Neurol. 2000;48:275–284. [PMC free article] [PubMed] [Google Scholar]
- 47.Rypma B, D’Esposito M. A subsequent-memory effect in dorsolateral prefrontal cortex. Brain Res Cogn Brain Res. 2003;16:162–166. doi: 10.1016/s0926-6410(02)00247-1. [DOI] [PubMed] [Google Scholar]